
Toward the next generation of sustainable aluminum-ion batteries: a review
Jiajin
Zhao
a,
Yan
Chen
a,
Ziqi
An
a,
Mengyan
Zhang
a,
Wenfeng
Wang
a,
Qiubo
Guo
b,
Yuan
Li
a,
Shumin
Han
 *a and
Lu
Zhang
*a
*a and
Lu
Zhang
*a
aHebei Key Laboratory of Applied Chemistry, State Key Laboratory of Metastable Materials Science and Technology, Yanshan University, PR China. E-mail: hanshm@ysu.edu.cn; zhanglu@ysu.edu.cn
bKey Laboratory for Soft Chemistry and Functional Materials, Nanjing University of Science and Technology, Ministry of Education, Nanjing 210094, Jiangsu, China
First published on 22nd November 2024
Abstract
Rechargeable aluminum-ion batteries (AIBs) are regarded as viable alternatives to lithium-ion battery technology because of their high volumetric capacity, low cost, and the rich abundance of aluminum. With the exploitation of high-performance electrode materials, electrolyte systems, and in-depth charge carrier storage mechanism investigation, the electrochemical performances of AIBs have been greatly enhanced; however, researches show that the cathode suffers from insufficient capacity, sluggish reaction kinetics, and poor cycling stability, and the anode also has challenges such as dendrites, passivation, and hydrogen evolution reaction side reactions. Herein, we review the strategies and progress of cathode materials for realizing the advantages in the literature according to the charge storage mechanism for AIBs. Current problems and possible solutions are discussed, and prospects are also proposed. In addition, we analyze recent anode electrode modification strategies and electrolyte modification strategies. Finally, we highlight the current problems and provide an outlook. This review could guide future research and development efforts toward more effective and efficient AIBs.
1. Introduction
Challenged by the vast consumption of fossil fuels and increased greenhouse gas emissions, most countries have set domestic targets to achieve zero carbon or carbon neutrality.1–3 Developing low-carbon energy, optimizing energy structure, and realizing a low-carbon economy have become realistic and imperative choices.4,5 Renewable energy sources, such as solar, tidal, and wind energy, are the most promising energy suppliers for the future.6–8 However, the utilization of intermittent and fluctuating renewable energy requires storage before integration into the grid to avoid power fluctuation.9 Among the various energy-storage technologies, rechargeable batteries present an effective solution for the large-scale grid-connected utilization of renewable energy.10Recently, lithium-ion batteries (LIBs) have been the main power source for electric vehicles (EVs) and portable electronic devices due to their high specific capacity and energy density, and continue to dominate the market.11,12 However, lithium is expensive, with resource depletion, and is in less than 0.1% of the Earth's crust.13,14 In addition, the flammability of the organic electrolytes used brings great safety concerns if the cell is short-circuited.15,16 The above issues hinder LIBs from meeting the calls for large-scale energy storage of low cost and high safety. Therefore, low-cost rechargeable batteries with high performance will be the next-generation batteries to meet the rigid requirements for commercialization. To achieve this goal, novel batteries based on Earth-abundant charge carriers have been expanded and developed, i.e. Na+,17 K+,18,19 NH4+,20,21 Zn2+,22–26 Mg2+,27 Ca2+,28,29 and Al3+.30–32
Among these batteries, aluminum-based batteries (ABs) have attracted much attention because of the high volumetric capacity (8046 mA h cm−3) of Al, which is approximately four times that of LIBs (2062 mA h cm−3) due to the unique three-electron redox reaction (Fig. 1).33–36 Albeit the redox potential of Al (−1.66 V vs. standard hydrogen electrode, SHE) is relatively high compared with Li (−3.04 V vs. SHE) and Na (−2.71 V vs. SHE), it still creates comparable energy to those battery systems using other metals, based on the higher capacity. Additionally, the rich abundance and low cost of Al also offer better opportunities for large-scale applications.
 | ||
| Fig. 1 Comparison of aluminum and other metal anodes in electrochemical systems in terms of gravimetric and volumetric capacities, abundance, and cost. | ||
Fig. 2 illustrates the history of the development of ABs. The origin of ABs can be traced back to 1972 when Holleck and Giner first reported the Al||Cl2 secondary battery with a NaCl/KCl/AlCl3 molten electrolyte (Fig. 2a).37 However, the molten electrolyte requires a high working temperature. The replacement of inorganic salts with organic salts in molten electrolytes can be a good solution to this problem. Due to the ionic flexibility and asymmetry of organic salts, the molecular forces in the electrolyte are significantly reduced, allowing the electrolyte to remain in a liquid state at room temperature, known as room temperature ionic liquids (ILs).38
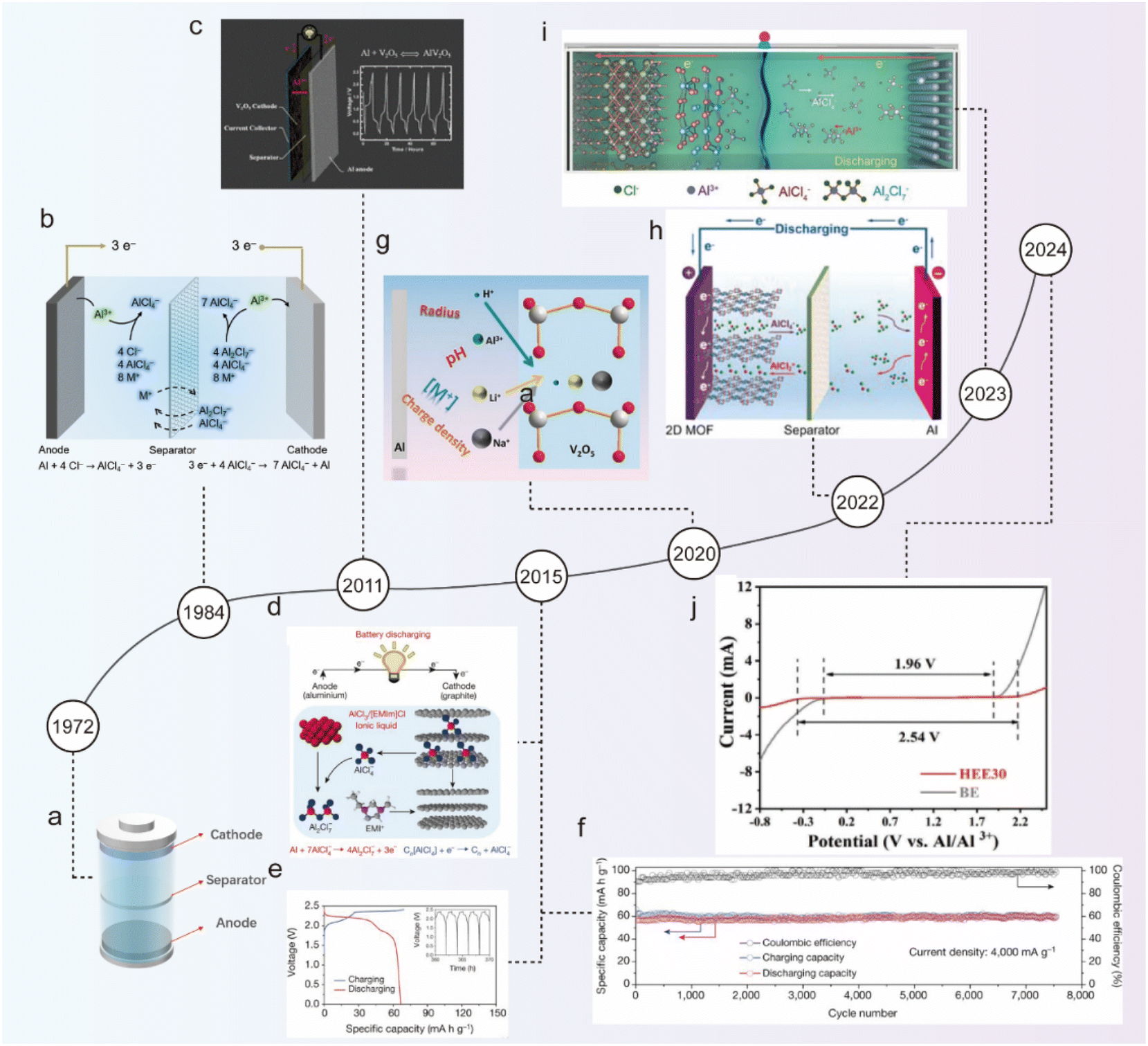 | ||
| Fig. 2 A brief history of AIBs. Schematic illustration of (a) an Al||Cl2 secondary battery with the NaCl/KCl/AlCl3 molten electrolyte, (b) an Al||Al battery with the AlCl3/MEICl ionic liquid electrolyte, and (c) an Al||V2O5 battery with the AlCl3/[EMIm]Cl ionic liquid electrolyte.40 Copyright 2011, The Royal Society of Chemistry. (d) Schematic illustration, (e) galvanostatic charge and discharge curves, and (f) long-term stability test of an Al||graphite battery with the AlCl3/[EMIm]Cl ionic liquid electrolyte.45 Copyright 2015, Springer Nature. (g) Schematic illustration of a reversibly co-inserted Al3+/H+ in an Al||V2O5 battery with the 2 M Al(OTf)3 aqueous electrolyte.46 Copyright 2020, Wiley-VCH. (h) Schematic illustration of an Al||MOF battery with the AlCl3/[EMIm]Cl ionic liquid electrolyte.47 Copyright 2022, Wiley-VCH. (i) Schematic illustration of an Al||CoSe2/MXene battery with the AlCl3/[EMIm]Cl ionic liquid electrolyte.48 Copyright 2023, Wiley-VCH. (j) LSV curves of the BE and HEE30 electrolytes at a scan rate of 10 mV s−1.49 Copyright 2024, Wiley-VCH. | ||
In 1984, Dymek et al. first achieved reversible Al3+ plating and stripping at room temperature using an ionic liquid composed of 1-methyl-3-ethylimidazolium chloride (MEICl) and aluminum chloride (AlCl3) as an electrolyte for ABs (Fig. 2b).39 The acidic ionic liquid can remove the surficial passivation film of the Al anode and promote the rapid transport of Al3+, successfully revolutionizing the electrolyte for ABs. In 2011, Jayaprakash et al. pioneered the use of layered V2O5 as a cathode material for Al, transferring Al3+, thus forming aluminum-ion batteries (AIBs) and extending the cycling life to 20 cycles (Fig. 2c).40 Although Wen et al. later confirmed that V2O5 was not stable in AlCl3/1-ethyl-3-methylimidazolium chloride ([EMIm]Cl) ionic liquid, the work still provided valuable insight for AIBs.41 Since then, numerous metal oxide cathode materials had emerged in the field, such as WO3,42 TiO2,43 MoO3,44etc.
In 2015, Dai's team developed and reported graphite-based materials as the cathode for AIBs with outstanding electrochemical performance, representing a breakthrough and indicating a bright future for AIBs.45 The graphite-based materials with three-dimensional foam structures allowed reversible (de)insertion of the [AlCl4]− anion charge carrier (Fig. 2d), providing a high discharge plateau of ∼2 V with a high capacity of 70 mA h g−1 when paired with Al foil anode (Fig. 2e). Furthermore, the battery achieved robust long-term cycling stability, nearly perfectly maintaining its specific capacity over 200 cycles with a coulombic efficiency (CE) of 98.1 ± 0.4% at a current rate of 66 mA g−1 (1 C) (Fig. 2f). Based on Dai's work, a series of cathode materials have been developed. Zhao et al. reported an aqueous Al-V2O5 battery, in which H+ and Al3+ were co-inserted into the V2O5 cathode by adjusting the pH and composition of the electrolyte.46 Further studies revealed that the cation (H+, Li+, Na+, etc.) (de)intercalation chemistry can be rationally designed (Fig. 2g). Guo et al. developed a metal–organic framework (MOF) cathode material with bipolar ligands, which provided reactive sites for both [AlCl4]− and [AlCl2]+ charge carriers.47 As a result, the battery doubled the capacity of the single-stage ligand to 184 mA h g−1 and achieved an energy density of 177 W h kg−1 (Fig. 2h). Yuan et al. further studied a CoSe2/MXene composite as a cathode for AIBs (Fig. 2i),48 and found that MXene as a support material reduced the size of CoSe2 growing on its surface, which effectively inhibited the lattice distortion caused by the interaction with [AlCl4]−, addressing the issues of poor reversibility, cycling instability, and low CE. Recently, Zhang et al. proposed a eutectic electrolyte HEE30 composed of Al(OTf)3, glycerol (Gly), sodium beta-glycerophosphate pentahydrate (SG), and H2O with a molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :8:1:30, in which the unique eutectic network significantly enhanced the hydrogen bonding between Gly and H2O, reducing the solvation interaction of Al3+ with active H2O, extending electrochemical windows and suppressing HER (Fig. 2j).49 Therefore, the Al||Prussian white cell exhibited capacity retention of 72% after 500 cycles at 0.5 A g−1, and maintained a high capacity of 109 mA h g−1 after 200 cycles at a low current rate of 0.1 A g−1.
:8:1:30, in which the unique eutectic network significantly enhanced the hydrogen bonding between Gly and H2O, reducing the solvation interaction of Al3+ with active H2O, extending electrochemical windows and suppressing HER (Fig. 2j).49 Therefore, the Al||Prussian white cell exhibited capacity retention of 72% after 500 cycles at 0.5 A g−1, and maintained a high capacity of 109 mA h g−1 after 200 cycles at a low current rate of 0.1 A g−1.
The working mechanisms of AIBs are currently classified into two main categories: the insertion and the conversion mechanisms. The insertion mechanism itself can be divided based on the type of guest ions involved, cations or anions. For the cation-based insertion mechanism, the guest ions include Al3+, [AlCl]2+, or [AlCl2]+, characteristic of the so-called rocking chair battery (RCB). For the anion-based insertion mechanism, the guest ions are [AlCl4]−/[Al2Cl7]−, representing the dual-ion battery (DIB). On the other hand, the conversion mechanism involves the conversion of elements, such as the sulfide and iodine materials, through interactions with Al3+.
For the insertion mechanism of AIBs, Al3+ can be reversibly inserted into most transition metal oxides, transition metal sulfides, etc. through the following processes:
Cathode:
| MxNy + nAl3+ + 3ne− ↔ AlnMxNy |
Anode:
| Al ↔ Al3+ + 3e− |
In addition, [AlCl]2+/[AlCl2]+ cations are usually found as the stored charge carrier in organic materials, such as conductive polymers, which is
Cathode:
| M + n[AlCl]2+ + 2ne− ↔ AlClnM |
Anode:
| 4[Al2Cl7]− + 3e− ↔ 7[AlCl4]− + Al |
On the other hand, [AlCl4]− can act as the host anion in graphite-based cathode materials through (de)insertion processes as follows, and the anode undergoes the conversion of [AlCl4]−/[Al2Cl7]− with Al, thereby forming the DIB:
Cathode:
| M + [AlCl4]− ↔ M[AlCl4] + e− |
Anode:
| 4[Al2Cl7]− + 3e− ↔ 7[AlCl4]− + Al |
In conversion materials, the valence change of the host materials usually accompany the formation of AlMx by binding with Al in the carrier, commonly using materials such as sulfur and iodine. The reaction mechanism can be expressed as follows:
Cathode:
| xM + 4[Al2Cl7]− + 3e− ↔ AlMx + 7[AlCl4]− |
Anode:
| 7[AlCl4]− + Al ↔ 4[Al2Cl7]− + 3e− |
Currently, almost all AIBs fall short of achieving high energy density due to the limited capacity of existing cathodes. Developing new high-capacity cathode materials and achieving a higher discharge voltage are crucial for energy density enhancement. Additionally, a stable cathode structure is essential for battery longevity. Although Al anodes provide adequate capacity and voltage, their low standard electrode potential makes them susceptible to HER, along with challenges like corrosion, passivation, and dendrite formation. This review offers a comprehensive overview of cathode material development for AIBs categorized by the working mechanism, anode modification, and electrolyte optimization. We also discuss the strategies to improve electrochemical performances and propose future perspectives toward advancing AIB technology.
2. Cathode materials
2.1 Transition metal oxides
Transition metal oxides (TMO), e.g. Mn-based oxides MnO2 and V-based oxides V2O5, are among the most commonly utilized cathodes in versatile energy storage devices. Table 1 shows the typical electrochemical performance of AIBs on the TMO cathodes. Identified as excellent hosts for the Al3+ cation, transition metal oxides are frequently employed as cathodes for AIBs, through a rocking chair mechanism. The specific process can be expressed as follows, where M represents a transition metal.| Cathode material | Electrolyte | Specific capacity (mA h g−1) | Cycle number | Coulombic efficiency | Ref. |
|---|---|---|---|---|---|
| AlxMnO2 | 2 M Al(OTF)3 aqueous electrolyte | 460 | 80 | 95% | 31 |
| AlxMnO2 nanosphere | AlCl3·6H2O:MnSO4·6H2O:H2O = 4:1:1 aqueous water-in-salt electrolyte |
285 | 500 | 95% | 77 |
| MnO2 | 4.4 M AlCl3:1 M MnCl2 saturated aqueous electrolyte |
493 | 1000 | 97% | 78 |
| δ-MnO2 nanofibers | [EMIm]Cl and AlCl3 (1:1 in weight) IL electrolyte |
59 | 100 | 99% | 79 |
| α-MnO2 | 2 M Al(CF3SO3)3 aqueous electrolyte | 380 | 100 | 96% | 80 |
| Vanadium-doped δ-MnO2 | 2 M Al(OTF)3 aqueous electrolyte | 518 | 400 | — | 51 |
| Bir-MnO2 (first cycle), AlxMn(1−-x)O2 (following cycles) | 2 M Al(OTF)3 + 0.5 M MnSO4 aqueous electrolyte | 554 | 65 | — | 81 |
| V2O5 nanofibers | AlCl3/[EMIm]Cl (molar ratio of 1.5:1) IL electrolyte |
105 | 50 | 81% | 82 |
| Interconnected sheet-like V2O5 | 0.5 M AlCl3 aqueous electrolyte | 140 | 1000 | 94% | 58 |
| AmorphousV2O5/C composite | AlCl3, dipropylsulfone and toluene (1:10:5) in mole ratio |
200 | 30 | — | 83 |
| Anatase TiO2 nanorods | AlCl3:[EMIm]Cl (2:1 molar ratios) IL electrolyte |
165 | 150 | 99.9% | 68 |
| WS2/WO3 | — | 282 | 100 | 97% | 84 |
| WO3−x nanorods | AlCl3/[EMIm]Cl (molar ratio of 1.3:1) IL electrolyte |
118.9 | 100 | 75% | 85 |
| WO3 | 1 M AlCl3 aqueous electrolyte | 170 | 500 | 100% | 86 |
| Co3O4@MWCNTs | AlCl3 and [EMIm]Cl (molar ratio of 1.3:1) IL electrolyte |
266.3 | 150 | 99.1% | 87 |
| Nanosphere-rod-like Co3O4 | AlCl3/[EMIm]Cl (molar ratio of 1.3:1) IL electrolyte |
490 | 100 | 80–90% | 71 |
| MoO3 | 1 M AlCl3 aqueous electrolyte | 680 | 350 | — | 44 |
| Porous CuO microsphere | AlCl3:[EMIm]Cl (molar ratio of 1.3:1) IL electrolyte |
250.1 | 100 | 99.5% | 72 |
Cathode:
| MxOy + nAl3+ + 3ne− ↔ AlnMxOy, |
Anode:
| Al ↔ Al3+ + 3e− |
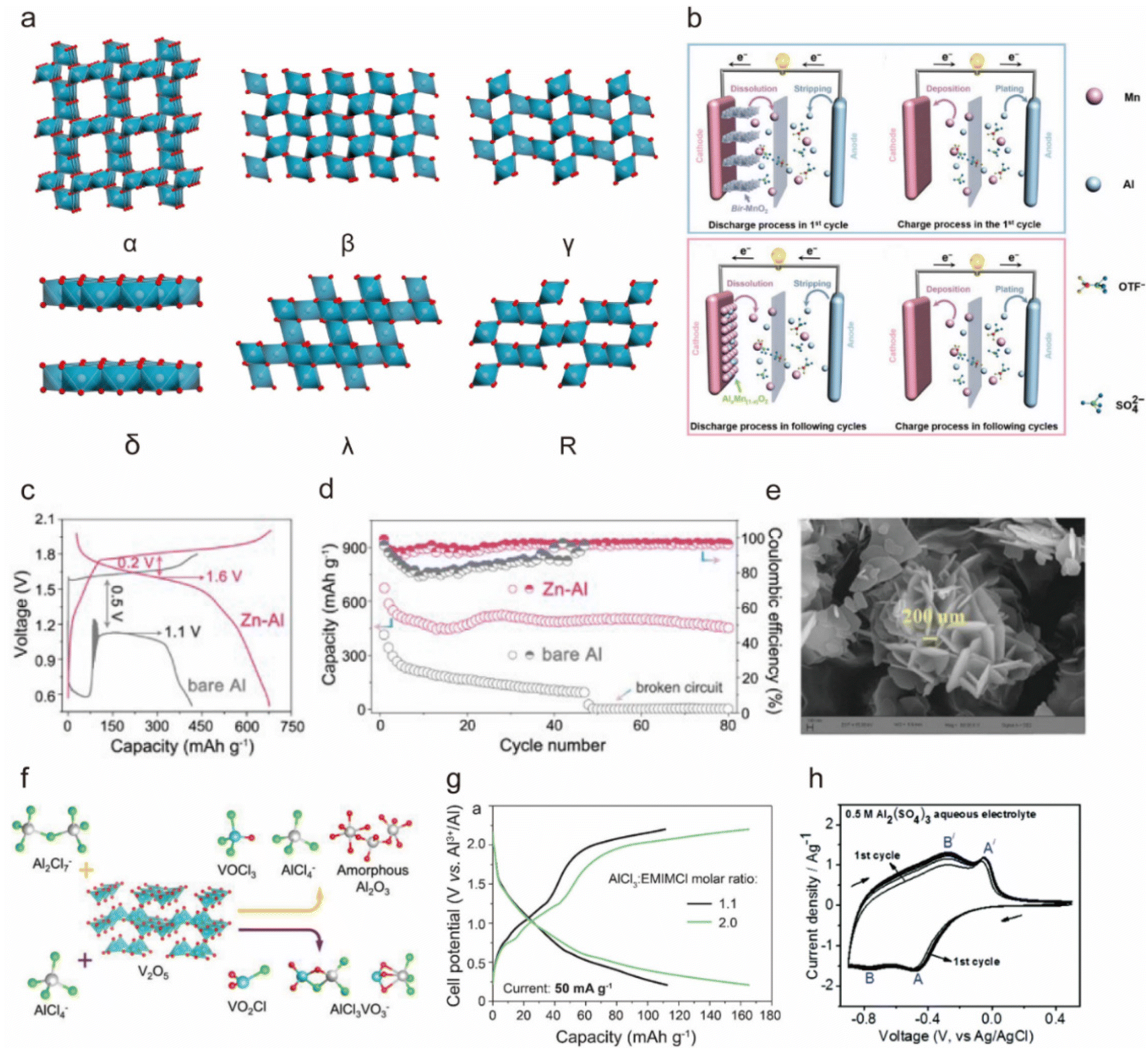 | ||
| Fig. 3 (a) Schematic of the crystal structure of different types of MnO2. (b) The schematic electrochemistry diagram of the Al||MnO2 batteries during the first discharge and discharge processes. Copyright 2019, Wiley-VCH. (c) First galvanostatic discharge and charge (GCD) curves, and (d) cycling performance at a current rate of 100 mA g−1 of the Al||AlxMnO2 and Zn-Al||AlxMnO2 batteries in the 2 M Al(OTF)3 electrolyte.31 Copyright 2020, American Chemical Society. (e) Scanning electron microscope (SEM) image of the synthesized V2O5 nanosheets.58 Copyright 2023, American Chemical Society. (f) Schematic illustration of the product of the reaction of V2O5 with the [AlCl4]− and [Al2Cl7]− charge carriers.41 Copyright 2019, American Chemical Society. (g) GCD curves of the TiO2 NRs at the 3rd cycle at a current rate of 50 mA g−1 in mild (1.1:1 molar ratio) and acidic (2.0:1 molar ratio) AlCl3/EMImCl ionic liquid electrolytes.68 Copyright 2019, American Chemical Society. (h) CV curves of WO3 in the 0.5 M Al2(SO4)3 aqueous electrolyte.69 Copyright 2019, The Royal Society of Chemistry. | ||
In 2018, Archer et al. proposed α-MnO2 nanorods as the cathode for an AIB in a 2 M Al(CF3SO3)3 aqueous electrolyte and reported that the one-dimensional nanostructure of α-MnO2 facilitated fast transport of Al3+, resulting in a specific capacity of ∼380 mA h g−1 and a high specific energy of 500 W h kg−1.52 However, the battery exhibited capacity degradation due to the dissolution of low-valence Mn discharge products from the cathode, necessitating further mechanistic explanation. To delve into the specific insertion process of Al3+ into α-MnO2, Alfaruqi et al. conducted first-principle calculation based on density functional theory (DFT) and pointed out that the insertion involved four stages and induced structural distortion of MnO2.53
To inhibit the dissolution of Mn-based materials, Yu's team utilized 0.5 M MnSO4 as an additive in 2 M Al(OTF)3 aqueous electrolyte,54 and AlxMn1−xO2 was produced during charging, acting as a reversible cathode active material in the subsequent cycles (Fig. 3b). As a result, the cell maintained a capacity of 320 mA h g−1 after 65 cycles, surpassing the cell without pre-added Mn2+. In 2020, Yan et al. also utilized AlxMnO2 as a cathode for AIBs by implementing a Zn–Al anode, and they revealed that the battery generated a high discharge voltage plateau of ∼1.6 V, a specific capacity of 460 mA h g−1, and prolonged cycling life to 80 cycles in an aqueous Al(OTF)3 electrolyte because the addition of Zn mitigated passivation and self-discharge on the anode side (Fig. 3c and d).31
In addition, ion doping can significantly strengthen MnO2 frameworks. Huang et al.51 reported that V-doped δ-MnO2 not only reinforced structural stability due to high bond dissociation energy with oxygen but also enhanced the cohesive energy of δ-MnO2, improved interaction with Al3+, and boosted electrical conductivity. As a result, the Al–Cu||V-δ-MnO2 in 2 M Al(OTf)3 delivered a high specific capacity of 518 mA h g−1 at 200 mA g−1 with remarkable cycling stability for 400 cycles and impressive rate capabilities of 468, 339, and 285 mA h g−1 at 0.5, 1, and 2 A g−1, respectively.
Albeit MnO2 employed as the cathode for AIBs presents high specific capacity and energy density, the dissolution of discharge products and structural collapse during cycling because of the crystal structure transformation of different types of MnO2 are still the key issues to be resolved. Therefore, future research still should focus on the improvement of the cycle life of MnO2.
Nanosizing has proved to be an effective method to enhance the crystal structure stability of V2O5. Chandra's team demonstrated that the interconnected sheet-like morphology of V2O5 nanosheets as a cathode for AIBs could increase specific surface area and support the robust crystal structure, leading to excellent storage capacity (Fig. 3e).58 As a result, the lattice expansion of the V2O5 electrode was nearly negligible over 20 cycles, and the battery delivered an output of ∼140 mA h g−1 at 0.5 A g−1, with excellent capacity retention of 96% even after 1000 cycles at 1 A g−1.
Composite materials can significantly enhance the stability of V2O5 and improve the efficiency of electrochemical reactions. Recently, Li et al. designed and constructed an exceptionally effective cathode featuring dual morphologies. In this innovative approach, two-dimensional (2D) layered MXene materials, known for their excellent electrical conductivity and hydrophilicity, served as substrates for the deposition of rod-shaped V2O5, thereby creating a three-dimensional (3D) cathode structure. Both Al3+ and H+ ions demonstrated rapid polynomial conduction along the ab-axis and c-axis of V2O5. The V2O5@MXene composite facilitated facile electrolyte access, which was crucial for efficient reactions. Leveraging this composite architecture, the Al||V2O5@MXene system in a 5 M Al(OTf)3 electrolyte achieved an outstanding initial specific capacity of 626 mA h g−1 at a current rate of 0.1 A g−1, coupled with stable cycling performance over 100 cycles.
Other than V2O5, other vanadium compounds such as VO2 and FeVO4 have also been utilized as the cathode materials for AIBs. Wang et al. reported that a 3D interconnected tunneling structure of strip VO2 facilitated rapid Al3+ (de)intercalation, giving a high specific capacity of 235 mA h g−1 at 200 mA g−1.59 Kumar et al. focused on FeVO4 and achieved a high specific capacity of 350 mA g−1. However, the FeVO4 underwent conversion reactions with Al3+ instead of the insertion mechanism, leading to the generation of the AlxVyO4 spinel phase and amorphous Fe–O–Al phase during charging.60
On the other hand, Reed et al. contested the feasibility of using V2O5 as a cathode material for AIBs because they found that V2O5 exhibited a lack of electrochemical activity for Al3+.61 They observed that the battery-like charge–discharge behavior of V2O5 in the acidic AlCl3/[EMIm]Cl chloride ionic liquid electrolyte was due to the redox reaction of iron and chromium on the current collector. In addition, Wen et al. studied the compatibility between V2O5 and chloroaluminate and concluded that they were skeptical about the application of V2O5 cathode material in AIBs.41 DFT calculation showed that V2O5 reacted chemically in AlCl3-[EMIm]Cl electrolyte both with the Lewis neutral substance [Al2Cl7]− to produce vanadium trichloro oxide (VOCl3), [AlCl4]−, and amorphous Al2O3, and with the Lewis acidic substance [AlCl4]− to produce vanadium oxychloride (VO2Cl) and a new species of metavanadate anion coordinated with aluminum chloride (AlCl3VO3−), as shown in Fig. 3f.
In summary, vanadium-based oxide assembly of AIBs has achieved certain results with the advantages of high specific discharge capacity and high energy density. Nonetheless, despite enhancement of the structural stability of V2O5 through nanosizing and composite strategies, performance is constrained by the material's tendency to dissolve at low current rates. Additionally, a lack of consensus exists regarding the viability of V2O5 for use in AIBs. Consequently, further investigation into the mechanism that could fundamentally enhance battery performance is warranted.
:EMIMCl in 1.1:1 and 2.0:1 electrolytes, respectively (Fig. 3g).
TMOs as the host of the Al3+ charge carrier show exciting capacity and energy density. However, the Al3+ insertion process remains a challenge in terms of large-scale applications due to the high charge density around Al3+ and poor cycling stability resulting from the structural collapse of the host material after repeated (de)intercation of charge carriers. To address the issues, ion doping and structural nanosizing are effective approaches which can improve structural stability. On the other hand, TMOs have low electrical conductivity, therefore they are usually combined with carbon materials to enhance the electrical conductivity to improve the electrochemical properties.
2.2 Transition metal sulfide
Compared with metal oxide, transition metal sulfides (MxSy) are easier to polarize than oxides, thus making Al3+ more favorable for transport due to the lower electronegativity and larger ionic radius of S.73,74 The (de)insertion of Al3+ in the MxSy host can be expressed as:Cathode:
| MxSy + nAl3+ + 3ne− ↔ AlnMxSy |
Anode:
| Al ↔ Al3+ + 3e− |
In 2016, Yu et al. reported a new hexagonal NiS nanobelt as the cathode of AIBs, which could deliver a capacity of ∼110 mA h g−1 in the AlCl3-[EMIm]Cl electrolyte and exhibited a capacity of 100 mA h g−1 after 100 cycles because the nanobelt structure facilitated the entry and diffusion of Al3+ (Fig. 4a and b).75 However, it presented a low discharge voltage plateau (∼1.15 V vs. Al3+/Al). Wang et al. composited Ni3S2 with graphene to improve the (de)intercalation of Al3+ and showed that the cathode showed an initial specific capacity of 350 mA h g−1 at 100 mA g−1 in the AlCl3/[EMIm]Cl ionic liquid electrolyte.76
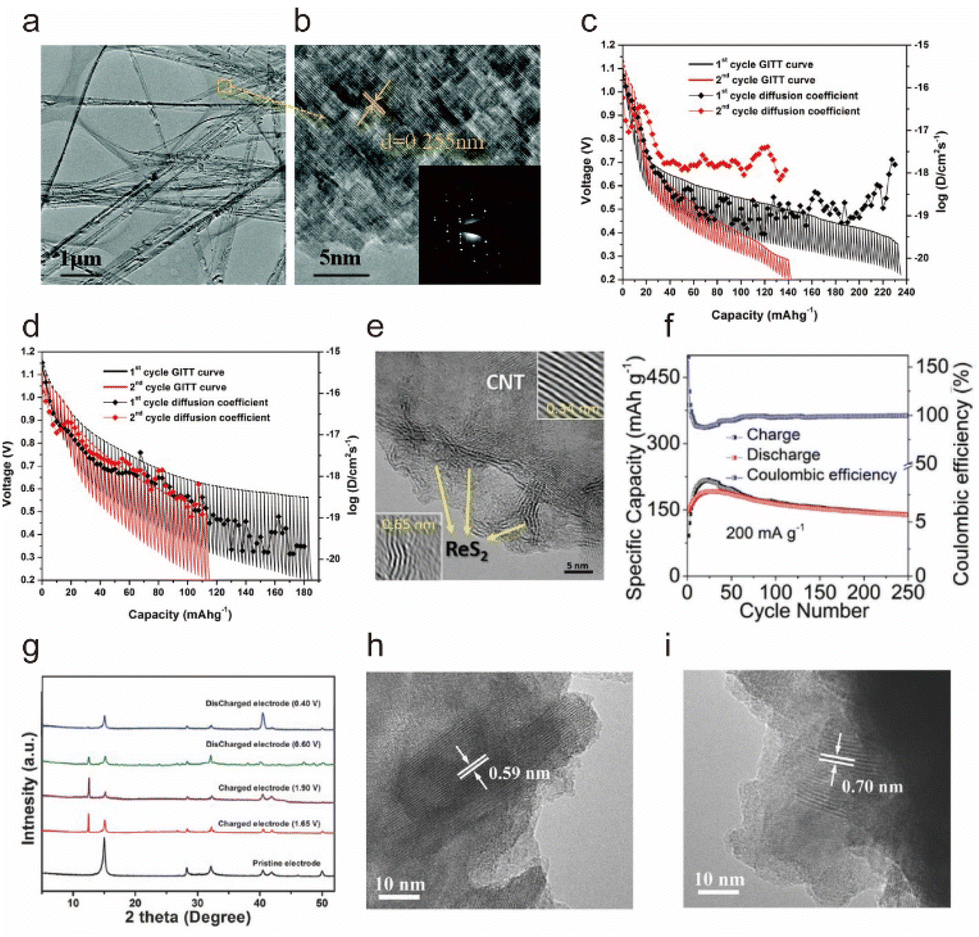 | ||
| Fig. 4 (a) Transmission electron microscopy (TEM) and (b) High-resolution transmission electron microscopy (HRTEM) images of NiS nanobelts and the corresponding selected area electron diffraction (SAED) pattern in the inset at the lower right corner.75 Copyright 2016, The Royal Society of Chemistry. Galvanostatic intermittent titration technique (GITT) curve of the (c) layered TiS2 and (d) cubic Cu0.31Ti2S4 at 50 °C.73 Copyright 2017, American Chemical Society. (e) HRTEM image of the ReS2@CNTs. (f) Cycling stability of the ReS2@CNTs at 0.2 A g−1.90 Copyright 2022, Wiley-VCH. (g) X-ray diffraction (XRD) patterns of the SnS2 electrodes at various electrochemical measurement stages: pristine, the end of charge platform (1.65 V), charged (1.90 V), the end of discharge plateau (0.60 V), and discharged (0.40 V) states. (h and i) HRTEM image of the electrode at pristine, and charged (1.90 V) states.91 Copyright 2017, Wiley-VCH. | ||
Geng et al. conducted extensive simulation speculations as well as an experimental demonstration of layered TiS2 and spinel Cu0.31Ti2S4 as the cathodes for AIBs, which showed capacities of 50 mA h g−1 and 95 mA h g−1, respectively.73 In addition, they inferred that the kinetic potential barrier of Al3+ diffusion in sulfide was caused by the strong Coulomb force attraction between Al3+ and sulfide anion skeleton (Fig. 4c and d), which was the main obstacle of Al3+ intercalation chemistry. Subsequently, Li et al. developed a porous Co3S4 microsphere to accommodate Al3+via the redox transition of the Co3+/Co2+ couple, showing a discharge capacity of ∼288 mA h g−1.88 It was reported that the (de)intercalation of Al3+ into the Co3S4 included surface (de)intercalation and bulk diffusion, of which the solid phase diffusion step was considered to be the key factor limiting the discharge ability at high discharge/charge current densities.
To alleviate the Coulomb force and facilitate Al3+ transition, Wu et al. constructed an interlayer-expanded MoS2/N-doped carbon (MNC) with a 3D hierarchical tremella structure with a large interlayer spacing up to 0.82 nm through a hydrothermal treatment and calcination, which reduced the diffusion path for Al3+, boosted the diffusion of Al3+ and provided more active sites.89 Meanwhile, the N-doped carbon promoted electronic conductivity and maintained structural integrity during cycles. The Al||MNC battery in an AlCl3/[EMIm]Cl (1.3:1) electrolyte presented a capacity of 127.5 mA h g−1 after 1700 cycles at 1 A g−1 with a CE of 99.5%. Zhang et al. also reported 2D ultra-thin ReS2 nanosheets on carbon nanotubes (CNTs) as an outstanding cathode (ReS2@CNTs) for the AIBs.90 As ReS2 featured a large interlayer spacing of ∼0.65 nm (Fig. 4e), the extremely weak interlayer coupling could effectively reduce the electrostatic repulsion with Al3+, adequately accommodating large amounts of Al3+ without significant volume expansion. The Al||ReS2@CNTs battery in an AlCl3/[EMIm]Cl (1.3:1) electrolyte delivered a capacity of 396.3 mA h g−1 and a CE of ∼100% after 250 cycles at a low current rate of 200 mA g−1 (Fig. 4f).
Noticeably, transition metal sulfides can not only host Al3+ cation charge carrier but also store [AlCl4]− anion with the following mechanism:
Cathode:
| M + n[AlCl4]− ↔ ne− + M[AlCl4]n |
Anode:
| 4[Al2Cl7]− + 3e− ↔ Al + 7[AlCl4]− |
Hu et al. reported that layered SnS2 nanosheets anchored on a 3D reduced graphene oxide network worked on the reversible (de)intercalate [AlCl4]− anion, as proved by XRD and HRTEM (Fig. 4g–i).91 The graphene-loaded SnS2 nanosheet not only offered high electronic conductivity but also fast kinetic diffusion pathways for [AlCl4]− (de)intercalation, which presented a high specific capacity (392 mA h g−1 at 100 mA g−1) and good rate performance (112 mA h g−1 at 1000 mA g−1). In addition, Liang et al. obtained a specific capacity of 406 mA h g−1 for self-supported SnS porous films as a cathode and focused on flexible batteries, and they found that the unique porous structure provided good cycling stability due to the reduced volume change during (dis)charge with a capacity decay rate of only 0.03% per cycle.92 It is interesting that the capacity of batteries with [AlCl4]− as the carrier is generally higher than that of Al3+ as the carrier, which may be caused by the incomplete utilization of Al3+ and the large Coulomb force.
In summary, MxSy have garnered significant attention in the field of secondary batteries due to their unique layered structure, large specific surface area, and fast ion diffusion properties. However, their energy storage mechanism is complex. The (de)insertion involving multivalent Al3+ theoretically offers high specific capacity and energy density. Nevertheless, the kinetics is limited by the strong Coulomb interaction between Al3+ and the host material, which creates a substantial-high energy potential barrier. On the other hand, the reaction based on single-valent [AlCl]4−, with its relatively large size, can damage the structure of the host material during the (de)insertion process, leading to poor cycling performance. Therefore, future research still should focus on modifying cathode materials.
2.3 Prussian blue analogs
Prussian blue analogs (PBAs), with a chemical formula of AxMy[B(CN)6]z·mH2O, where A and B are alkaline metals and M is a transition metal,93 are a class of materials with a cubic structure that contains a large number of tunnels and voids (4.6 Å in diameter), enabling rapid solid-state diffusion of various carrier ions.94,95 Moreover, the open framework structures undergo very little change upon insertion of ions, therefore the batteries with such materials as cathodes usually have good cycle life.96–98 PBAs are based on the redox reaction of transition metals M (such as Fe, Co, Mn) with reversible (de)intercalation of Li+, Na+, and K+ into these vacancies.99–101 However, it is more difficult for Al3+ to enter into the host because of the stronger Coulomb forces.In 2015, Liu et al. first reported copper hexacyanoferrate (CuHCF) nanoparticles featuring Al3+ insertion sites using Fe3+/Fe2+ as the redox couple (Fig. 5a), delivering a specific capacity of 62.9 mA h g−1 in a 0.5 M Al2(SO4)3 aqueous electrolyte.102 However, in aqueous electrolytes, only a single active site in the PBAs is activated and limited by the single electrochemical group Fe(CN)63−, resulting in an actual discharge capacity of only ∼60 mA h g−1, which is much lower than its theoretical capacity of 170 mA h g−1.103,104
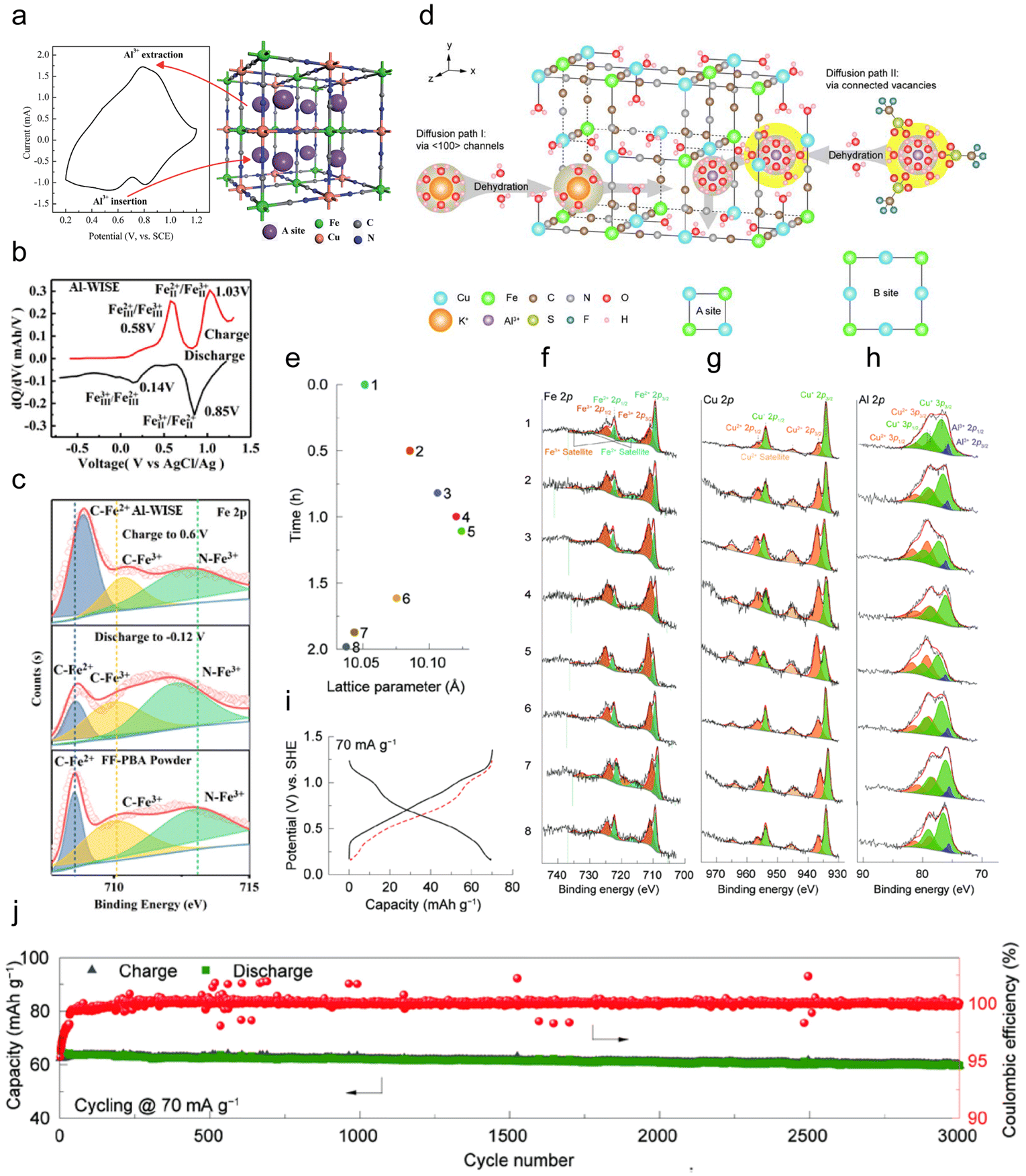 | ||
| Fig. 5 (a) CV curve and structure of the CuHCF nanoparticle.102 Copyright 2015, The Royal Society of Chemistry. (b) The differential profiles at the 50th cycle, and (c) XPS spectrum of the FeFe(CN)6 electrode in different charge–discharge states (pristine, discharge to −0.12 V and charge to 0.6 V) in the 5 M Al(CF3SO3)3 water-in-salt electrolyte.105 Copyright 2019, American Chemical Society. (d) Crystal structure of the CuHCFe with two possible diffusion paths. Diffusion path I was through 〈100〉 channels, and path II was through enlarged open channels by the connected ferro- and ferri-cyanide vacancies. (e–h) XPS patterns of the Fe 2p (f), Cu 2p (g), and Al 2p (h) for the electrodes. (i) GCPL curve at a current rate of 70 mA g−1. The red dash line was the flipped discharge curve. (j) Long-term stability at 70 mA g−1.106 Copyright 2023, Elsevier B.V. | ||
Zhou et al. synthesized FeFe(CN)6 from K3Fe(CN)6, featuring two active sites with Fe exhibiting variable valences at different positions as a cathode material for AIBs, which showed two pairs of redox peaks at 0.2 V/0.6 V and 0.8 V/1.1 V (Fig. 5b).105 X-ray photoelectron spectroscopy (XPS) demonstrated a significant binding energy shift to a lower position in the Fe 2p spectrum when discharged to −0.12 V, which returned to a higher position after charging to 0.6 V, indicating the valence change of Fe at both active sites (Fig. 5c). A high specific capacity of 116 mA h g−1 was obtained due to the removal of K from FeFe(CN)6, which provided more vacant sites for Al3+ storage. Zhang et al. also developed a graphite-coated Co[Fe(CN)6] cathode with two active sites, i.e. Co3+/Co2+ and Fe3+/Fe2+, achieving a superior discharge capacity of 372 mA h g−1 with excellent long-cycle performance in which only 0.7% capacity decayed per cycle with a CE of 94.1%.107
Recently, Li et al. proposed a defective copper CuHCFe with ferro- and ferri-cyanide vacancies, and proved multiple insertion pathways of Al3+, through the (100) channels along the x-axis with a one-step dehydration process, through the connected vacancies corresponding to several-step dehydration processes, or both (Fig. 5d).106 Furthermore, ex situ XPS demonstrated that the CuHCFe underwent a reversible redox reaction with Al3+ (de)intercalation (Fig. 5e–h). Fe2+ and Cu+ were oxidized to Fe3+ and Cu2+ during charge (states 1–4), respectively, resulting in a decrease in the intensity of Al3+. In the subsequent discharge (states 5–8), Fe3+ and Cu2+ were reduced to Fe2+ and Cu+, respectively, showing an enhancement of the Al3+ 2p peak. The results showed that the cathode delivered a reversible capacity of ∼70 mA h g−1 at 70 mA g−1, and presented remarkable cycling stability with negligible capacity loss over 3000 cycles (Fig. 5i and j).
To summarize, the three-dimensional structure PBAs provide notable capacity and cycling performance due to the following reasons: (1) the wide-channel nanostructure increases electrode–electrolyte interface contact, ensuring rapid ion conduction; (2) numerous tunnels and voids provide multiple active sites, boosting ion storage; (3) their distinct skeleton structure allows ample space for Al3+, averting structural collapse. Nevertheless, PBAs face challenges in AIB applications, such as the strong charge effect of Al3+ insertion limiting ion diffusion, and generally low capacity from incomplete site utilization. Developing PBAs with more active sites is promising, but current methods fall short of commercialization needs. However, given that research is still in its early stages, significant opportunities for advancement remain.
2.4 Organic compounds
Organic materials have sparked extensive research interest as electrode materials due to their accessibility from abundant precursor materials, potential for sustainable production, low cost, good cycling stability, and eco-friendliness.46,108–110 Unlike inorganic materials, the charge storage mechanism of organic materials relies on the charge states of their active functional groups.111 Based on the redox reaction mechanism, organic materials can be categorized into n-type, p-type, and bipolar-type. As shown in Fig. 6a, n-type materials undergo reduction reactions with cations (e.g. Al3+, [AlCl]2+, H+⋯), allowing them to function as anodes. Conversely, p-type materials participate in oxidation reactions with anions ([AlCl4]−), typically serving as cathodes due to their higher reaction potential compared with n-type materials. Bipolar-type materials exhibit characteristics of both n-type and p-type electrodes, capable of undergoing both oxidation and reduction reactions.112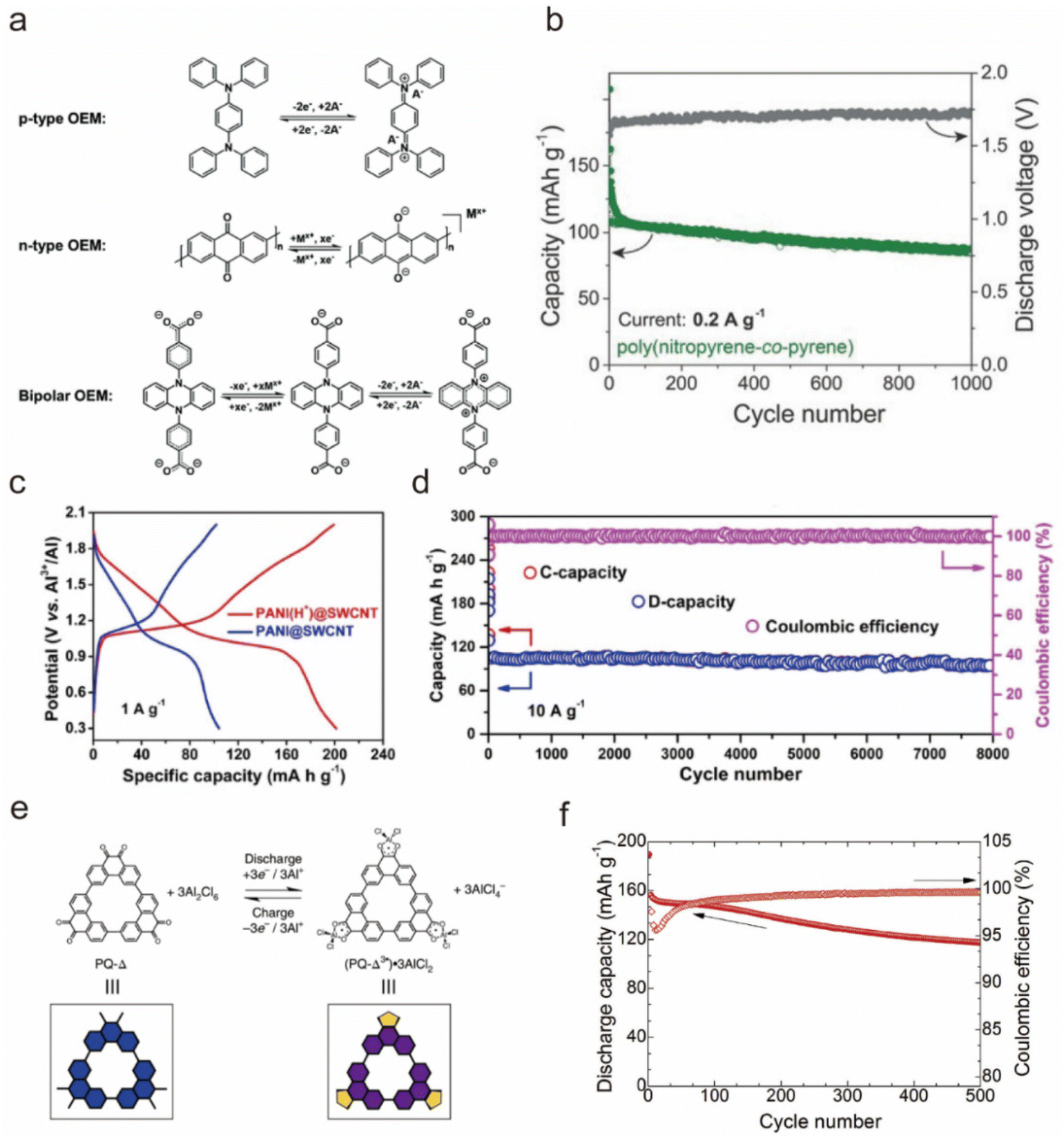 | ||
| Fig. 6 (a) The reaction mechanisms for n-type, p-type, and bipolar organic electrode materials.112 Copyright 2020, The Royal Society of Chemistry. (b) The capacity retention upon cycling and average discharge voltages of poly(nitropyrene-co-pyrene) as a cathode material for AIBs in the AlCl3/[EMIm]Cl IL electrolyte.118 Copyright 2018, Wiley-VCH. (c) First discharge/charge curves and (d) Cycling stability of an Al||PANI battery in the AlCl3/[EMIm]Cl IL electrolyte.119 Copyright 2020, Wiley-VCH. (e) Electrochemical redox chemistry of the PQ-Δ (blue) and its schematic representation in the AlCl3/ethyl-3-methylimidazolium tetrachloroaluminate (EMImAlCl4) electrolyte.121 (f) Discharge capacity and coulombic efficiency of the PAQS/MWCNT electrode in the AlCl3/[EMIm]Cl IL electrolyte at 0.5 C and 0.4–1.8 V.122 Copyright 2020, Elsevier B.V. | ||
:EMIC with a 1.5:1 molar ratio, achieving capacities close to theoretical values of 30–100 mA h g−1 with nearly 100% CE upon insertion of [AlCl4]−.117 Notably, the poly(polythiophene) cathode achieved an energy density of 44 W h kg−1, indicating potential for large-scale energy storage. Furthermore, Walter et al. compared the electrochemical properties of polypyrene and poly(nitropyrene-co-pyrene) cathodes, and revealed that poly(nitropyrene-co-pyrene) in the AlCl3-[EMIm]Cl ionic liquid electrolyte could run for 1000 cycles with an average capacity of 100 mA h g−1 at 200 mA g−1, higher than the neat polypyrene (70 mA h g−1) or crystalline pyrene (20 mA h g−1), and reached a high discharge voltage plateau of 1.7 V (Fig. 6b) with [AlCl4]− as the carrier.118
Conductive polymers have excellent electrical conductivity, mechanical strength, and chemical stability; however, the few effective redox sites result in a relatively low specific capacity. Therefore, it is crucial to effectively increase the active sites. Niu et al. utilized single-walled carbon nanotubes to prepare a composite film with polyaniline to enhance the active sites of polyaniline through the protonation of polyaniline nanorods (PANI).119 Protonating the polyaniline nanorods provided additional accommodation for the reversible (de)insertion of [AlCl2]+ cations in AlCl3/[EMIm]Cl electrolyte. The protonated polyaniline nanorods exhibited a specific capacity of ∼200 mA h g−1, which was twice that of the unprotonated one, and sustained 8000 cycles with only 0.003% capacity attenuation per cycle and an approximate CE of 100% (Fig. 6c and d). Zhang's team prepared bipolar conjugated poly(2,3-diaminophenazine) (PDAP) on carbon substrates via a straightforward electropolymerization method introduced as cathode materials for AIBs.120 The integration of n-type and p-type active units endowed PDAP with an increased number of sites for ion interaction. As a result, the Al||PDAP battery in a 1 M Al(ClO4)3 aqueous electrolyte exhibited a high capacity (338 mA h g−1 at 0.2 A g−1), extended cycle life (1000 cycles), and excellent high-rate capability (101 mA h g−1 at 5 A g−1).
Robert Dominko's team studied an anthraquinone (AQ) cathode for AIBs and reported that the AQ could deliver a specific capacity of 183 mA h g−1 and output a 1.1 V discharge plateau in the AlCl3/EMIMCl electrolyte as hosting the [AlCl2]+ carrier.122 However, the discharge capacity of AQ rapidly decreased to ∼80 mA h g−1 after 50 cycles, presumably due to the dissolution of anthraquinone in the electrolyte. To improve the cycling life, they further fabricated poly(antraquinonyl sulfide), an AQ-based polymer, through in situ polymerization in a multi-walled carbon nanotube suspension. Results showed that upon polymerization, more than 60% of the initial capacity was retained after cycling 500 times (Fig. 6f), and the CE gradually increased from ∼95% to ∼99%.
In summary, organic cathode materials, characterized by inherent low cost, long cycle life, and unique coordination chemistry provide significant advantages over conventional cathode materials. However, despite these promising attributes, challenges such as poor electrical conductivity, low discharge voltage, and low energy density hinder their practical applications. These issues can be effectively mitigated through strategies such as compounding organic materials with carbon-based substances, molecular polymerization, and continued research into novel organic materials.
2.5 Carbon material
Crystalline carbons, i.e. graphite, graphene, and carbon nanotube, have been proposed for use as the cathode materials for AIBs, allowing anion insertion,123 commonly the [AlCl4]− anion, thus forming Al-based DIBs (Table 2). The specific process can be expressed as follows:Cathode:
| Cn + [AlCl4]− ↔ Cn[AlCl4]− + e− |
Anode:
| 4[Al2Cl7]− + 3e− ↔ 7[AlCl4]− + Al |
| Cathode material | Electrolyte | Specific capacity (mA h g−1) | Cycle number | Coulombic efficiency | Ref. |
|---|---|---|---|---|---|
| Graphite paper | AlCl3:[EMIm]Cl (molar ratio of 1.3) IL electrolyte |
70 | 600 | 98.5% | 129 |
| Synthetic kish graphite | AlCl3:EMIMCl (molar ratio of 2) IL electrolyte |
142 | 200 | — | 130 |
| Natural graphite | AlCl3:[EMIm]Cl (molar ratio of 1.3) IL electrolyte |
110 | 6000 | 99% | 131 |
| Graphite | AlCl3:[EMIm]Cl (molar ratio of 1.3) IL electrolyte |
89 | 800 | 97% | 132 |
| Expanded graphite | 50 m ChCl:5 m AlCl3 aqueous electrolyte |
171 | 150 | 90% | 133 |
| Exfoliated graphite | 1 M AlCl3 aqueous electrolyte | 213 | 50 | — | 134 |
| Expanded graphite | AlCl3:triethylaminehydrochloride (mole ratio of 1.5:1) IL |
110 | 18000 |
98% | 135 |
| Graphene | AlCl3:[EMIm]Cl (molar ratio of 1.3) IL electrolyte |
120 | 250000 |
98% | 136 |
| Graphene aerogel | AlCl3:[EMIm]Cl (molar ratio of 1.3) IL electrolyte |
100 | 25000 |
98% | 137 |
| Graphene fabric | AlCl3:ET3NHCl (molar ratio of 1.5) IL electrolyte |
150 | 7000 | 98% | 138 |
| Crystal carbon@graphene microsphere | AlCl3:[EMIm]Cl (molar ratio of 1.3) IL electrolyte |
99.1 | 10000 |
100% | 139 |
| Three-dimensional graphene aerogels | AlCl3:[EMIm]Cl (molar ratio of 1.3) IL electrolyte |
245 | 5000 | 99.8% | 140 |
| Few-layer graphene nanosheets | AlCl3:[EMIm]Cl (molar ratio of 1.3) IL electrolyte |
173 | 10000 |
98% | 141 |
| Edge-rich graphene paper | AlCl3:[EMIm]Cl (molar ratio of 1.3) IL electrolyte |
128 | 20000 |
99.2 | 142 |
| Unzipped multi-walled carbon nanotubes | AlCl3:[EMIm]Cl (molar ratio of 1.3) IL electrolyte |
100 | 5600 | 98% | 143 |
| One-dimensional multi-walled carbon nanotubes | AlCl3:[EMIm]Cl (molar ratio of 1.3) IL electrolyte |
64 | 1000 | 99.5% | 144 |
| Graphitic multi-walled carbon nanotubes | AlCl3:[EMIm]Cl (molar ratio of 1.3) IL electrolyte |
65 | 90 | 100% | 145 |
| Waste-induced pyrolytic 3D-structured carbon nanotube forest | AlCl3:[EMIm]Cl (molar ratio of 1.3) IL electrolyte |
90.5 | 2500 | 99.4–99.8% | 146 |
In 2013, Rani et al. reported a fluorinated natural graphite cathode that achieved a specific capacity of 225 mA h g−1 in AlCl3-containing imidazolium-based ionic liquid electrolyte,125 but the cell failed after cycling only 40 times because the [AlCl4]− insertion process inevitably damaged the graphite layer structure due to the larger size of the [AlCl4]− anion (5.28 Å) than the planar spacing of graphite (3.35 Å).126 To enhance the cycling stability, it is necessary to provide more attachment points for ions and to address the graphite volume expansion issue.
In 2015, Dai's group achieved a breakthrough in the Al-DIB technology.45 Remarkably, the battery supported by a porous 3D graphite cathode achieved an ultra-long cycling life of 7500 cycles and maintained a CE of ∼98%, where the whiskers in the foam with a large space greatly reduced the diffusion length and energy barrier during (de)intercalation of the [AlCl4]− anion, as shown in Fig. 7a. In addition, the system delivered a high discharge voltage plateau of ∼2 V and a specific capacity of ∼70 mA h g−1 in the AlCl3/[EMIm]Cl ionic liquid electrolyte (Fig. 7b). Subsequently, Wang et al.127 introduced kish graphite as the cathode for Al-DIBs, characterized by its unique “cratered morphology” (Fig. 7c and d). This distinctive morphology provided additional ion insertion sites, enabling a specific capacity of 142 mA h g−1 with a high energy density of up to 65 W h kg−1 and a power density of 4363 W kg−1.
 | ||
| Fig. 7 (a) SEM image showing a graphitic foam with an open frame structure; scale bar, 300 μm. Inset, the photograph of graphitic foam; scale bar, 1 cm. (b) GCD curves of an Al||pyrolytic graphite (PG) Swagelok cell at a current rate of 66 mA g−1. Inset, charge, and discharge cycles.45 Copyright 2015, Springer Nature. (c and d) SEM images of the kish graphite flakes, characterized by small 5–10 μm-deep holes facilitating the penetration of the ionic liquid within the flake.127 Copyright 2017, American Chemical Society. SEM images of (e) NG and (f) SGN.128 Copyright 2020, The Royal Society of Chemistry. (g) The [AlCl4]− diffusivities (D) in graphite and six-, five-, four-, three-, and two-layer graphene films at T = 300 K, with film thicknesses of 2.56, 2.24, 1.97, 1.64, and 1.33 nm, respectively.147 Copyright 2016, American Chemical Society. (h) Schematic of the defect-free design.148 Copyright 2017, Wiley-VCH. Schematic diagrams of (i) multi-armed carbon nanotubes and (j) unzipped multi-walled carbon nanotubes. Multi-walled carbon nanotubes that cannot store chloroaluminate anions due to a lack of active intercalation sites. The unzipped multi-walled carbon nanotubes provide numerous active intercalation sites to store [AlCl4]−, and the core carbon nanotubes are responsible for the rapid transportation of electrons to the active sites and maintain structural integrity.143 Copyright 2019, Elsevier B.V. (k) Long-term charge/discharge cycling stability test of the Al||G-MWCNT-1 cell at 1200 mA g−1.144 Copyright 2020, Elsevier B.V. | ||
Recently, a new strategy for graphite processing has been proposed by Hu et al. who obtained small graphite nanoflakes (SGN) from natural graphite (NG) by lithiation followed by reaction with an AlCl3/[EMIm]Cl ionic liquid electrolyte.128Fig. 7e and f show that the increased edge plane and enlarged edge plane in the SGN, owing to its reduced size, provide more active sites for [AlCl4]− (de)intercalation and promote the rate performance of SGN. Consequently, the SGN cathode achieved a specific capacity of 115 mA h g−1 at 500 mA g−1 and maintained >97% capacity after 1000 cycles.
Graphite cathode materials possess characteristics such as a high discharge platform, good multiplicity performance, and good cycling stability, but also present the challenge of volumetric expansion during ion insertion. This issue can be mitigated through the strategic modulation of the material's structure, such as expanding the channel dimensions, incorporating porous architectures, and minimizing ion diffusion pathways. These approaches may provide valuable directions for future research endeavors.
000 cycles without capacity loss, as well as superior high-temperature performance.154
Han's group proposed ultrafast Al-DIBs featuring reversible (de)intercalation of [AlCl4]− in graphite foam cathodes and revealed that the rapid charging and discharging capabilities of graphene were due to decreased elastic stiffness and increased free space for [AlCl4]− diffusion as the number of graphene layers was reduced, leading to a steep increase in diffusion rate through first-principles calculations (Fig. 7g).147 The defect-free graphene produced based on a fewer-layered structure can further improve the performance of graphene. Chen et al. proposed the defect-free principle of graphene-based cathodes, demonstrating that the cathode exhibited a capacity of 100 mA h g−1 at a current rate of 5 A g−1 and a capacity retention rate of 97% after 25000 cycles.148 The schematic diagram for the defect-free graphene structure design is shown in Fig. 7h, where a mature high-temperature annealing technology has been used to repair these defects. Through comparative experiments, the authors identified three key factors contributing to the degradation of battery performance due to defects: (1) defects do not serve as active sites for the intercalation of [AlCl4]−, as confirmed by in situ Raman spectroscopy; (2) barrier-like defects obstruct the rapid intercalation of [AlCl4]− into graphene layers; and (3) defects diminish the electrical conductivity of the electrode.
In summary, graphene-based Al-DIBs have attracted widespread attention due to their good rate performance and excellent cycling stability. Fewer-layered and non-defective structures can greatly improve the charge and discharge performance. However, as an emerging material, graphene's production process remains underdeveloped, and the preparation of defect-free and highly crystalline graphene is a significant obstacle to the commercialization of these cathode materials.
CNTs can be divided into single-walled carbon nanotubes (SWCNTs) and multi-walled carbon nanotubes (MWCNTs). Bhauriyal et al. used first-principle calculation to investigate the adsorption positions of [AlCl4]− on carbon nanotubes of different diameters and proved the feasibility of SWCNTs as cathodes for Al-DIBs.156 Nonetheless, the lack of experimental verification has limited the advancement of SWCNTs. Generally, the non-defective surface of CNTs hinders the insertion of [AlCl4]−, thus appropriately introducing defects or altering the ion storage sites could address the energy storage limitation of CNTs and enhance the capacity.104
Zhang et al. combined graphene nanoribbons and CNTs to prepare flexible MWCNT membranes with more defects, where CNTs transported electrons while graphene nanoribbons provided more active sites for storing [AlCl4]− (Fig. 7i and j).143 This effectively resolved the kinetic issues, achieving a high capacity of 100 mA h g−1 at a current rate of 2000 mA g−1. Recently, Lin's group further confirmed that [AlCl4]− was inserted in the nanotube walls of MWCNTs during charging rather than simply adsorbed on the surface, providing a reference for future experiments, which delivered a specific capacity of 64 mA h g−1 at 200 mA g−1, with the discharge capacity maintained at ∼58 mA h g−1 after 1000 cycles (Fig. 7k).144 The excellent electrical conductivity and the unique tubular structure of CNTs are attracting increasing attention. However, due to the limited capacity achieved when used as an electrode material alone, CNTs are often combined with other materials to enhance both the conductivity and the capacity of the battery,157–159 which is also an area of significant interest.
Carbon-based materials are among the most common cathode materials for AIBs because of their stable structure and long cycling stability. In addition, during the insertion of [AlCl4]− anion in the cathode, the single-electron transfer minimizes the strong coulombic forces that occur when the Al3+ cation is inserted into the cathode material.160 However, the advantage of AIBs lies in the multi-electron reaction of Al3+. Since the cathode hosts the [AlCl4]− anion rather than the Al3+ forming Al-DIBs, the energy density is reduced, which contradicts our initial goals. Meanwhile, carbon materials as cathodes typically exhibit low capacity when hosting the [AlCl4]− anion in AIBs. Therefore, increasing the specific surface area of carbon-based materials to provide more active sites is a very effective approach for overcoming this limitation.
2.6 Sulfur and iodine
The working mechanism of Al–S batteries is a typical conversion reaction mechanism. As shown in Fig. 8a, the S monomer cathode gains electrons and converts to the S2− state during discharge, and reversibly during the charging process,167 which can be expressed by the following equations:
Cathode:
| 8[Al2Cl7]− + 6e− + 3S ↔ Al2S3 + 14[AlCl4]− |
Anode:
| 2Al + 14[AlCl4]− ↔ 8[Al2Cl7]− + 6e− |
 | ||
| Fig. 8 (a) Schematic diagram of an Al-S battery.167 Copyright 2023, Wiley-VCH. (b) Galvanostatic discharge curves of Al||S battery at a current density of 30 mA g−1 in various molar ratios of EMImCl: AlCl3 ionic liquid electrolytes.169 Copyright 2015, Elsevier B.V. (c) The TEM images of nanospheres carbon-S material.170 Copyright 2021, Elsevier B.V. (d and e) Energy profiles of the dissociation reactions of [Al2Cl7]− and [Al2Cl6Br]− anions. (f) Comparison of cycling performance of S@Cu1Co1@NC, S@Cu@NC, S@Co@NC, and S@NC at a current density of 1.5 A g−1.171 Copyright 2023, Wiley-VCH. (g) Schematic illustration and (h) SEM of the synthesis process of I2@ZIF-8-C. (i) Cycling performance of the flexible Al||I2@ZIF-8-C battery.172 Copyright 2021, Wiley-VCH. | ||
The first Al–S battery could be traced back to 1993, which was proposed by Stuart Licht et al., in which an alkaline aqueous electrolyte allowed the battery to output an open-circuit potential of 1.3 V with an energy density of 110 W h g−1.168 Although they provided the Al–S battery model, its development has since been exceptionally slow due to the severe side reactions occurring at the Al anode in the aqueous electrolyte. In 2015, Cohn et al. proposed a novel non-aqueous Al–S battery in an ionic liquid of AlCl3/[EMIm]Cl electrolyte and obtained an ultra-high specific capacity of over 1500 mA h g−1 (close to the theoretical specific capacity of sulfur, 1675 mA h g−1) and an energy density of 1700 W h kg−1 (Fig. 8b).169 Unfortunately, its capacity rapidly decayed to less than 200 mA h g−1 at the second cycle caused by the dissolution of the produced sulfur-containing substances. To improve cycling stability, efforts must focus on inhibiting the dissolution of discharge products.
Compositing the S cathode with other materials can increase structural stability and improve battery life. Recently, Zhang et al. proposed using hollow CNTs as the host for sulfur in a non-aqueous Al–S battery,170 where the cage-like hollow CNTs can provide the necessary spaces to accommodate sulfur expansion during reactions (Fig. 8c) and store sulfur and polysulfides, thereby mitigating the sulfur shuttle effect and improving cycling stability. Huang et al. proposed sulfur-anchored cobalt/nitrogen co-doped graphene (S@CoNG) as a cathode material for AIBs and an ionic liquid-impregnated metal–organic framework (IL@MOF) as the electrolyte.173 The IL@MOF stabilized reversible sulfur conversion and inhibited the shuttle effect of polysulfides, resulting in a high specific capacity of 820 mA h g−1 at the first cycle and 78% capacity retention beyond 300 cycles. In addition, the S@CoNG cathode facilitated continuous Al3+ dissociation from [AlxCly]− ions and the breaking of S–S bonds, while the IL@MOF with IL ions enabled fast active ion transport, greatly accelerating sulfur reaction kinetics. Additionally, the cycling stability of Al–S batteries can be improved via the formation of a solid electrolyte interface (SEI). Xu et al. enhanced the cycling performance of Al–S batteries by the addition of alkaline-earth metal chlorides to the AlCl3/EmimCl electrolyte.174 Using NaCl as an additive resulted in a thick SEI containing NaxAlyO2 on the Al anode, which reduced the deposition of polysulfides. As a result, a capacity of 473 mA h g−1 was obtained compared with only 313 mA h g−1 without the additive. Similar results were obtained with the addition of KCl and LiCl. In addition, Yu et al. leveraged the synergistic advantages of adsorptive Co and the catalytic properties of a conductive nitrogen-doped carbon matrix, designated as Cu1Co1@NC, to mitigate the shuttle effect, while Co facilitated the S/Al2S3 conversion reaction.171 The combined effects of Cu and cobalt Co endowed the Al–S cell with remarkable cycling stability and reaction kinetics, achieving a high capacity of 317.5 mA h g−1 after 320 cycles at 1.5 A g−1 (Fig. 8d), an ultra-long lifespan exceeding 10000 h, and excellent reversibility with a CE of 99.8–99.9%.
Another challenge in Al–S batteries is the slow reaction kinetics. In 2018, Yang et al. proposed that this sluggishness could be attributed to the inevitable dissociation of [Al2Cl7]− into free Al3+. In contrast, the use of [Al2Cl6Br]− anions resulted in reaction kinetics that was 15 times faster, yielding four times higher sulfur utilization and five times greater current density, owing to the significantly lower dissociation energy of [Al2Cl6Br]− compared with [Al2Cl7]− (Fig. 8e and f).175
:1, and they demonstrated that, based on the I3−/I− redox chemistry with high reversible storage of Al3+, the Al–I2 battery provided a high capacity above 200 mA h g−1 at 0.2 C and high stability for more than 150 cycles at 1 C.179 However, the poor conductivity of iodine, poor thermal stability, and the shuttle effect of multiple iodides impede the practical application of iodine electrodes in AIBs.
The stability of the iodine cathode can be enhanced by physically confining iodine in a porous carbon or metal–organic framework (MOF). Yang et al. used MOF-derived N-doped microporous carbon polyhedra as the host material for iodine (I2@ZIF-8-C) and assembled AIBs with a water-in-salt electrolyte composed of LiTFSI and AlCl3 (Fig. 8g and h).172 They demonstrated that the I2@ZIF-8-C electrode exhibited a high specific capacity of 219.8 mA h g−1 at 2 A g−1 and a high-rate performance of 102.6 mA h g−1 at 8 A g−1. This performance was due to the confined liquid–solid conversion of iodine within the hierarchical nitrogen-doped microporous carbon polyhedron, as well as the improved reaction kinetics of the aqueous electrolyte facilitated by the conversion of I3− and I5− intermediates. Furthermore, the flexible battery showed reasonable stability, retaining a capacity of 145.6 mA h g−1 after 100 cycles, with a capacity decay of 0.46% per cycle (Fig. 8i).
Moreover, the shuttling problem associated with polyiodides can be effectively suppressed by chemical bonding. Han et al. complexed active carbon cloth with polyvinylpyrrolidone (PVPI) to serve as the cathode for an Al-I2 battery in an AlCl3/[EMIm]Cl ionic liquid electrolyte, which featured a high capacity of 180.1 mA h g−1 at 0.2 C and maintained a stable capacity of 127 mA h g−1 after 500 cycles at 0.6 C.180 The hydrogen bonding interactions between PVP and iodine in PVPI guaranteed the suppression of the shuttle effect of polyiodides, thus extending the cycling life to 1050 cycles.
In conclusion, Al–S and Al–I2 batteries based on conversion chemistry have been a hot research topic in recent years due to their low cost and high theoretical capacity. However, poor cycling performance due to dissolution and the shuttle effect, as well as slow reaction kinetics, are urgent problems to be solved. Improving the sulfur and iodine cathodes by combining them with other materials and constructing SEI films are currently effective solutions. Meanwhile, developing new electrolytes suitable for the current mainstream cathode materials is necessary to improve the electrochemical performance of Al–S or Al–I2 cells.
3. Anode
The application of Al metal as an anode material for batteries dates back to 1972 when Holleck and Giner first reported the Al||Cl2 secondary battery with a NaCl/KCl/AlCl3 molten electrolyte. Al metal is widely used in various battery technologies such as Al–air batteries, Al fuel cells, and Al-ion batteries.37 However, Al is prone to react with oxidizing substances to generate Al2O3 passivation and delay the electrochemical reaction activity, hindering normal electrochemical reactions. In addition, the Al anode is susceptible to electrochemical corrosion during charge and discharge processes, especially in the environment containing chloride ions, which causes the expansion or even explosion of the battery. How to solve the above scientific problems remains a major challenge for researchers.3.1 Functional interface layer regulation
Constructing a stable functionalized interfacial layer on the surface of the Al anode can promote uniform nucleation of Al3+ and alleviate side reactions of corrosion and HER, thus improving cycling stability. A qualified interfacial layer should have one or more of the following properties: (1) high ionic conductivity and low electronic conductivity, (2) mechanical stability to alleviate the volume change during the cycling process, (3) physical restriction or electrostatic interaction to limit the two-dimensional diffusion of Al3+, and (4) abundant polar groups, which can interact with metal ions.Dense thin films of metals or compounds can be formed by in situ chemical deposition. Yan et al. constructed an amorphous Al (a-Al) coating layer via in situ lithium-ion alloying and dealloying on a low-strength Al metal substrate (Al@a-Al) (Fig. 9a).181 This amorphous structure significantly lowered the energy barrier for Al nucleation, facilitating Al3+ plating in competition with the electron-consuming HER, thus enhancing charge transfer kinetics (Fig. 9b). Simultaneously, by inhibiting the HER, passivation was reduced, which improved interfacial ion transfer kinetics and enabled stable Al3+ plating/stripping for 800 h in a symmetric cell (Fig. 9c). The Al@a-Al||KNHCF battery maintained 91% of its capacity after 200 cycles in a 0.5M Al2(SO4)3 electrolyte. Similarly, Yu et al. in situ deposited an MXene-based hybrid ion/electronic conductor interfacial layer on an Al substrate to regulate Al3+ flux and electric field distribution, thereby enhancing the Al3+ stripping and plating.182 This modification enabled the Al anode to achieve an impressive cycling life of over 5000 h at an ultra-high current density of 50 mA cm−2 in the Al||Al symmetric battery. The MXene layer with appropriate electronic conductivity can homogenize the electric field on the electrode surface, which is conducive to the smooth deposition of Al3+. In addition, the high mechanical flexibility of the 2D MXene layer can withstand large volume changes and inhibit the growth of dendrites. The assembled Al||pyrene-4,5,9,10-tetraone cell in the [EMIm]Cl/AlCl3 (molar ratio of 1.3:1) electrolyte operated for 200 cycles with a 100% capacity retention.
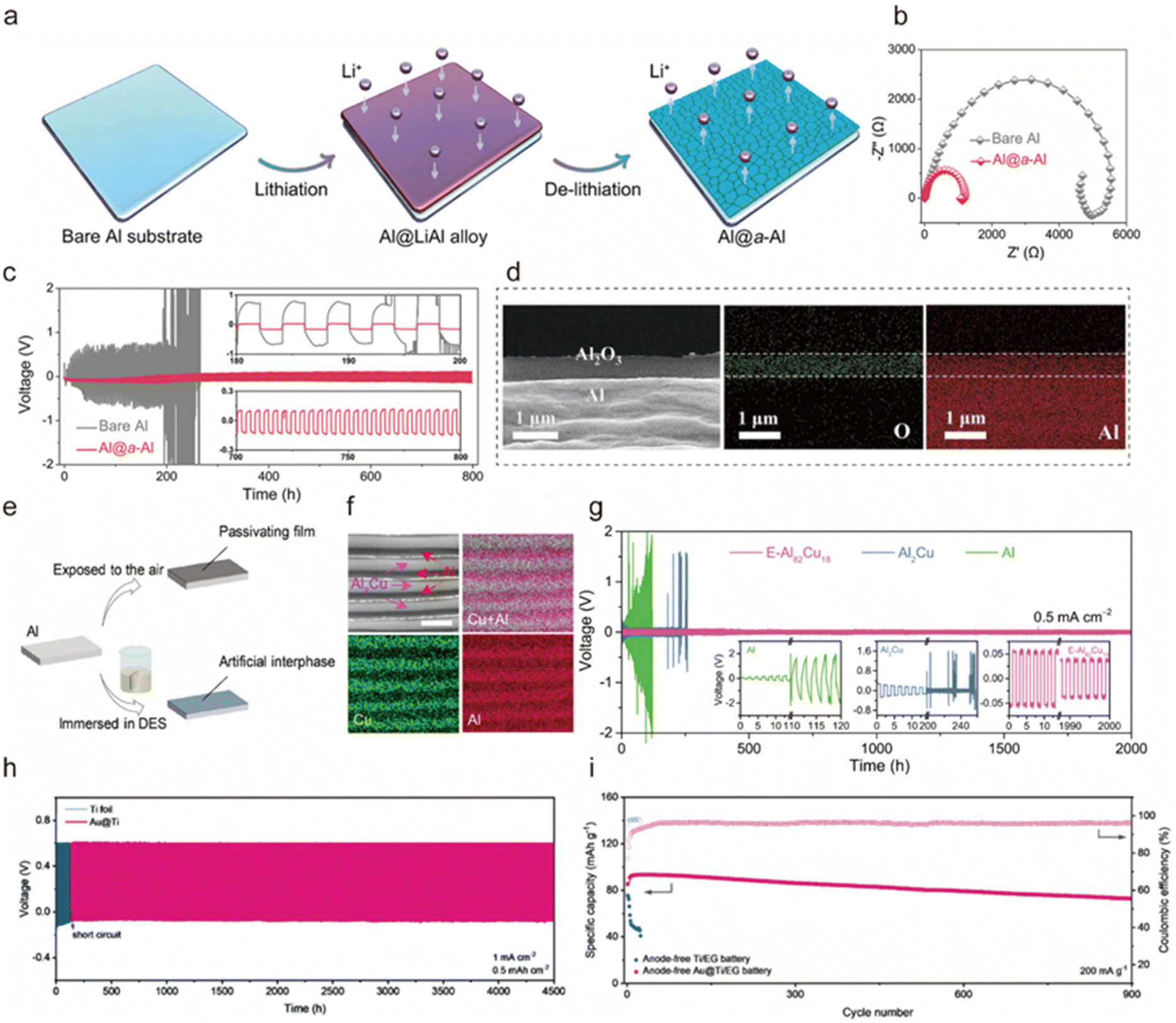 | ||
| Fig. 9 (a) Schematic illustration of the design and preparation of the Al@a-Al anode. (b) Comparative Nyquist plots of bare Al and Al@a-Al anode. (c) Comparison of voltage profiles for symmetric cells based on bare Al and Al@a-Al anodes in the 0.5 M Al2(SO4)3 electrolyte cycling at 0.05 mA cm−2 for 2 h in each half-cycle. Inset: the magnification of selected periods in (c).181 Copyright 2022, American Chemical Society. (d) SEM image of the Al2O3/Al electrode cross-section and elemental maps of Al and O from an EDS analysis of the electrode section.184 Copyright 2023, Elsevier B.V. (e) Schematic diagram of the formation of artificial interphase/passivating layer.187 Copyright 2021, Elsevier B.V. (f) SEM backscattered electron image of the E-Al82Cu18 with different contrasts corresponding to α-Al and intermetallic Al2Cu lamellas, as well as the corresponding EDS elemental mapping of Cu (in green) and Al (in red). Scale bar, 1 μm. (g) Long-term cycling stability of Al plating/stripping for symmetric cells based on the E-Al82Cu18 (pink line), Al2Cu (blue line), and Al (green line) electrodes at 0.5 mA cm−2 in the 2 M Al(OTF)3 aqueous electrolyte with CO2 = 0.13 mg L−1. Inset: voltage evolutions for the Al (left), Al2Cu (middle), and E-Al82Cu18 (right).192 Copyright 2022, Springer Nature. (h) Long-term stability of Al||Au@Ti cell and Al||Ti cell at 0.5 mA h cm−2 and 1 mA cm−2. (i) Cycling stability of anode-free Au@Ti||EG battery and Ti||EG battery at 200 mA g−1.194 Copyright 2023, Wiley-VCH. | ||
Exploitation of physical treatment is one of the common surface treatment techniques. Tang et al. employed direct current magnetron sputtering to deposit amorphous carbon nanofilms (ACNI) on the Al foil acting as an artificial SEI layer to improve the structural stability of the Al anode.183 The assembled ACNI/Al||graphite cell in a 4.0 M LiPF6/ethyl methyl carbonate with 5 wt% vinylene carbonate electrolyte demonstrated significantly improved performance with a specific capacity of 115 mA h g−1, achieving capacity retention of ∼94% after 1000 cycles at 200 mA h g−1. Xie et al. designed an Al2O3/Al electrode by laser etching and anodizing, which not only increased the effective active area of the electrode but also formed a stable electrode/electrolyte interface to induce uniform plating of Al3+ (Fig. 9d).184 The Al2O3/Al anode exhibited excellent cycling performance and rate performance, which retained the area specific capacity of 5 mA h cm−2 after cycling 1400 h. In addition, the 480 mA h pouch cell with the Al2O3/Al anode could maintain an energy density of 170 W h kg−1 with an energy efficiency of 90% (calculated based on the positive active component), indicating strong performance scalability. Li et al. developed a polyvinylidene fluoride (PVDF) coating for the Al anode to inhibit the presence of free H2O/O2, thereby alleviating corrosion issues.185 This coating demonstrated excellent anodic properties, attributed to the strong interaction between PVDF and Al3+ through the F–Al bond, which promoted uniform Al3+ plating. Consequently, the PVDF-Al||K2CoFe(CN)6 cell, utilizing a 1 M Al(OTF)3 electrolyte, achieved a CE of 98.2% after 400 cycles at a current rate of 100 mA g−1. Archer's team also confirmed the role played by chemical bonding in Al anodes.186 By eliminating fragile electrons and promoting strong oxygen-mediated chemical bonding between deposited Al and the substrate, they achieved fine control of the morphology of Al, realizing non-planar deposition of Al3+. As a result, the anode exhibited excellent reversibility (99.6–99.8%) and sustained unusually long cycling over 3600 h.
The formation of a stable SEI at the anode can effectively prevent passivation and enhance electron and ion mobility. Tang et al. obtained a treated Al anode (TAl) by immersing polished Al foil in an AlCl3/acetamide (molar ratio of 1.3:1) deep eutectic solvent (DES) for 24 h (Fig. 9e).187 This treatment effectively inhibited surface passivation and improved the chemical properties of the interface. The assembled symmetric cell with the TAl was able to cycle for over 300 h while maintaining a low overpotential of ∼0.2 V. Additionally, the TAl||FeFe(CN)6 cell in a 2 M Al(CF3SO3)3 electrolyte provided a high capacity of 85 mA h g−1, and maintained 58 mA h g−1 after 150 cycles, with an average CE of 97.1% at a current rate of 100 mA g−1. Similarly, Srinivasan et al. engineered an artificial protective barrier layer on Al using AlCl3 and urea eutectic coating formulations (UTAl).188 Benefiting from the coating, the conductivity and kinetics at the anode–electrolyte interface were improved, and the overpotential for stripping and plating was reduced. The assembled UTAl||FeHCF cell in a 2 M Al(OTf)3 electrolyte achieved a stable battery performance of ∼60 W h kg−1 over 100 cycles.
3.2 Alloying
Aluminum-based alloys play a crucial role in mitigating HER, passivation, and corrosion, showing superior plating and stripping reversibility as well as extended cycle life compared with bare Al anodes. However, careful consideration must be given to the selection of alloying elements to ensure they possess the following characteristics, as inappropriate additions can exacerbate HER and corrosion issues: (1) the alloying elements should be nobler than Al to activate the passivation layer and suppress the HER; (2) they should have a more positive electrode potential than Al to facilitate the underpotential deposition process; and (3) they should exhibit significant solid solubility in the Al matrix to ensure a homogeneous composition and morphology.189,190To date, Zn, Cu, Ce, and Sn have been applied to create intermetallic alloys with Al as anode materials. Yan et al. prepared a Zn-Al alloy by depositing Al on Zn in a 2 M Al(OTF)3 solution in a Zn symmetric cell, which was subsequently employed as the anode for ABs.31 The specialized alloy interface layer effectively mitigated passivation and suppressed dendrite growth, ensuring long-term stability in Al3+ plating and stripping for over 1500 h. The Zn–Al||AlxMnO2 cell demonstrated a high reversible capacity of 460 mA h g−1 at 100 mA g−1 after 80 cycles, a high discharge voltage plateau of 1.6 V, and an impressive rate capability of 100 mA h g−1 at 3 A g−1. Park et al. directly assembled cells with Zn foils in a 0.8 M Al(OTF)3 acetonitrile-H2O hybridization electrolyte, with an H2O to Al(OTF)3 ratio of 2:1, to in situ generate the Zn–Al alloy.191 This setup presented weak Al3+–solvent interactions and fast Al3+ transfer kinetics, achieving an ultra-long time cycling for more than 8000 h of plating and stripping at the anode. The assembled Zn–Al||VO2/carbon nanotubes cell achieved a high capacity of 183 mA h g−1 and 1.08 mA h cm−2, along with remarkable cycling stability of 45000 cycles.
Optimization of the alloy structure can inhibit passivation, HER side reactions, etc., thereby improving anode reversibility. Jiang et al. reported a eutectic Al–Cu alloy (E-Al82Cu18) with a lamellar heterostructure composed of alternating α-Al and Al2Cu nanoflakes (Fig. 9f), wherein the more noble Al2Cu lamellas acted as electron transfer pathways to facilitate Al3+ stripping from the less-noble Al lamellas, and they served as nanopatterns guiding subsequent dendrite-free plating, thus improving reversibility at low potentials.192 Consequently, the E-Al82Cu18 electrode exhibited a CE of ∼100% for over 2000 h with an overpotential as low as ∼53 mV (Fig. 9g). When paired with an AlxMnO2 cathode, the cell achieved a high specific energy of ∼670 W h kg−1 and energy density of 815 W h L−1 at 100 mA g−1, retaining 83% capacity after 400 cycles. Furthermore, they also reported a eutectic Al–Ce (E-Al97Ce3) alloy in situ grafted with uniform ultrathin MXene (MXene/E-Al97Ce3) as a flexible, reversible, and dendrite-free anode for AIBs.193 The E-Al97Ce3 alloy comprised alternating symbiotic α-Al metal and intermetallic Al11Ce3 nanolamellas, providing Al3+ sources and serving as a 2D nanopattern to direct Al3+ plating and stripping. This design mitigated passivation from native oxide and curtailed side reactions, enabling the MXene/E-Al97Ce3 hybrid electrode to exhibit dendrite-free and highly reversible Al3+ plating and stripping, with low voltage polarization of ±54 mV and a high energy efficiency of ∼99.2% for over 1000 h in a 2 M Al(OTF)3 aqueous electrolyte with ultralow oxygen concentration. Moreover, the MXene/E-Al97Ce3||AlxMnO2 pouch cell showed a high coulombic efficiency of up to ∼99.5%, excellent rate performance, and cycling stability, maintaining ∼85% of the initial discharge capacity after 500 cycles at a current density of 1 A g−1.
Yan et al. employed a scalable folding and rolling method to prepare Sn–Al laminate electrodes (Sn@Al). The choice of Sn, with a suitable redox standard potential (−0.13 V) and work function (4.42 eV), complemented Al to facilitate Al underpotential deposition and improve anode reversibility.195 Sn and Al demonstrated strong interfacial adhesion, creating a tightly bonded, layered, interlaced configuration where the spaces and gaps between layers increased the electrode's specific surface area and enhanced ion transport. In this structure, the metallic Al network served as a source pool for Al3+ within the electrode, while the Sn skeleton provided numerous active sites for the underpotential plating of Al3+ over HER. Additionally, the combination of Sn and Al formed localized Al/Sn galvanic couples, which effectively promoted Al stripping, thereby reducing internal resistance and improving charge transfer kinetics. The Sn@Al electrode demonstrated stable cycling for over 900 h in a symmetric cell in a 0.5 M Al2(SO4)3 electrolyte. To further suppress HER significantly, the PVDF solution was coated onto Sn@Al to form p-Sn@Al, and the p-Sn@Al||KNHCF cell showed a capacity retention of 82% after 700 cycles.
3.3 Anode-free
Anode-free Al cells are considered a promising strategy for metal plating and stripping to mitigate the effects of HER, passivation, and corrosion. In this novel design, ions are stored directly on the surface of the collector by electrochemical deposition during charging; during discharge, ions detach from the collector surface and return to the electrolyte or cathode. This seemingly simple change offers several significant advantages: first, removing the anode reduces the battery's weight and size, increasing overall energy density. This means a battery of the same size can store more energy, or a battery of the same capacity can be made smaller and lighter. Second, the simplified structure lowers production costs, which is critical for large-scale commercialization. However, the anode-free strategy faces serious challenges, such as the lack of excess Al anode replenishment during cycling, leading to poor cycling stability. Additionally, ensuring good contact between the electrolyte and collector is crucial for efficient ion transport.To address the above issues, the choice of collector is critical, as it directly regulates metal deposition morphology by affecting deposition kinetics and the crystallographic growth behaviors of metal anodes.194 Archer et al.196 reported using two-dimensional gold nanosheets with strong diffraction from (111) facets and low lattice mismatch as an anode collector for anode-free AIBs. They verified that Au coatings sustain stable cell operations for over 500 cycles with a high CE of over 99%. The battery, with Au nanosheets as the anode substrate and graphene as the cathode, exhibited capacity retention of 80% after 1000 cycles and 74% after 2000 cycles in the AlCl3/[EMIm]Cl (molar ratio of 1.5:1) electrolyte. However, insightful explanations and mechanistic probes regarding collector selection are still lacking. Jin et al.197 conducted in-depth monitoring of a variety of carbon and metal-based materials used as anode collectors (ACCs) for anode-free AIBs and investigated their corrosion resistance to Cl− in the AlCl3-based ionic liquid. The results indicated that electrochemically stable ACCs, such as graphite paper and Mo foils, exhibited better cycling stability and higher CEs comparable to the Al anode. In contrast, metallic ACCs susceptible to corrosion by Cl− ions, such as Cu, Ag, Ni, steel, and Mg foils, exhibited relatively lower specific capacities and rapid capacity fade during long-term cycling. Chen et al.194 designed an ultrathin lattice-matching layer (LML) to investigate the nucleation and growth mechanisms of Al3+ on the Au@Ti collector. The LML prolonged the nucleation process of Al, increased nucleation density, and reduced the average particle size. Evenly distributed Au significantly enhanced the nucleation density and reduced the average particle size, tailoring the morphology of Al toward anode-free Al anodes without dendrite formation. As a result, stable Al3+ plating and striping occurred on the Au@Ti substrate for over 4500 h, achieving an excellent CE of 99.92%. Furthermore, the anode-free Au@Ti||expanded graphite (EG) cell with an AlCl3/[EMIM]Cl (molar ratio of 1.3:1) electrolyte exhibited significantly longer cycling stability, exceeding 900 cycles with a capacity retention rate of 80%.
Wei et al. first reported a proof-of-concept of anode-free aqueous AIBs, in which Al2TiO5, as a cathode pre-aluminum additive (Al source), could replenish Al loss by over cycling.198 The Cu collector surface forms a uniform AlCu alloy layer via the charging process as a means of maintaining reversible plating/stripping of hydrated Al3+ on the Cu foil surface. The Cu||PANI@G-Al2TiO5 cell in the 0.5 M Al2(SO4)3 battery delivered a high initial discharge capacity of 175 mA h g−1 and power density of 410 W h L−1 with a capacity retention of 60% after 1000 cycles.
4. Electrolyte
The electrolyte is one of the important and indispensable components of the battery, and plays many key roles such as: (1) transferring ions and realizing electrochemical reactions. The electrolyte serves as a medium for ion transfer, and in the process of battery charging and discharging, redox reactions will occur between the cathode and anode materials, and these reactions require the participation of ions. The electrolyte salt in the electrolyte can dissociate into cation and anion, and these ions migrate between the cathode and the anode through the electrolyte under the action of the electric field, thus realizing the electrochemical reaction; (2) maintaining the stability of the battery performance. The composition and concentration of the electrolyte have a direct impact on the performance of the battery. A suitable electrolyte formula can ensure that the battery maintains a stable voltage, capacity, and cycle life during charging and discharging.Electrolytes can be broadly categorized into aqueous and non-aqueous types. Aqueous electrolytes, commonly composed of compounds such as AlCl3, Al2(SO4)3, and Al(OTF)3, are recognized for their environmental friendliness, cost-effectiveness, intrinsic safety, and high conductivity. However, several scientific challenges persist: the narrow electrochemical stabilization window (ESW) of aqueous electrolytes restricts the voltage and energy density of the battery; the low decomposition voltage of H2O leads to the anodic HER occurring before the Al3+ reduction reaction; and the high reactivity of H2O at the electrode surface results in detrimental side reactions that cause corrosion and passivation. In contrast, non-aqueous electrolytes are less susceptible to HER due to the absence of water, offering a wider electrochemical stability window along with excellent thermal and chemical stability, making them suitable for high-performance applications. Other than the widely used ionic liquid electrolytes, types of new electrolyte systems include water-in-salt electrolyte, aqueous and non-aqueous DES electrolyte, molten salts electrolyte, and gel polymer electrolyte. Despite these advantages, it is crucial to consider the costs and environmental impacts associated with the disposal of certain solvents used in non-aqueous systems.
4.1 Water-in-salt electrolyte
To inhibit the occurrence of side reactions such as HER in AIBs, one effective approach is to reduce the activity of H2O by increasing electrolyte concentration, therefore water-in-salt electrolytes and aqueous deep eutectic solvent (DES) electrolytes have been developed. For instance, Wu et al. proposed a water-in-bi-salt electrolyte composed of 1 M Al(OTF)3 and 15 M LiOTF, wherein the traditional octahedral solvation sheath structure of Al3+ in dilute solutions, i.e. Al(H2O)63+, transformed to mixed octahedral Al(OTF)x(H2O)6−x (x ≥ 0) and tetrahedral Al(OTF)3OH− ion pairs (Fig. 10a).199 This significantly reduced water activity and expanded the electrochemical window to 4.35 V while maintaining a low overpotential (Fig. 10b). Consequently, it suppressed cathode dissolution, alleviated self-discharge behavior, and stabilized the interface at both the cathode and anode. As a result, the AlxMnO2 cathode achieved a discharge capacity of 160 mA h g−1 after 150 cycles with a CE of ∼95%. Luo et al. also proposed a WiSE electrolyte containing 2 M Al(OTf)3 + 20 M LiTFSI. In the electrolyte, the H2O molecules were confined within the Li+ solvation structures, which reduced the Al3+–H2O interaction, thus essentially eliminating the hydrolysis effect, effectively protecting the Al anode from corrosion. As a result, long-term Al plating and stripping could be achieved over 1800 h.200 | ||
| Fig. 10 (a) FTIR spectra of electrolytes with varying different concentrations. (b) The schematic diagram of Al(OTF)3OH− and Al(OTF)x(H2O)6−x.199 Copyright 2022, Elsevier B.V. (c) LSV curves of the BE and HEE30 electrolytes at a scan rate of 10 mV s−1. (d) Cycling stability of Al||PW full cells with the BE and HEE30 electrolytes at 0.1 A g−1 at 25 °C.49 Copyright 2024, Wiley-VCH. SEM images of Al anodes cycled in the (e) AU and (f) AUdF.203 Copyright 2024, Wiley-VCH. (g) Long-term cycling stability of the PA, PA450 and PA650 electrodes in molten salt electrolytes at 10 A g−1.208 Copyright 2024, Elsevier B.V. (h) Schematic illustration of the in situ preparation of cross-linked PEA-GPE and the pouch cell assembly procedure. (i) Cycling stability of the Al||cross-linked PEA-GPE||graphite battery with 11 mg cm−2 graphite loading at 100 mA g−1.209 Copyright 2023, Elsevier B.V. (j) The cycling performance at a current rate of 0.5 A g−1.210 Copyright 2024, Wiley-VCH. | ||
4.2 Aqueous DES electrolyte
Aqueous DES electrolytes combine the high ionic conductivity of aqueous electrolytes with the electrochemical stability of DES electrolytes and effectively inhibit side reactions. Zhang et al. proposed a eutectic electrolyte named HEE30, formulated with an optimal molar ratio of 1:8:1:30 for Al(OTf)3, glycerol (Gly), sodium beta-glycerophosphate pentahydrate (SG), and H2O.49 The unique eutectic network significantly enhanced the hydrogen bonding between Gly and H2O, reducing the solvation interaction of Al3+ with active H2O, thereby lowering the freezing point, extending electrochemical windows, and suppressing HER. Consequently, the Al symmetric cell achieved an extended lifespan of 1000 h at 0.05 mA cm−2 and even demonstrated prolonged cycling stability exceeding 500 h and 1000 h at temperatures of −20 and 60 °C, respectively. Moreover, the Al||Prussian white full cell exhibited a capacity retention of 72% after 500 cycles at 0.5 A g−1 while maintaining a high capacity of 109 mA h g−1 at a low current rate of 0.1 A g−1 after 200 cycles. In addition, their group also designed a hydrated eutectic electrolyte (AATH40) composed of Al(OTf)3, acetonitrile (AN), triethyl phosphate (TEP), and H2O.201 Molecular dynamics simulations and spectroscopy analysis revealed that AATH40 possessed a less-water-solvated structure, presented as [Al(AN)2(TEP)(OTf)2(H2O)]3+, which effectively inhibited side reactions, decreased the freezing point, and extended the electrochemical window of the electrolyte. Consequently, the Al symmetrical cell demonstrated cycling stability for 520 h under 0.05 mA cm−2 at 25 °C, 450 h at −10 °C, and 1500 h at 50 °C. Furthermore, the Al||polyaniline (PANI) full cell delivered a capacity of 129 mA h g−1 over 100 cycles at 200 mA g−1.
4.3 Non-aqueous DES electrolyte
Non-aqueous DES are commonly used as electrolytes in addition to ionic liquids, which can avoid the air sensitivity and high cost of ionic liquids. Yan et al. proposed a new low-cost and environmentally friendly DES composed of AlCl3 and guanidine hydrochloride (GdnHCl) with a molar ratio of 1.8:1.202 The assembled Al||NG battery exhibited a capacity of 128 mA h g−1 at 100 mA g−1 and a capacity 124.6 mA h g−1 at 1000 mA g−1, achieving a CE of 99.5% over 1000 cycles. Passerini et al. proposed a locally concentrated DES consisting of AlCl3 and urea in a molar ratio of 1.3:1, combined with the non-solvating co-solvent 1,2-difluorobenzene (dFBn).203 The inclusion of dFBn effectively enhanced fluidity and ion transport without compromising ionic dynamics in the electrolyte. Moreover, dFBn modified the solid electrolyte interphase that formed on the anode, which reduced the interfacial resistance and promoted a more homogeneous Al3+ plating and stripping, thereby improving the cycling stability of the Al anode (Fig. 10e and f). As a result, the lifespan of the Al symmetric cell was extended from 210 h to 2000 h, while the cell polarization decreased from 0.36 to 0.14 V at 1.0 mA cm−2.
4.4 Molten salt electrolyte
Molten salt electrolytes are known for fast reaction kinetics due to high ionic concentration and excellent electrical conductivity, as well as good thermal and chemical stability, and have been developed for AIBs but at high temperatures. Jiao et al. developed an AlCl3-NaCl-LiCl-KCl (molar ratio of 1.31:0.43:0.42:0.15) quaternary inorganic molten salt electrolyte with the lowest eutectic temperature of less than 75 °C.204 The Al||graphite battery showed a stable specific capacity of 114.9 mA h g−1 at 200 mA g−1 over 1500 cycles at 90 °C. Wu et al. reported an intermediate-temperature AB featuring a nickel disulfide-based cathode and a NaCl-AlCl3-Al2S3 molten salt electrolyte, in which NaCl, AlCl3, and S powders were fully mixed with a molar ratio of 1.05:1.0:0.12.205 The Al||NiS2 cell delivered a capacity of 320 mA h g−1 at 240 °C and a current rate of 2000 mA g−1 with negligible capacity fade after 2000 cycles. In addition, AlCl3-NaCl-KCl molten salt electrolyte has been widely used.206–208 Mai et al. proposed a novel and efficient Al-organic battery with ultrafast reaction kinetics by utilizing a molten salt electrolyte (AlCl3–NaCl–KCl molar ratio of 61:26:13) and designing a strongly interacting organic cathode PTCDA (perylene-3,4,9,10-tetracarboxylic dianhydride, PA450).208 The Al||PA450 molten salt battery demonstrated exceptional electrochemical performance, showcasing a highly reversible capacity of 135 mA h g−1 and outstanding cyclability for up to 2000 cycles at 10 A g−1 (Fig. 10g).
4.5 Gel polymer electrolyte
Gel polymer electrolytes serve as a bridge between liquid and solid electrolytes, comprising a liquid electrolyte and a polymer matrix, which combine the high ionic conductivity of liquid electrolytes with the safety benefits of solid electrolytes. Lee et al. developed a novel gel polymer electrolyte using ethyl acrylate (EA) as a monomer, AlCl3/[EMIm]Cl (molar ratio of 1.7) ionic liquid as the electrolyte, 2,2-azobisisobutyronitrile (AIBN) as the initiator, and N, N′-methylene-bis-acrylamide (MBAA) as the cross-linking agent.211 The assembled Al||graphite battery delivered a capacity of 90 mA h g−1 at 200 mA g−1 with a high-capacity retention of 95% over l500 cycles. Du et al. proposed a gel polymer electrolyte via in situ cross-linking polymerization of EA monomer and pentaerythritol acrylate (PETEA) crosslinker in a high molar ratio of AlCl3/EMIC ionic liquid (Fig. 10h).209 The gel polymer electrolyte exhibited an ionic conductivity of 1.46 × 10−3 S cm−1 and a wide electrochemical window up to 3.0 V (vs. Al3+/Al), along with alleviated moisture sensitivity and appreciable interfacial stability at room temperature. Consequently, the assembled solid-state Al||graphite battery with the gel polymer electrolyte delivered a stable capacity of ∼90 mA h g−1 for 1000 cycles at 100 mA g−1, both at ambient temperature and a low temperature of −10 °C. Even with a high-loading graphite cathode of 11 mg cm−2, a capacity of ∼60 mA h g−1 could still be obtained for 300 cycles (Fig. 10i).4.6 Electrolyte additive
Electrolyte additives are commonly employed to assist in the formation of a stable SEI layer on the anode surface, which can effectively avoid HER, corrosion, dendrites, passivation, etc. Seh et al. first reported a novel electrolyte combination based on Al(OTf)3 with tetrabutylammonium chloride (TBAC) additive in diglyme (0.5 M Al(OTf)3 + 0.1 M TBAC in diglyme solvent).14 They found that TBAC reduced the charge transfer resistance and the surface activation energy at the anode surface and also augmented the dissociation of Al(OTf)3 to generate the SEI components. Consequently, the reduced anodic overpotential for cells ran for 1300 cycles in Al plating and stripping tests. Du et al. proposed electrolyte interfacial engineering involving the nonionic surfactant (F127) as an interfacial optimizer in ILs (molar ratio of AlCl3:Et3NHCl = 1.5:1, F127 added at 0.5 wt%, F127-0.5), simultaneously modulating the anode–electrolyte and cathode–electrolyte interfaces.210 F127 preferentially adsorbs on the electrode surface, forming a dense and uniform adsorption layer, which can effectively mitigate the corrosion of ILs on the Al anode and regulate the current density to achieve uniform Al deposition. The Al||FG full battery assembled with F127 modifying ILs (F127-0.5) was able to retain a specific capacity of 104.9 mA h g−1 after 1600 cycles, which was higher than ILs (69.0 mA h g−1) (Fig. 10j).
5. Conclusions and future outlook
As one of the promising alternatives for the next generation of energy storage devices, AIBs have received widespread attention and achieved significant research results in recent years. This paper focuses on the progress of different types of cathode materials for AIBs, based on the classification of the charge storage mechanism in terms of charge carriers, to reveal the electrochemical performance. Then the current development status and modification methods of anodes and electrolytes in recent years are reviewed, and an outlook is presented.Although marked progress has been made in the development of cathode materials for AIBs, several problems remain to be solved. Currently, transition metal oxides exhibit high capacity via hosting the Al3+ cation based on the valence change of transition metals, which can transfer multiple electrons. However, the structures of transition metal oxides are prone to collapsing during repeated Al3+ (de)insertion, and they also easily dissolve into the electrolyte, leading to poor cycling stability. Prussian blue analogs can accommodate the (de)insertion of Al3+ due to their unique framework structure. Nevertheless, as a trivalent ion, Al3+ possesses the highest charge density, leading to high electrostatic interaction between the intercalated Al3+ and the host frameworks, as well as poor diffusion kinetics. In addition, the lack of sites available for Al3+ insertion in the main material leads to a low specific capacity. The unique coordination chemistry due to the diverse functional groups of organic compounds provides abundant charge storage. However, the low potential and poor conductivity of organic materials and the problem of cathode material dissolution limit their applications. Carbon materials have good electrical conductivity and stable structure, but only [AlCl4]− anion can be stored, resulting in a low capacity. Sulfur and iodine cathode have high theoretical capacity, high energy density, and low cost, but the dissolution of cathode materials leads to poor cycling stability, and poor conductivity results in poor kinetics.
In view of the above problems, finding new materials or developing new strategies to optimize the cathode materials of the existing AIBs is crucial for large-scale applications. Several strategies for optimizing battery performance by improving cathode materials are summarized: (1) reasonable structure regulation and optimization strategies, such as pre-embedded ions to expand the layer spacing to improve the ion migration rate, altering the phase structure through in situ electrochemical transformations, and designing three-dimensional, core–shell, defective, nanostructures, and polymerized structures, etc.; (2) material composite synergistic strategies, such as compositing with carbon materials to enhance the electrical conductivity and combining with MOF materials to improve the stability of the structure, etc.; (3) theoretical calculations to predict the feasibility analysis of new materials. It can be believed that through in-depth research on AIBs, the realization of AIBs for large-scale energy storage and other fields of widespread application is just around the corner.
For the anode, Al metal can provide satisfactory capacity; however, corrosion, passivation, and dendrites have emerged as troublesome problems. In this regard, we summarize the approaches for anode modification in recent years and make recommendations: (1) functional interfacial layer modulation on the surface of Al anode is considered as a possible solution to the dilemma of the Al anode. Although this approach is widely used at present, the issue still needs to be thoroughly investigated and analyzed. In general, the SEI film should have bi-directional conductivity, i.e., allow Al3+ to shuttle freely and electron not; be thermally and chemically stable; have good mechanical strength and be able to withstand the volume change of the battery during charging and discharging; (2) the utilization of alloy anodes resists passivation as well as corrosion. The doping of various metal elements in the alloy can change the atomic arrangement, thus changing the grain structure and crystalline phase to achieve high anode efficiency and power output to explore new Al-alloy-based anodes with simultaneous Al activation sites and low corrosion; (3) anode-free is the emerging strategy in recent years, characterized by high energy density, low cost, and high safety. However, attention still needs to be paid to the instability of anode morphology change and the reaction problem at the anode–liquid electrolyte interface. In addition, the utilization of advanced theoretical analysis and artificial intelligence in the assembly of batteries may guide the development of anodes for AIBs.
The electrolyte plays a key role in conducting ions between the cathode and anode, which affects the performance of the battery. AIBs moving from the laboratory to commercialization face many challenges in terms of electrolytes. For aqueous battery electrolytes, how to improve their ESW is an urgent issue. Researchers have adopted the method of increasing the electrolyte concentration to improve the ESW, such as the development of water-in-salt electrolytes, aqueous deep eutectic electrolytes, and so on. However, it should be noted that these electrolytes have poor conductivity and there is the need to control the cost compared with dilute solutions. Non-aqueous electrolytes have a natural advantage in terms of ESW, but attention needs to be paid to their sensitivity to air, which limits their application. Ionic liquids are currently the most commonly used electrolytes; in addition, non-aqueous deep eutectic electrolytes, molten salt electrolytes, and gel polymer electrolytes have also been developed in recent years.
In summary, AIBs are still in the early stages. The choice of electrode materials and electrolyte and system matching are scientific issues that need to be emphasized, which usually require a variety of methods to cooperate to realize high-performance AIBs. Although there are still many problems to be solved for AIBs, their advantages of high theoretical capacity, high voltage, and low cost still give them high research value.
Author contributions
Jiajin Zhao: collected papers related to the topic, investigation, and writing of the original draft. Yan Chen, Ziqi An, Mengyan Zhang, Wenfeng Wang, Qiubo Guo: collected papers related to the topic. Yuan Li: writing – reviewing and editing. Shumin Han: project administration, funding acquisition. Lu Zhang: supervision, writing – reviewing, and editing funding acquisition.Data availability
Data availability is not applicable to this article as no new data were created or analyzed in this study.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge financial support from the Science Research Project of Hebei Education Department (No. BJK2022033), National Natural Science Foundation of China (No. 52371239, 52201282 and 52071281), Natural Science Foundation of Hebei Province (No. E2024203037), and Basic Innovation Research Project in Yanshan University (No. 2022LGZD004).References
- J. Lin, X. D. Zhang, E. R. Fan, R. J. Chen, F. Wu and L. Li, Energy Environ. Sci., 2023, 16, 745–791 RSC.
- Z. Liu, Z. Deng, G. He, H. Wang, X. Zhang, J. Lin, Y. Qi and X. Liang, Nat. Rev. Earth Environ., 2022, 3, 141–155 CrossRef.
- J. P. Dees, W. J. Sagues, E. Woods, H. M. Goldstein, A. J. Simon and D. L. Sanchez, Green Chem., 2023, 25, 2930–2957 RSC.
- C. Feng, X. Jiang, Q. Zhou, T. Li, Y. Zhao, Z. Niu, Y. Wu, H. Zhou, M. Wang, X. Zhang, M. Chen, L. Ni, G. Diao and Y. Wei, J. Mater. Chem. A, 2023, 11, 18029–18045 RSC.
- X. Peng, Y. Li, F. Kang, X. Li, Z. Zheng and L. Dong, Small, 2024, 20, 2305547 CrossRef CAS PubMed.
- Q. Zhou, Y. Zheng, D. Wang, Y. Lian, C. Ban, J. Zhao and H. Zhang, Ceram. Int., 2020, 46, 26454–26465 CrossRef CAS.
- Y. Q. Yang, S. Bremner, C. Menictas and M. Kay, Renewable Sustainable Energy Rev., 2018, 91, 109–125 CrossRef.
- B. Diouf and R. Pode, Renewable Energy, 2015, 76, 375–380 CrossRef.
- W. Wang, B. Q. Yuan, Q. Sun and R. Wennersten, J. Energy Storage, 2022, 52, 104812 CrossRef.
- Z. Yang, X. Huang, P. Meng, M. Jiang, Y. Wang, Z. Yao, J. Zhang, B. Sun and C. Fu, Angew. Chem., Int. Ed., 2023, 62, e202216797 CrossRef CAS PubMed.
- M. Tao, J. Chen, H. Lin, Y. Zhou, D. Zhao, P. Shan, Y. Jin and Y. Yang, J. Energy Chem., 2024, 96, 226–248 CrossRef CAS.
- G. Harper, R. Sommerville, E. Kendrick, L. Driscoll, P. Slater, R. Stolkin, A. Walton, P. Christensen, O. Heidrich, S. Lambert, A. Abbott, K. Ryder, L. Gaines and P. Anderson, Nature, 2019, 575, 75–86 CrossRef CAS PubMed.
- R. Buckingham, T. Asset and P. Atanassov, J. Power Sources, 2021, 498, 229762 CrossRef CAS.
- S. Kumar, P. Rama, G. Yang, W. Y. Lieu, D. Chinnadurai and Z. W. Seh, Nano-Micro Lett., 2022, 15, 21 CrossRef PubMed.
- A. L. Phan, C. Jayawardana, P. M. L. Le, J. X. Zhang, B. Nan, W. R. Zhang, B. L. Lucht, S. Y. Hou and C. S. Wang, Adv. Funct. Mater., 2023, 33, 2301177 CrossRef CAS.
- X. G. Miao, H. Y. Wang, R. Sun, C. X. Wang, Z. W. Zhang, Z. Q. Li and L. W. Yin, Energy Environ. Sci., 2020, 13, 3780–3822 RSC.
- M. I. Jamesh and A. S. Prakash, J. Power Sources, 2018, 378, 268–300 CrossRef CAS.
- H. Kim, J. C. Kim, M. Bianchini, D. H. Seo, J. Rodriguez-Garcia and G. Ceder, Adv. Energy Mater., 2018, 8, 1702384 CrossRef.
- G. Shao, H. Liu, L. Chen, M. Wu, D. Wang, D. Wu and J. Xia, Chem. Sci., 2024, 15, 3323–3329 RSC.
- Z. M. Zhao, Y. J. Lei, L. Shi, Z. N. Tian, M. N. Hedhili, Y. Khan and H. N. Alshareef, Angew. Chem., Int. Ed., 2022, 61, e202212941 CrossRef CAS PubMed.
- X. Y. Wu, Y. T. Qi, J. J. Hong, Z. F. Li, A. S. Hernandez and X. L. Ji, Angew. Chem., Int. Ed., 2017, 56, 13026–13030 CrossRef CAS PubMed.
- Z. Chen, Q. Yang, F. Mo, N. Li, G. Liang, X. Li, Z. Huang, D. Wang, W. Huang, J. Fan and C. Zhi, Adv. Mater., 2020, 32, 2001469 CrossRef CAS PubMed.
- Z. W. Tie and Z. Q. Niu, Angew. Chem., Int. Ed., 2020, 59, 21293–21303 CrossRef CAS PubMed.
- F.-D. Yu, Z.-J. Yi, R.-Y. Li, W.-H. Lin, J. Chen, X.-Y. Chen, Y.-M. Xie, J.-H. Wu, Z. Lan, L.-F. Que, B.-S. Liu, H. Luo and Z.-B. Wang, J. Energy Chem., 2024, 91, 245–253 CrossRef CAS.
- G. Zhang, J. Zhu, L. Lin, Y. Liu, S. Li, Q. Li, X.-X. Liu and X. Sun, Chem. Sci., 2024, 15, 3545–3551 RSC.
- S. Yang, Z. Xu, S. Wang, J. Sun, D. Zhao, B. Cao and X. Wang, Green Chem., 2024, 26, 7293–7301 RSC.
- I. A. Rodríguez-Pérez, Y. Yuan, C. Bommier, X. Wang, L. Ma, D. P. Leonard, M. M. Lerner, R. G. Carter, T. Wu, P. A. Greaney, J. Lu and X. Ji, J. Am. Chem. Soc., 2017, 139, 13031–13037 CrossRef PubMed.
- S. Gheytani, Y. Liang, F. Wu, Y. Jing, H. Dong, K. K. Rao, X. Chi, F. Fang and Y. Yao, Adv. Sci., 2017, 4, 1700465 CrossRef PubMed.
- Q. Wei, L. Zhang, X. Sun and T. L. Liu, Chem. Sci., 2022, 13, 5797–5812 RSC.
- D.-Y. Wang, C.-Y. Wei, M.-C. Lin, C.-J. Pan, H.-L. Chou, H.-A. Chen, M. Gong, Y. Wu, C. Yuan, M. Angell, Y.-J. Hsieh, Y.-H. Chen, C.-Y. Wen, C.-W. Chen, B.-J. Hwang, C.-C. Chen and H. Dai, Nat. Commun., 2017, 8, 14283 CrossRef CAS PubMed.
- C. S. Yan, C. Lv, L. G. Wang, W. Cui, L. Y. Zhang, K. N. Dinh, H. T. Tan, C. Wu, T. P. Wu, Y. Ren, J. Q. Chen, Z. Liu, M. Srinivasan, X. H. Rui, Q. Y. Yan and G. H. Yu, J. Am. Chem. Soc., 2020, 142, 15295–15304 CrossRef CAS PubMed.
- H. Liang, Y. Liu, F. Zuo, C. Zhang, L. Yang, L. Zhao, Y. Li, Y. Xu, T. Wang, X. Hua, Y. Zhu and H. Li, Chem. Sci., 2022, 13, 14191–14197 RSC.
- S. Das, S. S. Manna and B. Pathak, ACS Omega, 2021, 6, 1043–1053 CrossRef CAS PubMed.
- W.-D. Pan, C. Liu, M.-Y. Wang, Z.-J. Zhang, X.-Y. Yan, S.-C. Yang, X.-H. Liu, Y.-F. Wang and D. Y. C. Leung, Rare Met., 2021, 41, 762–774 CrossRef.
- T. Dutta and J. Mary Gladis, J. Energy Storage, 2024, 86, 111287 CrossRef.
- X. Yuan, Z. Lin, Y. Duan, Z. Chen, L. Fu, Y. Chen, L. Liu, X. Yuan and Y. Wu, Batteries Supercaps, 2024, 7, e202400263 CrossRef CAS.
- G. L. Holleck and J. Giner, J. Electrochem. Soc., 1972, 119, 1161 CrossRef CAS.
- F. Wu, H. Yang, Y. Bai and C. Wu, Adv. Mater., 2019, 31, e1806510 CrossRef PubMed.
- J. C. J. Dymek, J. L. Williams, D. J. Groeger and J. J. Auborn, J. Electrochem. Soc., 1984, 131, 2887–2892 CrossRef.
- N. Jayaprakash, S. K. Das and L. A. Archer, Chem. Commun., 2011, 47, 12610 RSC.
- X. Wen, Y. Liu, A. Jadhav, J. Zhang, D. Borchardt, J. Shi, B. M. Wong, B. Sanyal, R. J. Messinger and J. Guo, Chem. Mater., 2019, 31, 7238–7247 CrossRef CAS.
- M. Maczka and P. Pasierb, Ceram. Int., 2019, 45, 11041–11049 CrossRef CAS.
- S. Liu, J. J. Hu, N. F. Yan, G. L. Pan, G. R. Li and X. P. Gao, Energy Environ. Sci., 2012, 5, 9743 RSC.
- H. Lahan and S. K. Das, J. Power Sources, 2019, 413, 134–138 CrossRef CAS.
- M. C. Lin, M. Gong, B. Lu, Y. Wu, D. Y. Wang, M. Guan, M. Angell, C. Chen, J. Yang, B. J. Hwang and H. Dai, Nature, 2015, 520, 325–328 CrossRef PubMed.
- Q. Zhao, L. Liu, J. Yin, J. Zheng, D. Zhang, J. Chen and L. A. Archer, Angew. Chem., Int. Ed., 2020, 59, 3048–3052 CrossRef CAS PubMed.
- Y. Guo, W. Wang, H. Lei, M. Wang and S. Jiao, Adv. Mater., 2022, 34, 2110109 CrossRef CAS PubMed.
- Z. Yuan, Q. Lin, Y. Li, W. Han and L. Wang, Adv. Mater., 2023, 2211527 CrossRef CAS PubMed.
- X. Zhang, R. Wang, Z. Liu, Q. Ma, H. Li, Y. Liu, J. Hao, S. Zhang, J. Mao and C. Zhang, Adv. Energy Mater., 2024, 14, 2400314 CrossRef CAS.
- Q. Feng, H. Kanoh, Y. Miyai and K. Ooi, Chem. Mater., 1995, 7, 1722–1727 CrossRef CAS.
- S. Chen, Y. Kong, C. Tang, N. A. Gadelhak, A. K. Nanjundan, A. Du, C. Yu and X. Huang, Small, 2024, 2312229 CrossRef CAS PubMed.
- Q. Zhao, M. J. Zachman, W. I. Al Sadat, J. X. Zheng, L. F. Kourkoutis and L. Archer, Sci. Adv., 2018, 4, eaau8131 CrossRef CAS PubMed.
- M. H. Alfaruqi, S. Islam, J. Lee, J. Jo, V. Mathew and J. Kim, J. Mater. Chem. A, 2019, 7, 26966–26974 RSC.
- S. He, J. Wang, X. Zhang, J. Chen, Z. Wang, T. Yang, Z. Liu, Y. Liang, B. Wang, S. Liu, L. Zhang, J. Huang, J. Huang, L. A. O'Dell and H. Yu, Adv. Funct. Mater., 2019, 29, 1905228 CrossRef CAS.
- F. W. Ming, H. F. Liang, Y. J. Lei, S. Kandambeth, M. Eddaoudi and H. N. Alshareef, ACS Energy Lett., 2018, 3, 2602–2609 CrossRef CAS.
- X. H. Rui, Z. Y. Lu, H. Yu, D. Yang, H. H. Hng, T. M. Lim and Q. Y. Yan, Nanoscale, 2013, 5, 556–560 RSC.
- A. M. Diem, B. Fenk, J. Bill and Z. Burghard, Nanomaterials, 2020, 10, 247 CrossRef CAS PubMed.
- P. De, J. Halder, S. Priya, A. K. Srivastava and A. Chandra, ACS Appl. Energy Mater., 2023, 6, 753–762 CrossRef CAS.
- H. L. Wang, Y. Bai, S. Chen, X. Y. Luo, C. Wu, F. Wu, J. Lu and K. Amine, ACS Appl. Mater. Interfaces, 2015, 7, 80–84 CrossRef CAS PubMed.
- S. Kumar, R. Satish, V. Verma, H. Ren, P. Kidkhunthod, W. Manalastas and M. Srinivasan, J. Power Sources, 2019, 426, 151–161 CrossRef CAS.
- L. D. Reed and E. Menke, J. Electrochem. Soc., 2013, 160, A915–A917 CrossRef CAS.
- E. H. M. Salhabi, J. L. Zhao, J. Y. Wang, M. Yang, B. Wang and D. Wang, Angew. Chem., Int. Ed., 2019, 58, 9078–9082 CrossRef CAS PubMed.
- Y. E. Zhu, L. P. Yang, J. Sheng, Y. N. Chen, H. C. Gu, J. P. Wei and Z. Zhou, Adv. Energy Mater., 2017, 7, 1701222 CrossRef.
- N. Zhu, F. Wu, Z. H. Wang, L. M. Ling, H. Y. Yang, Y. N. Gao, S. N. Guo, L. M. Suo, H. Li, H. J. Xu, Y. Bai and C. Wu, J. Energy Chem., 2020, 51, 72–80 CrossRef.
- S. K. Das, T. Palaniselvam and P. Adelhelm, Solid State Ionics, 2019, 340, 115017 CrossRef CAS.
- M. Kazazi, P. Abdollahi and M. Mirzaei-Moghadam, Solid State Ionics, 2017, 300, 32–37 CrossRef CAS.
- H. Lahan, R. Boruah, A. Hazarika and S. K. Das, J. Phys. Chem. C, 2017, 121, 26241–26249 CrossRef CAS.
- S. Wang, K. V. Kravchyk, S. Pigeot-Rémy, W. Tang, F. Krumeich, M. Wörle, M. I. Bodnarchuk, S. Cassaignon, O. Durupthy, S. Zhao, C. Sanchez and M. V. Kovalenko, ACS Appl. Nano Mater., 2019, 2, 6428–6435 CrossRef CAS.
- H. Lahan and S. K. Das, Dalton Trans., 2019, 48, 6337–6340 RSC.
- M. Mączka, M. Mosiałek and P. Pasierb, Electrochim. Acta, 2022, 424, 140606 CrossRef.
- J. Liu, Z. Li, X. Huo and J. Li, J. Power Sources, 2019, 422, 49–56 CrossRef CAS.
- X. Zhang, G. Zhang, S. Wang, S. Li and S. Jiao, J. Mater. Chem. A, 2018, 6, 3084–3090 RSC.
- L. Geng, J. Scheifers, C. Fu, J. Zhang, B. P. T. Fokwa and J. Guo, ACS Appl. Mater. Interfaces, 2017, 9, 21251–21257 CrossRef CAS PubMed.
- L. Geng, G. Lv, X. Xing and J. Guo, Chem. Mater., 2015, 27, 4926–4929 CrossRef CAS.
- Z. J. Yu, Z. P. Kang, Z. Q. Hu, J. H. Lu, Z. Zhou and S. Q. Jiao, Chem. Commun., 2016, 52, 10427–10430 RSC.
- S. Wang, Z. Yu, J. Tu, J. Wang, D. Tian, Y. Liu and S. Jiao, Adv. Energy Mater., 2016, 6, 1600137 CrossRef.
- W. Pan, J. Mao, Y. Wang, X. Zhao, K. W. Leong, S. Luo, Y. Chen and D. Y. C. Leung, Small Methods, 2021, 5, 2100491 CrossRef CAS PubMed.
- Y. Xu, J. Ma, T. Jiang, H. Ding, W. Wang, M. Wang, X. Zheng, J. Sun, Y. Yuan, M. Chuai, N. Chen, Z. Li, H. Hu and W. Chen, Energy Storage Mater., 2022, 47, 113–121 CrossRef.
- P. Almodóvar, D. A. Giraldo, J. Chancón, I. Álvarez-Serrano and M. L. López, ChemElectroChem, 2020, 7, 2102–2106 CrossRef.
- Q. Zhao, M. J. Zachman, W. I. Al Sadat, J. Zheng, L. F. Kourkoutis and L. Archer, Sci. Adv., 2018, 4, eaau8131 CrossRef CAS PubMed.
- S. He, J. Wang, X. Zhang, J. Chen, Z. Wang, T. Yang, Z. Liu, Y. Liang, B. Wang, S. Liu, L. Zhang, J. Huang, J. Huang, L. A. O'Dell and H. Yu, Adv. Funct. Mater., 2019, 29, 1905228 CrossRef CAS.
- A. M. Diem, J. Bill and Z. Burghard, ACS Appl. Energy Mater., 2020, 3, 4033–4042 CrossRef CAS.
- M. Chiku, H. Takeda, S. Matsumura, E. Higuchi and H. Inoue, ACS Appl. Mater. Interfaces, 2015, 7, 24385–24389 CrossRef CAS PubMed.
- S. Lu, T. Lu, W. Luo, Z. Chao, Y. Liu, Z. Zhang and J. Fan, Energy Fuels, 2022, 36, 7890–7897 CrossRef CAS.
- J. Tu, H. Lei, Z. Yu and S. Jiao, Chem. Commun., 2018, 54, 1343–1346 RSC.
- R. Roy, M. K. Ganesha, P. Dutta, D. Pal and A. K. Singh, ACS Appl. Energy Mater., 2023, 6, 11683–11693 CrossRef CAS.
- X. Xiao, M. Wang, J. Tu, Y. Luo and S. Jiao, ACS Sustainable Chem. Eng., 2019, 7, 16200–16208 CrossRef CAS.
- H. Li, H. Yang, Z. Sun, Y. Shi, H.-M. Cheng and F. Li, Nano Energy, 2019, 56, 100–108 CrossRef CAS.
- S. Guo, H. Yang, M. Liu, X. Feng, H. Xu, Y. Bai and C. Wu, ACS Appl. Energy Mater., 2021, 4, 7064–7072 CrossRef CAS.
- K. Gao, X. Lin, W. Yu, X. Cheng, S. Zhang, S. Li and Z. Zhang, Adv. Mater. Interfaces, 2022, 9, 2200635 CrossRef CAS.
- Y. X. Hu, B. Luo, D. L. Ye, X. B. Zhu, M. Q. Lyu and L. Z. Wang, Adv. Mater., 2017, 29, 1606132 CrossRef PubMed.
- K. Liang, L. Ju, S. Koul, A. Kushima and Y. Yang, Adv. Energy Mater., 2019, 9, 1802543 CrossRef.
- Y. Xu, S. Zheng, H. Tang, X. Guo, H. Xue and H. Pang, Energy Storage Mater., 2017, 9, 11–30 CrossRef.
- Y. Moritomo, M. Takachi, Y. Kurihara and T. Matsuda, Appl. Phys. Express, 2012, 5, 041801 CrossRef.
- N. R. D. Tacconi, K. Rajeshwar and R. O. Lezna, Chem. Mater., 2003, 15, 3046–3062 CrossRef.
- T. Matsuda and Y. Moritomo, J. Nanotechnol., 2012, 2012, 1–8 CrossRef.
- H. Yi, R. Qin, S. Ding, Y. Wang, S. Li, Q. Zhao and F. Pan, Adv. Funct. Mater., 2020, 31, 2006970 CrossRef.
- R. Tao, C. Gao, E. Xie, B. Wang and B. Lu, Chem. Sci., 2022, 13, 10066–10073 RSC.
- S. Qiu, Y. Xu, X. Wu and X. Ji, Electrochem. Energy Rev., 2022, 5, 242–262 CrossRef CAS.
- X. Lamprecht, F. Speck, P. Marzak, S. Cherevko and A. S. Bandarenka, ACS Appl. Mater. Interfaces, 2022, 14, 3515–3525 CrossRef CAS PubMed.
- M. Xia, X. Zhang, T. Liu, H. Yu, S. Chen, N. Peng, R. Zheng, J. Zhang and J. Shu, Chem. Eng. J., 2020, 394, 124923 CrossRef CAS.
- S. Liu, G. L. Pan, G. R. Li and X. P. Gao, J. Mater. Chem. A, 2015, 3, 959–962 RSC.
- X.-H. Liu, J. Peng, W.-H. Lai, Y. Gao, H. Zhang, L. Li, Y. Qiao and S.-L. Chou, Adv. Funct. Mater., 2022, 32, 2108616 CrossRef CAS.
- J. Qian, C. Wu, Y. Cao, Z. Ma, Y. Huang, X. Ai and H. Yang, Adv. Energy Mater., 2018, 8, 1702619 CrossRef.
- A. X. Zhou, L. W. Jiang, J. M. Yue, Y. X. Tong, Q. Q. Zhang, Z. J. Lin, B. H. Liu, C. Wu, L. M. Suo, Y. S. Hu, H. Li and L. Q. Chen, ACS Appl. Mater. Interfaces, 2019, 11, 41356–41362 CrossRef CAS PubMed.
- X. Li, A. Wu, C. Gao, Z. Li and S. W. Lee, Mater. Today Energy, 2023, 31, 101205 CrossRef CAS.
- K. Q. Zhang, T. H. Lee, B. Bubach, H. W. Jang, M. Ostadhassan, J. W. Choi and M. Shokouhimehr, Sci. Rep., 2019, 9, 13665 CrossRef PubMed.
- M. Armand and J. M. Tarascon, Nature, 2008, 451, 652–657 CrossRef CAS PubMed.
- K. C. Kim, Ind. Eng. Chem. Res., 2017, 56, 12009–12023 CrossRef CAS.
- S. Muench, A. Wild, C. Friebe, B. Häupler, T. Janoschka and U. S. Schubert, Chem. Rev., 2016, 116, 9438–9484 CrossRef CAS PubMed.
- A. V. Desai, R. E. Morris and A. R. Armstrong, ChemSusChem, 2020, 13, 4866–4884 CrossRef CAS PubMed.
- K. Q. Qin, J. H. Huang, K. Holguin and C. Luo, Energy Environ. Sci., 2020, 13, 3950–3992 RSC.
- P. G. Pickup and R. A. Osteryoung, J. Am. Chem. Soc., 1984, 106, 2294–2299 CrossRef CAS.
- L. Janiszewska and R. A. Osteryoung, J. Electrochem. Soc., 1987, 134, 2787–2794 CrossRef CAS.
- J. Tang and R. A. Osteryoung, Synth. Met., 1991, 45, 1–13 CrossRef CAS.
- L. M. Goldenberg and R. A. Osteryoung, Synth. Met., 1994, 64, 63–68 CrossRef CAS.
- N. S. Hudak, J. Phys. Chem. C, 2014, 118, 5203–5215 CrossRef CAS.
- M. Walter, K. V. Kravchyk, C. Böfer, R. Widmer and M. V. Kovalenko, Adv. Mater., 2018, 30, e1705644 CrossRef PubMed.
- S. Wang, S. Huang, M. Yao, Y. Zhang and Z. Niu, Angew. Chem., Int. Ed., 2020, 59, 11800–11807 CrossRef CAS PubMed.
- W. Wang, S. Zhang, L. Zhang, R. Wang, Q. Ma, H. Li, J. Hao, T. Zhou, J. Mao and C. Zhang, Adv. Mater., 2024, 36, 2400642 CrossRef CAS PubMed.
- D. J. Kim, D.-J. Yoo, M. T. Otley, A. Prokofjevs, C. Pezzato, M. Owczarek, S. J. Lee, J. W. Choi and J. F. Stoddart, Nat. Energy, 2018, 4, 51–59 CrossRef.
- J. Bitenc, N. Lindahl, A. Vizintin, M. E. Abdelhamid, R. Dominko and P. Johansson, Energy Storage Mater., 2020, 24, 379–383 CrossRef.
- C. Li, P. C. Rath, S.-X. Lu, J. Patra, C.-Y. Su, D. Bresser, S. Passerini and J.-K. Chang, Chem. Eng. J., 2021, 417, 129131 CrossRef CAS.
- P. R. Gifford and J. B. Palmisano, J. Electrochem. Soc., 1987, 134, 610–614 CrossRef CAS.
- J. V. Rani, V. Kanakaiah, T. Dadmal, M. S. Rao and S. Bhavanarushi, J. Electrochem. Soc., 2013, 160, A1781–A1784 CrossRef CAS.
- Y. Kong, C. Tang, X. Huang, A. K. Nanjundan, J. Zou, A. Du and C. Yu, Adv. Funct. Mater., 2021, 31, 2010569 CrossRef CAS.
- S. T. Wang, K. V. Kravchyk, F. Krumeich and M. V. Kovalenko, ACS Appl. Mater. Interfaces, 2017, 9, 28478–28485 CrossRef CAS PubMed.
- H. Y. Hu, T. H. Cai, P. Bai, J. Xu, S. H. Ge, H. Hu, M. B. Wu, Q. Z. Xue, Y. F. Yan, X. L. Gao and W. Xing, Chem. Commun., 2020, 56, 1593–1596 RSC.
- S. Wang, S. Jiao, W.-L. Song, H.-S. Chen, J. Tu, D. Tian, H. Jiao, C. Fu and D.-N. Fang, Energy Storage Mater., 2018, 12, 119–127 CrossRef.
- S. Wang, K. V. Kravchyk, F. Krumeich and M. V. Kovalenko, ACS Appl. Mater. Interfaces, 2017, 9, 28478–28485 CrossRef CAS PubMed.
- D.-Y. Wang, C.-Y. Wei, M.-C. Lin, C.-J. Pan, H.-L. Chou, H.-A. Chen, M. Gong, Y. Wu, C. Yuan, M. Angell, Y.-J. Hsieh, Y.-H. Chen, C.-Y. Wen, C.-W. Chen, B.-J. Hwang, C.-C. Chen and H. Dai, Nat. Commun., 2017, 8, 14283 CrossRef CAS PubMed.
- J. Li, K. S. Hui, S. Ji, C. Zha, C. Yuan, S. Wu, F. Bin, X. Fan, F. Chen, Z. Shao and K. N. Hui, Carbon Energy, 2021, 4, 155–169 CrossRef.
- J. Zhao, C. Zhao, C. Zou, Y. Wang, W. Wang, Q. Peng, Y. Li, S. Han and L. Zhang, ChemSusChem, 2024, 17, e202301109 CrossRef CAS PubMed.
- S. Nandi and S. K. Das, ACS Sustainable Chem. Eng., 2019, 7, 19839–19847 CrossRef CAS.
- X. Dong, H. Chen, H. Lai, L. Wang, J. Wang, W. Fang and C. Gao, J. Energy Chem., 2022, 66, 38–44 CrossRef CAS.
- H. Chen, H. Xu, S. Wang, T. Huang, J. Xi, S. Cai, F. Guo, Z. Xu, W. Gao and C. Gao, Sci. Adv., 2017, 3, eaao7233 CrossRef PubMed.
- H. Chen, F. Guo, Y. Liu, T. Huang, B. Zheng, N. Ananth, Z. Xu, W. Gao and C. Gao, Adv. Mater., 2017, 29, 1605958 CrossRef PubMed.
- H. Xu, H. Chen, H. Lai, Z. Li, X. Dong, S. Cai, X. Chu and C. Gao, J. Energy Chem., 2020, 45, 40–44 CrossRef CAS.
- Z. Liu, J. Wang, X. Jia, W. Li, Q. Zhang, L. Fan, H. Ding, H. Yang, X. Yu, X. Li and B. Lu, ACS Nano, 2019, 13, 10631–10642 CrossRef CAS PubMed.
- H. Huang, F. Zhou, X. Shi, J. Qin, Z. Zhang, X. Bao and Z.-S. Wu, Energy Storage Mater., 2019, 23, 664–669 CrossRef.
- H. Huang, F. Zhou, P. Lu, X. Li, P. Das, X. Feng, K. Müllen and Z.-S. Wu, Energy Storage Mater., 2020, 27, 396–404 CrossRef.
- Q. Zhang, L. Wang, J. Wang, C. Xing, J. Ge, L. Fan, Z. Liu, X. Lu, M. Wu, X. Yu, H. Zhang and B. Lu, Energy Storage Mater., 2018, 15, 361–367 CrossRef.
- E. Zhang, J. Wang, B. Wang, X. Yu, H. Yang and B. Lu, Energy Storage Mater., 2019, 23, 72–78 CrossRef.
- M. Han, Z. Lv, L. Hou, S. Zhou, H. Cao, H. Chen, Y. Zhou, C. Meng, H. Du, M. Cai, Y. Bian and M.-C. Lin, J. Power Sources, 2020, 451, 227769 CrossRef CAS.
- L. Hou, H. Cao, M. Han, Z. Lv, S. Zhou, H. Chen, H. Du, M. Cai, Y. Zhou, C. Meng, Y. Bian and M.-C. Lin, ChemistryOpen, 2020, 9, 812–817 CrossRef CAS PubMed.
- S. Ha, J. C. Hyun, J. H. Kwak, H.-D. Lim, B. S. Youn, S. Cho, H.-J. Jin, H.-K. Lim, S. M. Lee and Y. S. Yun, Chem. Eng. J., 2022, 437, 135416 CrossRef CAS.
- S. C. Jung, Y.-J. Kang, D.-J. Yoo, J. W. Choi and Y.-K. Han, J. Phys. Chem. C, 2016, 120, 13384–13389 CrossRef CAS.
- H. Chen, F. Guo, Y. J. Liu, T. Q. Huang, B. N. Zheng, N. Ananth, Z. Xu, W. W. Gao and C. Gao, Adv. Mater., 2017, 29, 1605958 CrossRef PubMed.
- A. G. Olabi, M. A. Abdelkareem, T. Wilberforce and E. T. Sayed, Renewable Sustainable Energy Rev., 2021, 135, 110026 CrossRef CAS.
- Y. B. Zhou, J. T. Wang, P. He, S. M. Chen, Z. Chen, Y. Q. Zang, Y. Li and Y. Duan, J. Electron. Mater., 2022, 51, 2766–2785 CrossRef CAS.
- H. Xu, T. Bai, H. Chen, F. Guo, J. Xi, T. Huang, S. Cai, X. Chu, J. Ling, W. Gao, Z. Xu and C. Gao, Energy Storage Mater., 2019, 17, 38–45 CrossRef.
- S. K. Das, Angew. Chem., Int. Ed., 2018, 57, 16606–16617 CrossRef CAS PubMed.
- L. Zhang, L. Chen, H. Luo, X. Zhou and Z. Liu, Adv. Energy Mater., 2017, 7, 1700034 CrossRef.
- X. Z. Yu, B. Wang, D. C. Gong, Z. Xu and B. G. Lu, Adv. Mater., 2017, 29, 1604118 CrossRef PubMed.
- A. A. Eliseev, L. V. Yashina, M. M. Brzhezinskaya, M. V. Chernysheva, M. V. Kharlamova, N. I. Verbitsky, A. V. Lukashin, N. A. Kiselev, A. S. Kumskov, R. M. Zakalyuhin, J. L. Hutchison, B. Freitag and A. S. Vinogradov, Carbon, 2010, 48, 2708–2721 CrossRef CAS.
- P. Bhauriyal, A. Mahata and B. Pathak, Chem. – Asian J., 2017, 12, 1944–1951 CrossRef CAS PubMed.
- D. Q. Kong, H. D. Fan, X. F. Ding, H. Y. Hu, L. Zhou, B. Li, C. L. Chi, X. N. Wang, Y. S. Wang, X. H. Wang, D. D. Wang, Y. X. Shen, Z. J. Qiu, T. H. Cai, Y. P. Cui, Y. G. Ren, X. J. Li and W. Xing, Electrochim. Acta, 2021, 395, 139212 CrossRef CAS.
- J. Y. Zhang, L. Zhang, Y. L. Zhao, J. S. Meng, B. H. Wen, K. M. Muttaqi, M. R. Islam, Q. Cai and S. J. Zhang, Adv. Energy Mater., 2022, 12, 2200959 CrossRef CAS.
- K. Gao, X. Lin, W. Yu, X. Cheng, S. Zhang, S. Li and Z. Zhang, Adv. Mater. Interfaces, 2022, 9, 2200635 CrossRef CAS.
- J. B. Goodenough, J. Solid State Electrochem., 2012, 16, 2019–2029 CrossRef CAS.
- Y. X. Yin, S. Xin, Y. G. Guo and L. J. Wan, Angew. Chem., Int. Ed., 2013, 52, 13186–13200 CrossRef CAS PubMed.
- R. P. Fang, S. Y. Zhao, Z. H. Sun, W. Wang, H. M. Cheng and F. Li, Adv. Mater., 2017, 29, 1606823 CrossRef PubMed.
- Y. Z. Wang, D. Zhou, V. Palomares, D. Shanmukaraj, B. Sun, X. Tang, C. S. Wang, M. Armand, T. Rojo and G. X. Wang, Energy Environ. Sci., 2020, 13, 3848–3879 RSC.
- H. Zhang, M. Wang, X.-L. Huang, S. Lu, K. Lu and X. Wu, CCS Chem., 2024, 6, 2289–2304 CrossRef.
- W. Zhang, B. Song, M. Wang, T. Miao, X.-L. Huang, E. Zhang, X. Zhan, Y. Yang, H. Zhang and K. Lu, Energy Environ. Sci., 2024, 17, 5273–5282 RSC.
- Sungjemmenla, C. B. Soni and V. Kumar, Nanoscale Adv., 2021, 3, 1569–1581 RSC.
- J. Zhang, R. He, L. Jia, C. You, Y. Zhang, M. Liu, N. Tian, H. Lin and J. Wang, Adv. Funct. Mater., 2023, 33, 2305674 CrossRef CAS.
- S. Licht and D. Peramunage, J. Electrochem. Soc., 1993, 140, L4–L7 CrossRef CAS.
- G. Cohn, L. Ma and L. A. Archer, J. Power Sources, 2015, 283, 416–422 CrossRef CAS.
- Y. Zhang, L. Ma, R. Tang, X. Zheng, X. Wang, Y. Dong, G. Kong, F. Zhao and L. Wei, Int. J. Hydrogen Energy, 2021, 46, 4936–4946 CrossRef CAS.
- J. Zheng, H. Zhang, T. Xu, S. Ju, G. Xia and X. Yu, Adv. Funct. Mater., 2024, 34, 2307486 CrossRef CAS.
- S. Yang, C. Li, H. Lv, X. Guo, Y. Wang, C. Han, C. Zhi and H. Li, Small Methods, 2021, 5, 2100611 CrossRef CAS PubMed.
- Z. Huang, W. Wang, W. L. Song, M. Wang, H. Chen, S. Jiao and D. Fang, Angew. Chem., Int. Ed., 2022, 134, e202202696 CrossRef.
- C. Xu, T. Diemant, X. Liu and S. Passerini, Adv. Funct. Mater., 2023, 33, 2214405 CrossRef CAS.
- H. C. Yang, L. C. Yin, J. Liang, Z. H. Sun, Y. Z. Wang, H. C. Li, K. He, L. P. Ma, Z. Q. Peng, S. Y. Qiu, C. H. Sun, H. M. Cheng and F. Li, Angew. Chem., Int. Ed., 2018, 57, 1898–1902 CrossRef CAS PubMed.
- Z. Zhang, W. Ling, N. Ma, J. Wang, X. Chen, J. Fan, M. Yu and Y. Huang, Adv. Funct. Mater., 2024, 34, 2310294 CrossRef CAS.
- J. Zhao, Y. Chen, M. Zhang, Z. An, B. Nian, W. Wang, H. Wu, S. Han, Y. Li and L. Zhang, Adv. Sci., 2024, 2410988 CrossRef PubMed.
- S. Bi, H. Wang, Y. Zhang, M. Yang, Q. Li, J. Tian and Z. Niu, Angew. Chem., Int. Ed., 2023, 62, e202312982 CrossRef CAS PubMed.
- H. Tian, S. Zhang, Z. Meng, W. He and W.-Q. Han, ACS Energy Lett., 2017, 2, 1170–1176 CrossRef CAS.
- S. Zhang, X. Tan, Z. Meng, H. Tian, F. Xu and W.-Q. Han, J. Mater. Chem. A, 2018, 6, 9984–9996 RSC.
- C. Yan, C. Lv, B.-E. Jia, L. Zhong, X. Cao, X. Guo, H. Liu, W. Xu, D. Liu, L. Yang, J. Liu, H. H. Hng, W. Chen, L. Song, S. Li, Z. Liu, Q. Yan and G. Yu, J. Am. Chem. Soc., 2022, 144, 11444–11455 CrossRef CAS PubMed.
- L. Yao, S. Ju, T. Xu, W. Wang and X. Yu, ACS Nano, 2023, 17, 25027–25036 CrossRef CAS PubMed.
- S. Peng, X. Zhou, S. Tunmee, Z. Li, P. Kidkhunthod, M. Peng, W. Wang, H. Saitoh, F. Zhang and Y. Tang, ACS Sustainable Chem. Eng., 2021, 9, 3710–3717 CrossRef CAS.
- S. Wang, Y. Guo, X. Du, L. Xiong, Z. Huang, X. Li, M. Ma, Z. Liang and Y. Xie, Energy Storage Mater., 2023, 60, 102826 CrossRef.
- Q. Hao, F. Chen, X. Chen, Q. Meng, Y. Qi and N. Li, Electrochim. Acta, 2022, 421, 140495 CrossRef CAS.
- J. Zheng, D. C. Bock, T. Tang, Q. Zhao, J. Yin, K. R. Tallman, G. Wheeler, X. Liu, Y. Deng, S. Jin, A. C. Marschilok, E. S. Takeuchi, K. J. Takeuchi and L. A. Archer, Nat. Energy, 2021, 6, 398–406 CrossRef CAS.
- R. Bai, J. Yang, G. Li, J. Luo and W. Tang, Energy Storage Mater., 2021, 41, 41–50 CrossRef.
- S. Kumar, T. Salim, V. Verma, W. Manalastas and M. Srinivasan, Chem. Eng. J., 2022, 435, 134742 CrossRef CAS.
- H. Yu, C. Lv, C. Yan and G. Yu, Small Methods, 2024, 8, 2300758 CrossRef CAS PubMed.
- D. R. Egan, C. Ponce de León, R. J. K. Wood, R. L. Jones, K. R. Stokes and F. C. Walsh, J. Power Sources, 2013, 236, 293–310 CrossRef CAS.
- Q. Dou, N. Yao, W. K. Pang, Y. Park, P. Xiong, X. Han, H. H. Rana, X. Chen, Z.-H. Fu, L. Thomsen, B. Cowie, Y. Kang, Q. Liu, D. H. Min, Y. M. Jung, Z. Guo, Q. Zhang and H. S. Park, Energy Environ. Sci., 2022, 15, 4572–4583 RSC.
- Q. Ran, H. Shi, H. Meng, S.-P. Zeng, W.-B. Wan, W. Zhang, Z. Wen, X.-Y. Lang and Q. Jiang, Nat. Commun., 2022, 13, 576 CrossRef CAS PubMed.
- Q. Ran, S. P. Zeng, M. H. Zhu, W. B. Wan, H. Meng, H. Shi, Z. Wen, X. Y. Lang and Q. Jiang, Adv. Funct. Mater., 2022, 33, 2211271 CrossRef.
- Y. Meng, J. Wang, M. Wang, Q. Peng, Z. Xie, Z. Zhu, Z. Liu, W. Wang, K. Zhang, H. Liu, Y. Ma, Z. Li and W. Chen, Adv. Energy Mater., 2023, 13, 2301322 CrossRef CAS.
- B.-E. Jia, E. Hu, Z. Hu, J. J. Liew, Z. Hong, Y. Guo, M. Srinivasan, Q. Zhu, J. Xu, J. Chen, H. Pan and Q. Yan, Energy Storage Mater., 2024, 65, 103141 CrossRef.
- Q. Zhao, J. Zheng, Y. Deng and L. Archer, J. Mater. Chem. A, 2020, 8, 23231–23238 RSC.
- L. Wang, X. Song, Y. Hu, W. Yan, Z. Tie and Z. Jin, Energy Storage Mater., 2022, 44, 461–468 CrossRef.
- C. Lu, F. Zhao, B. Tao, Z. Wang, Y. Wang, J. Sheng, G. Tang, Y. Wang, X. Guo, J. Li and L. Wei, Small, 2024, 20, 2402025 CrossRef CAS PubMed.
- Y. Gao, Y. Li, H. Yang, L. Zheng, Y. Bai and C. Wu, J. Energy Chem., 2022, 67, 613–620 CrossRef CAS.
- W. Tang, L. Deng, L. Guo, S. Zhou, Q. Jiang and J. Luo, Green Energy Environ., 2024, 9, 1183–1191 CrossRef CAS.
- X. Luo, R. Wang, L. Zhang, Z. Liu, H. Li, J. Mao, S. Zhang, J. Hao, T. Zhou and C. Zhang, ACS Nano, 2024, 18, 12981–12993 CrossRef CAS PubMed.
- L. Kang, Q. Li, K. Luo, S. Zhong and D. Yan, ACS Sustainable Chem. Eng., 2023, 11, 7334–7343 CrossRef CAS.
- C. Xu, T. Diemant, X. Liu and S. Passerini, Adv. Mater., 2024, 36, 2400263 CrossRef CAS PubMed.
- J. Tu, J. Wang, H. Zhu and S. Jiao, J. Alloys Compd., 2020, 821, 153285 CrossRef CAS.
- K. Wang, K. Liu, C. Yang, Z. Chen, H. Zhang, Y. Wu, Y. Long, Y. Jin, X. He, M. Li and H. Wu, Energy Storage Mater., 2022, 48, 356–365 CrossRef.
- J. Meng, X. Yao, X. Hong, L. Zhu, Z. Xiao, Y. Jia, F. Liu, H. Song, Y. Zhao and Q. Pang, Nat. Commun., 2023, 14, 3909 CrossRef CAS PubMed.
- K. Wang, Y. Wu, C. Yang, M. Yu, C. Lei, Y. Zhang, T. Ding, Y. Long, K. Liu, M. Li and H. Wu, Adv. Energy Mater., 2024, 14, 2400589 CrossRef CAS.
- K. Han, X. Qiao, X. Wang, M. Huang, Z. Zhong, Q. Zhang, C. Niu, J. Meng and L. Mai, Nano Energy, 2024, 129, 110085 CrossRef CAS.
- S. Zhang, Z. Liu, R. Liu, L. Du, L. Zheng, Z. Liu, K. Li, M.-C. Lin, Y. Bian, M. Cai and H. Du, J. Power Sources, 2023, 575, 233110 CrossRef CAS.
- Y. Xie, Y. Meng, M. Liu, S. Wang, Y. Guo, Z. Liang and X. Du, Adv. Funct. Mater., 2024, 34, 2411395 CrossRef CAS.
- I. Kim, S. Jang, K. H. Lee, Y. Tak and G. Lee, Energy Storage Mater., 2021, 40, 229–238 CrossRef.
| This journal is © The Royal Society of Chemistry 2025 |