
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
PepMNet: a hybrid deep learning model for predicting peptide properties using hierarchical graph representations†
Daniel Garzon Otero ,
Omid Akbari and
Camille Bilodeau‡
*
,
Omid Akbari and
Camille Bilodeau‡
*
University of Virginia, Chemical Engineering Department, 385 McCormick Road, Charlottesville, VA 22903, USA. E-mail: cur5wz@virginia.edu
First published on 11th December 2024
Abstract
Peptides are a powerful class of molecules that can be applied to a range of problems including biomaterials development and drug design. Currently, machine learning-based property prediction models for peptides primarily rely on amino acid sequence, resulting in two key limitations: first, they are not compatible with non-natural peptide features like modified sidechains or staples, and second, they use human-crafted features to describe the relationships between different amino acids, which reduces the model's flexibility and generalizability. To address these challenges, we have developed PepMNet, a deep learning model that integrates atom-level and amino acid-level information through a hierarchical graph approach. The model first learns from an atom-level graph and then generates amino acid representations based on the atomic information captured in the first stage. These amino acid representations are then combined using graph convolutions on an amino acid-level graph to produce a molecular-level representation, which is then passed to a fully connected neural network for property prediction. We evaluated this architecture by predicting two peptide properties: chromatographic retention time (RT) as a regression task and antimicrobial peptide (AMP) activity as a classification task. For the regression task, PepMNet achieved an average R2 of 0.980 across eight datasets, which spanned different dataset sizes and three liquid chromatography (LC) methods. For the classification task, we developed an ensemble of five models to reduce overfitting and ensure robust classification performance, achieving an area under the receiver operating curve (AUC-ROC) of 0.978 and an average precision of 0.981. Overall, our model illustrates the potential for hierarchical deep learning models to learn peptide properties without relying on human engineering amino acid features.
Design, System, ApplicationPepMNet, a hierarchical graph neural network, addresses the limitations of current peptide property prediction models by integrating atomic and amino acid-level information using a multilevel graph convolutional architecture. Specifically, existing peptide models primarily rely on human-engineered amino acid features which are not compatible with non-natural featuressuch as staples or non-natural sidechains and introduce bias into the learning process. In contrast, PepMNet learns amino acid features from their atomic structure and then subsequently learns global peptide properties from these features. We demonstrate the versatility of PepMNet by evaluating its ability to predict peptide chromatographic retention time (a regression task) and antimicrobial activity (a classification task). The immediate applications of this model lie in peptide-based drug discovery, where rapid and accurate prediction of properties such as retention time and antimicrobial activity are valuable for the development of high throughput discovery assays and the identification of drug candidates, respectively. In the future, PepMNet's design can straightforwardly be extended to other biopolymer or synthetic polymer systems, providing a powerful framework for predicting properties across a wide variety of molecular systems. |
1. Introduction
Machine learning (ML) is a powerful tool for enabling rapid molecular design and discovery.1–4 In recent years, two parallel sub-fields have emerged within the field of molecular property prediction, one focused on predicting the properties of small molecules5–7 and a second focused on predicting the properties of peptides.1,4,8–25 For small molecule property prediction, it is necessary to learn properties directly from atom-level information and one of the most common methods of doing this is by representing the molecule as an atomic graph, where atoms are represented by nodes and bonds are represented by edges. In contrast, for peptide property prediction, it is more common to represent the peptide as a sequence of amino acids,26,27 with no atom-level information being given directly to the model. While amino acid-based peptide models have had some successes,4 they face two major problems: 1) they are not compatible with non-natural features, such as the inclusion of non-natural sidechains, staples, or peptoid units, and 2) they include human-crafted peptide features which introduce bias into the model.To address these limitations, we have developed PepMNet, a hybrid, deep learning approach which incorporates both atom-level and amino acid-level information via a hierarchical graph model. The first stage of our model borrows from commonly used deep learning architectures for small molecule property prediction with the model learning directly from the atom-level graph. We then compute the representations of each amino acid based on information about their constituent atoms, resulting in a coarse-grained molecular graph where nodes represent amino acids and edges represent adjacencies between them. Finally, we perform graph convolutions on the amino acid-level graph and sum over amino acid features to obtain a molecular-level representation which can be used for peptide property prediction. Importantly, by learning amino acid features from atom-level information, our model can learn the relationships between the atomic configurations of amino acids allowing it to better represent natural amino acids. Theoretically, this method can be straightforwardly extended to incorporate any peptide chemical groups such as non-natural sidechains or nonlinear peptide features, such as staples or cycles. In this way, the proposed model offers a more flexible and less biased alternative compared with current state-of-the-art property prediction models.
We evaluated our model by applying it to predict two model peptide properties: chromatographic retention time as a model regression task and antimicrobial peptide classification as a model classification task. Liquid chromatography (LC) is one of the most common techniques for identifying and quantifying the composition of peptide mixtures and plays a key role in most peptide discovery workflows.28,29 Different types of LC, such as strong cation exchange (SCX), reversed-phase LC (RPLC), and hydrophilic interaction LC (HILIC), are commonly utilized to effectively separate and analyze peptide samples.28 Chromatographic retention time (RT) is defined as the time required for a peptide to elute from a chromatography column and is determined by the strength of non-covalent interactions (e.g. charge, hydrophobicity, or hydrogen bonding) between the peptide and the stationary phase. Here, we evaluate our model's ability to predict RT for a variety of chromatographic modes because 1) an accurate RT prediction model can be used to facilitate the development of analytical and preparative peptide purification methods, and 2) multiple, high quality, publicly available datasets exist for model training.
We additionally evaluated our model using antimicrobial peptide (AMP) classification as a model classification task. AMPs are short, positively charged, amphipathic peptides that present a promising alternative to traditional antibiotics for addressing microbial resistance.30 AMPs offer a broad range of activity, low toxicity, and minimal development of microbial resistance, making them a valuable tool in the fight against resistant pathogens.30 ML-based AMP classification has gained interest in recent years as a strategy for reducing the time and resource intensive experiments required to screen new candidate peptides.2,31,32 In this way, AMP classification is a good model classification problem because 1) an accurate AMP classification model can be used to design new AMPs, and 2) there exist public AMP datasets for model training (albeit with fewer datapoints than RT datasets).
Previous studies have explored the development of various shallow and deep learning approaches for predicting both RT and antimicrobial activity. For example, DeepRT utilizes deep learning techniques to encode amino acid vectors within peptides, enabling accurate prediction of peptide retention times for various LC types.28 Similarly, AmPEP converts peptide sequences into a feature vector derived from physicochemical descriptors, which serves as input for a random forest model used to classify AMPs.1 As noted earlier, each of these methods relies on amino acid-level features instead of atom-level features making them less flexible than atomic models. A notable exception to this trend is the AMP-Net developed by Ruiz et al. which learns antimicrobial activity directly from the atom-level graph, which, when combined with peptide physicochemical properties, facilitates classification into AMPs or Non-AMPs.33 While AMP-Net provides a flexible alternative to previous methods, it sacrifices predictive power, failing to outperform random forest classifiers such as AmPEP.
In this work, we build upon these previous methodologies to develop PepMNet, a deep hierarchical graph model for peptide property prediction and we evaluate our model on two model tasks, chromatographic RT prediction and AMP classification. To quantify the impact of our hierarchical strategy we compare our model with non-hierarchical models trained on only atom-level or amino acid-level graphs. We additionally explore the impact of a series of deep learning strategies including graph convolutional layer choice, amino acid feature concatenation, and ensembling and we benchmark our model against current, state-of-the-art models. Finally, we explore how trends in the training datasets used impact molecular properties and model predictions. Overall, the resulting model achieves on par or better performance than other peptide property prediction models, while also offering greater flexibility in its ability to incorporate non-natural features. To make it straightforward to reproduce our results and repurpose our models, we have made our code publicly available on GitHub (https://github.com/danielgarzonotero/PepMNet.git).
2. Methods
2.1 Dataset curation and splitting
To evaluate PepMNet's ability to predict chromatographic retention time, we used a set of eight datasets curated previously by Ma et al.28 As shown in Table 1, these datasets were diverse, representing three modes of chromatography (RPLC,34–36 SCX,37 and HILIC38) and encompassing a wide range of dataset sizes spanning from 1170 peptides to 146![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 587 peptides. We trained and tested our model on each dataset separately using random splits of 90% and 10% for training and testing respectively. Instead of employing k-fold cross-validation, we opted for a random 90%/10% split for training and testing due to the large number (eight) and varied sizes of the datasets. This approach aims to balance computational feasibility with performance estimation. To account for prediction uncertainty, we performed the training process in triplicate for each dataset, and the final average performance metrics for each training and test set are reported in Table 1. We note that all datasets contain naturally occurring peptides without any synthetic modifications and most derived from digests of protein or peptide mixtures, except for the HeLa dataset, which includes peptides with modified amino acids, such as oxidized methionine, phosphorylated serine, phosphorylated threonine, and phosphorylated tyrosine, which were removed before model training.28
587 peptides. We trained and tested our model on each dataset separately using random splits of 90% and 10% for training and testing respectively. Instead of employing k-fold cross-validation, we opted for a random 90%/10% split for training and testing due to the large number (eight) and varied sizes of the datasets. This approach aims to balance computational feasibility with performance estimation. To account for prediction uncertainty, we performed the training process in triplicate for each dataset, and the final average performance metrics for each training and test set are reported in Table 1. We note that all datasets contain naturally occurring peptides without any synthetic modifications and most derived from digests of protein or peptide mixtures, except for the HeLa dataset, which includes peptides with modified amino acids, such as oxidized methionine, phosphorylated serine, phosphorylated threonine, and phosphorylated tyrosine, which were removed before model training.28
| Dataset | LC type | No. peptides | R2 training set | R2 testing set |
|---|---|---|---|---|
| HeLa | RPLC | 1170 | 0.9894 ± 0.0041 | 0.9427 ± 0.0045 |
| Yeast | RPLC | 14361 |
0.9927 ± 0.0032 | 0.9825 ± 0.0043 |
| Misc | RPLC | 146587 |
0.9919 ± 0.0005 | 0.9885 ± 0.0006 |
| SCX | SCX | 30482 |
0.9962 ± 0.0014 | 0.9942 ± 0.0012 |
| Luna HILIC | HILIC | 36271 |
0.9922 ± 0.0014 | 0.9841 ± 0.0017 |
| Xbridge | HILIC | 40290 |
0.9928 ± 0.0020 | 0.9876 ± 0.0023 |
| Atlantis silica | HILIC | 39091 |
0.9891 ± 0.0009 | 0.9809 ± 0.0008 |
| Luna silica | HILIC | 37110 |
0.9905 ± 0.0047 | 0.9829 ± 0.0048 |
For AMP classification, we used the datasets recently curated by Ruiz et al.33 which contains 23919 peptides, with 13334 classified as AMPs and 10585 as non-AMPS (Table 2). To assess the performance and differences between the proposed hierarchical and non-hierarchical graph models, a split of 80% and 20% was used for training and validation. To facilitate comparison across models, classification performance was evaluated on the same test dataset used previously by Ruiz et al. (Table 2). Additionally, we compared our model's performance against previous machine learning approaches, AMPEP and AMPepPy.1,14,33 We implemented 5-fold cross-validation during training to ensure robust evaluation and mitigate overfitting. The final model is an ensemble of the models from each fold.
| Dataset | AMP | Non-AMP | Total |
|---|---|---|---|
| Training and validation | 10667 |
8466 | 19133 |
| AMP testing | 2667 | 2119 | 4786 |
2.2 Model architecture
To predict peptide properties, our model represents peptides as hierarchical graphs, with a coarse-grained graph representing the amino acid composition and connectivity and a fine-grained graph representing the atomic composition and connectivity, as illustrated in Fig. 1. The key advantage of representing peptides in this manner is that the neural network can learn amino acid-level features from the underlying atomic structure of the amino acid, rather than requiring separate featurization such as one-hot encoding or composition, transition, and distribution descriptors.26 Based on this representation, PepMNet employs a multi-stage graph convolutional architecture as shown in Fig. 2.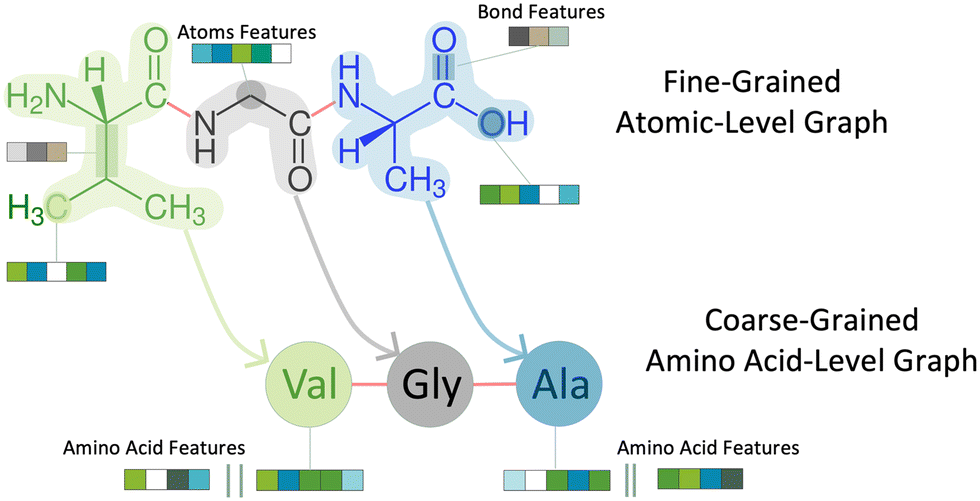 | ||
| Fig. 1 Construction of atomic-level graph from amino acid sequence. | ||
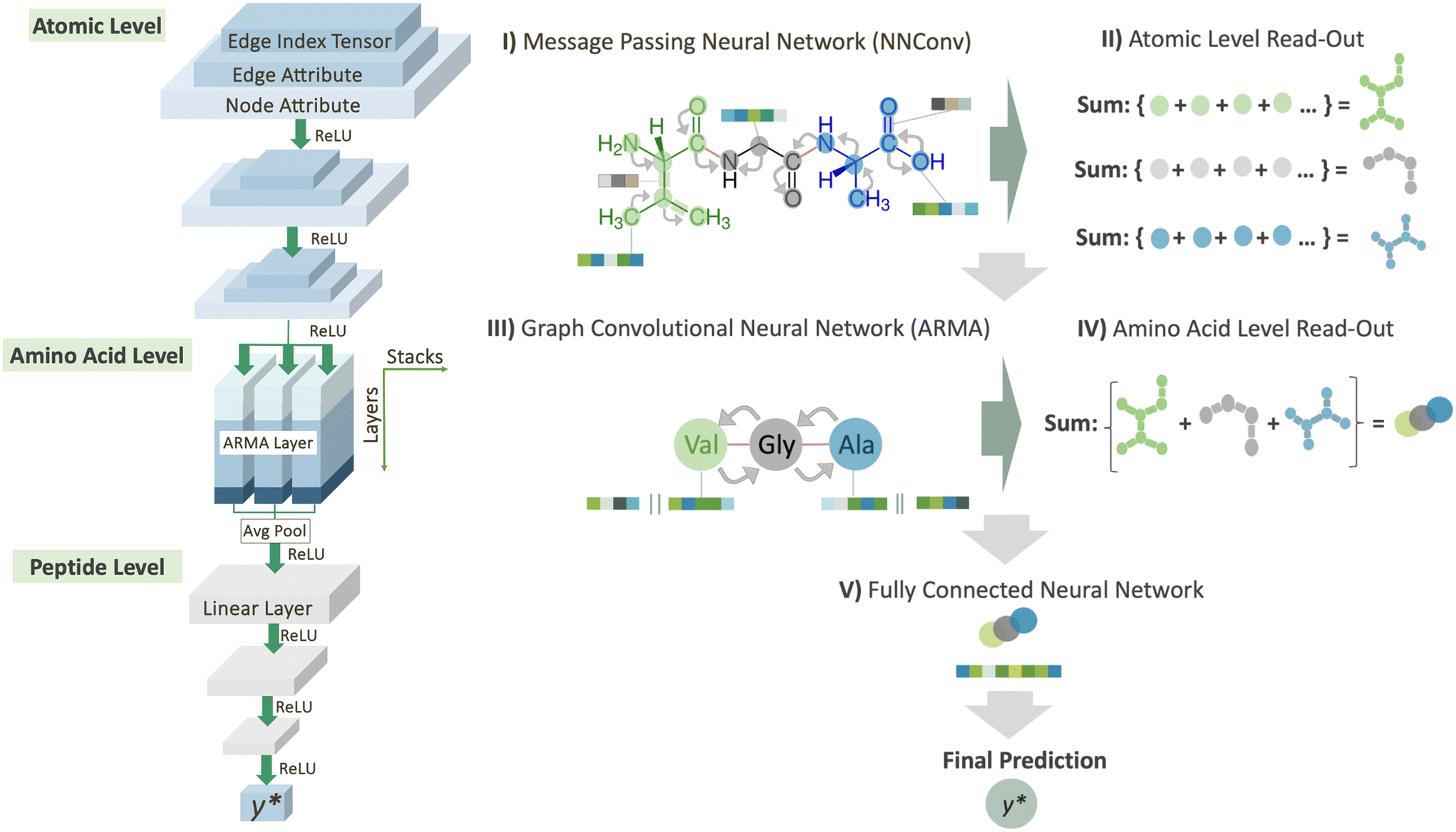 | ||
| Fig. 2 Workflow from atom-level information to final peptide representation. I) To fully utilize all available information within a molecule, including atom details, bond information, and their distribution throughout the molecule, we implemented message passing neural networks (NNConv). II) A readout was performed on the specific atoms of each amino acid in a peptide following the NNConv layers. III) After the readout phase, the resulting graph consists of nodes representing amino acid features, which are derived from atomic-level information and are used to capture the overall structure of the peptide trough a graph convolutional layer (ARMA). IV) Following the amino acid stage, we conducted an aggregation process over the amino acids in the peptide, aiming to obtain a final peptide representation that accounts for the relevance of each amino acid in the sequence. V) This final representation was further processed through linear layers to obtain a single, comprehensive representation of the peptide. | ||
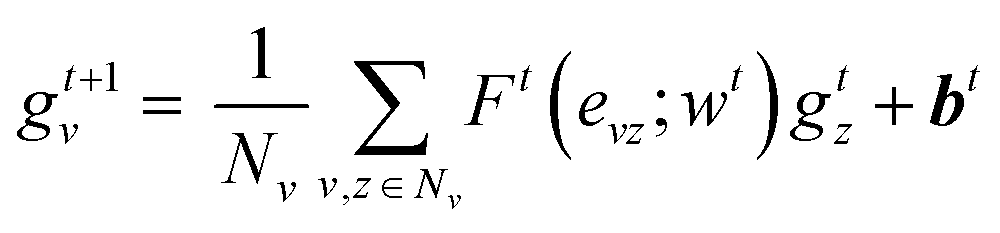 | (1) |
 | (2) |
| H0 = a = R({gTv|v ∈ A}) | (3) |
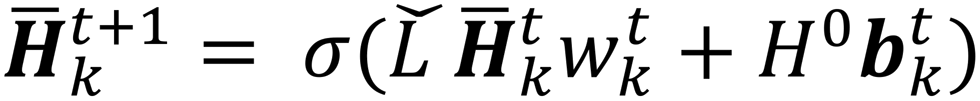 | (4) |
 is the modified Laplacian matrix, K is the number of parallel stacks, and T is the number of timesteps.41 In this way, during stage III, the information of each amino acid, represented by H, is sharing along the coarse-grained graph through ARMA spectral convolution. This convolution stage allows the model to learn more complex relationships between different amino acids. Here, after T timesteps, the feature matrices for each skip were averaged over the K stacks, to obtain a single feature matrix,
is the modified Laplacian matrix, K is the number of parallel stacks, and T is the number of timesteps.41 In this way, during stage III, the information of each amino acid, represented by H, is sharing along the coarse-grained graph through ARMA spectral convolution. This convolution stage allows the model to learn more complex relationships between different amino acids. Here, after T timesteps, the feature matrices for each skip were averaged over the K stacks, to obtain a single feature matrix, 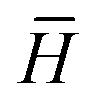 :
:
 | (5) |
 | (6) |
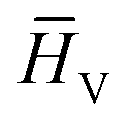 refers to the feature vector for amino acid V in peptide P and y* refers to the full peptide feature vector. For models where additional amino acid-level features were introduced, these features were concatenated to H0 before performing convolutions. Finally, in stage V, y* is passed to a linear fully connected neural network (FCNN) to yield the final prediction (shown in Fig. 2V).
refers to the feature vector for amino acid V in peptide P and y* refers to the full peptide feature vector. For models where additional amino acid-level features were introduced, these features were concatenated to H0 before performing convolutions. Finally, in stage V, y* is passed to a linear fully connected neural network (FCNN) to yield the final prediction (shown in Fig. 2V).2.3 Atomic and amino acid features
Atom-level features were designed based on the features used previously in ChemProp, a graph convolutional model for small molecule property prediction developed previously by Yang et al.42 and further developed by Heid and coworkers.43 The atomic and bond features selected are listed in Table 3, with each feature encoded using a one-hot encoding. Additionally, at the amino acid stage we tested our model with and without additional amino acid features as illustrated in Fig. 2. These features include molecular weight, aromaticity,44 hydrophobicity,45 net charge at pH 7, isoelectric point, logP,46 and number of atoms, calculated using the BioPython47 and RDKit48 library, as listed in Table 3. To process the peptide sequences in the datasets, we implemented the HELM notation, as described by Zhang et al.49 Each peptide was transformed into HELM notation and then converted into a molecule using the RDKit library.48| Feature | Type feature | Description | Size |
|---|---|---|---|
| Atom type | Atom | Type of atom by atomic number | 4 |
| Aromaticity | Atom | Whether this atom is part of an aromatic system | 2 |
| Number of bonds | Atom | Number of bonds the atom is involved in | 3 |
| Number of H2 bonds | Atom | Number of bonded hydrogen atoms | 4 |
| Hybridization | Atom | sp, sp2, sp3, sp3d, or sp3d2 | 2 |
| Implicit valence | Atom | Number of implicit H2 on the atom | 4 |
| Bond type | Bond | Single, double, triple, or aromatic | 3 |
| In ring | Bond | Whether the bond is part of a ring | 2 |
| Conjugated | Bond | Whether the bond is conjugated | 2 |
| Aromaticity bond | Bond | Whether the bond is aromatic | 2 |
| Valence contribution i | Bond | Contribution of the bond to the valence of atom i | 2 |
| Valence contribution f | Bond | Contribution of the bond to the valence of atom f | 2 |
| W amino acid | Amino acid | Amino acid molecular weight | 1 |
| Aromaticity | Amino acid | Aromaticity | 2 |
| Hydrophobicity | Amino acid | GRAVY | 1 |
| Net charge | Amino acid | Charge at pH 7 | 1 |
| Isoelectric point | Amino acid | Isoelectric point | 1 |
| LogP |
Amino acid | Octanol–water partition coefficient | 1 |
| Atoms number | Amino acid | Number of atoms in the amino acid | 1 |
2.4 Non-hierarchical graph models
To measure the contribution of the hierarchical architecture to model performance, we compared PepMNet with two types of non-hierarchical graph models, one using only atomic information and one using only amino acid information. For the atomic, non-hierarchical graph models, graph convolutions were performed over the atomic level graph only, after which the features of all atoms were summed to obtain the final peptide representation. We evaluated a variety of convolutional layers including those that operate in the spatial domain (over nodes and their neighborhoods) and those in the spectral domain (using the graph Laplacian spectrum). The spatial layers included NNConv,6,39 SAGEConv,50 TransformerConv,51 GATConv,52 and EGConv,53 and the spectral layers included GCNConv54 and ARMAConv.41 All convolutional layers were implemented using the PyTorch Geometric40 library with equal layer size.Similarly, we compared PepMNet with non-hierarchical graph models using only amino acid information. To obtain initial representations of the amino acids, we summed the features of the atoms within each amino acid, effectively removing the atomic graph convolutions performed in stages I and II in PepMNet. We additionally concatenated the amino acid features listed in Table 3. Each of the layers tested for the atomic non-hierarchical graph was also tested for the amino acid non-hierarchical graph except NNConv, which was not included because it requires bond features (not present in the amino acid graph).
3. Results
The objective of this work is to develop a hierarchical graph neural network architecture that uses feature extraction at the atomic-level and the amino acid-level to learn peptide properties. We evaluated the performance of our model and compared it to multiple non-hierarchical approaches for two model problems: 1) retention time (RT) prediction, which served as a model regression problem, and 2) antimicrobial peptide (AMP) identification, which served as a model classification problem. Both tasks are good model problems because they each play a role in peptide design and discovery2,30,55,56 and are supported by well-established datasets and databases, enabling robust model training and evaluation.3.1 Regression task-retention time prediction
The primary innovation of PepMNet is that the model learns properties directly from their atomic and amino acid compositions, providing a model that is not constrained by predefined amino acid property classifications and is adaptable enough to capture peptide characteristics by leveraging both atomic and amino acid features. To evaluate the performance of our hierarchical strategy, we compared PepMNet with two types of non-hierarchical models: one that extracts features from the atomic-level graph (Fig. 3a) and a second that extracts features from an amino acid-level graph (Fig. 3b). The hyperparameter configuration for PepMNet and the non-hierarchical model are listed in Tables S1 and S2,† respectively. To robustly compare our model to the non-hierarchical benchmarks, we trained and tested each model using eight different chromatographic retention time datasets, which vary in terms of their size and composition. Additionally, because the performance of the non-hierarchical models is expected to change depending on the type of graph convolutional layer used, we trained each non-hierarchical model with a range of layer types. Specifically, we trained non-hierarchical models with a series of spatial graph architectures, NNConv,6,39 SAGEConv,50 TransformerConv,51 GATConv,52 and EGConv,53 and two spectral graph architectures, GCNConv54 and ARMA.41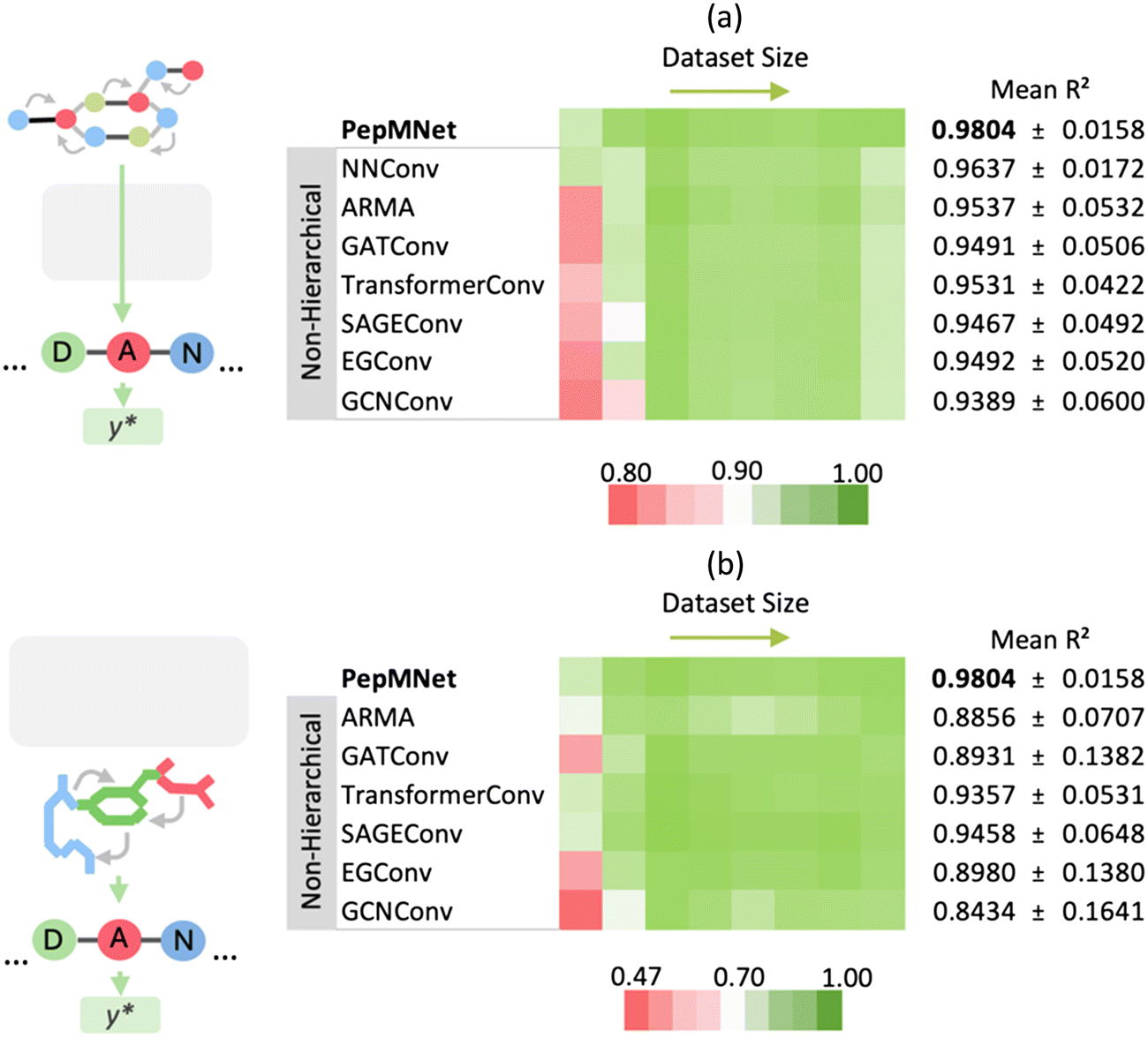 | ||
| Fig. 3 Performance comparison of PepMNet vs. non-hierarchical graph models trained on the (a) atomic-level graph and (b) amino acid-level graph for retention time prediction. PepMNet, which integrates both atomic- and amino acid-level feature extraction, outperformed models trained on either level alone . The final PepMNet architecture used NNConv for atomic-level and ARMA for amino acid-level graphs. | ||
As shown in Fig. 3, PepMNet outperformed all models trained on either the atomic-level graph alone (Fig. 3a) or the amino acid-level graph alone (Fig. 3b), regardless of the type of graph convolutional layer used, achieving a mean R2 of 0.9804 across the RT datasets. Further, PepMNet outperformed each non-hierarchical model, regardless of dataset size. This illustrates that integrating both atomic- and amino acid-level feature extraction leads to more robust model development than learning from either the atomic- or amino acid-level graphs alone. For the final PepMNet architecture we selected NNConv6,39 at the atomic level for its ability to incorporate bond features and ARMA41 layer at the amino acid stage which provided the best performance at this stage after hyperparameter optimization (Table S3†). The training for each dataset was performed in triplicate, and the results of each training along with the scatter plots for each RT dataset are shown in Table S4† and Fig. S1,† respectively.
Throughout the seven graph convolutional layers tested, the ARMA layer, a spectral layer that uses autoregressive filters to update node embeddings,41 exhibited the second best performance for the atomic-level graph, achieving a mean R2 of 0.9537 after the NNConv layers (R2 of 0.9637). Conversely, for the amino acid-level graph model, the SAGEConv layer, a spatial graph convolutional layer, demonstrated superior performance with a mean R2 of 0.9458.50 Overall, the relative performance of models trained on different layer types varied depending on the training dataset used, such that there were no clear “winners” among the graph convolutional layers. This highlights that while there may be advantages to using specific layer types in specific contexts, it is unclear whether in practice there are significant advantages to using one over another.
Fig. 4 illustrates the parity plots for PepMNet and each non-hierarchical model (using the ARMA layer) on two chromatographic retention time datasets, a large dataset called misc (containing 146587 peptides) and a small dataset called HeLa (containing 1170 peptides). Interestingly, prediction errors for the non-hierarchical models varies with retention time, such that peptides with high retention times are predicted with lower accuracy than peptides with lower retention times. In contrast, PepMNet makes robust retention time predictions regardless of where the peptide falls in the retention time distribution.
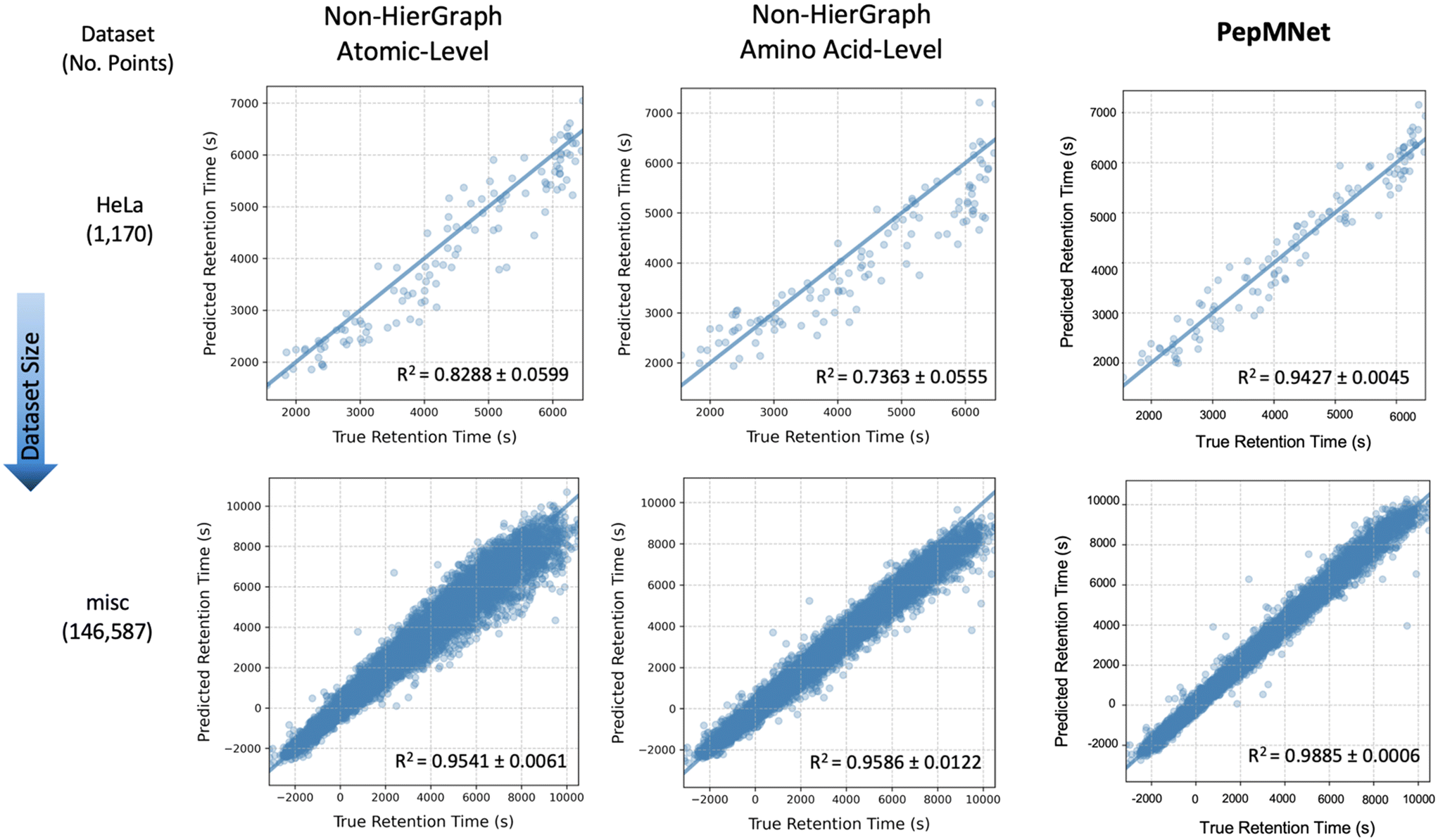 | ||
| Fig. 4 Parity plots comparing PepMNet and non-hierarchical models (using the ARMA layer) on two RT datasets: a large misc dataset (146587 peptides) and a small HeLa dataset (1170 peptides). PepMNet consistently outperforms non-hierarchical models, providing accurate RT predictions across the full RT distribution, while non-hierarchical models show increased prediction errors at extreme RT values, especially with smaller datasets. This highlights the advantage of integrating both atomic- and amino acid-level features. | ||
For the non-hierarchical atomic-level model, the scatter plots reveal different behaviors depending on the dataset size. In the smallest dataset (HeLa), the model produces imprecise predictions for RT at the high ends of the distribution, with the predictions becoming increasingly dispersed for high RT values. For the largest dataset (misc), this dispersion was also evident for greater RT values. In this way, atomic-level features alone may struggle to capture the full range of retention times values. Similarly, the non-hierarchical amino acid-level model exhibits analogous trends to a lesser extent, suggesting that amino acid-level features alone may not fully encapsulate the nuanced interactions governing peptide retention. Overall, the performance gap between non-hierarchical models and PepMNet was most pronounced for the smallest dataset, HeLa. This suggests that non-hierarchical models alone, especially when the availability of information is limited as in smaller datasets, may struggle to capture the full range of retention times.
In contrast, the hierarchical model, which integrates both atomic and amino acid-level features achieves consistently low error independent of retention time, resulting in the highest R2 among the three models. This indicates that the hierarchical approach benefits from capturing multi-scale information, effectively combining the detailed atomic interactions with the broader sequence-level context provided by amino acid sequence. As a result, the hierarchical model expressed a better generalization across datasets of varying sizes and retention time ranges, leading to more accurate predictions. The results of the non-hierarchical models at the atomic and amino acid levels, with each training performed in triplicate, are listed in Tables S5 and S6,† respectively.
3.2 Classification task-AMP classification
The second application we tested our model on was antimicrobial peptide (AMP) identification, a binary classification task. As with the regression task, we quantified the impact of our hierarchical methodology on model performance by comparing PepMNet with a series of non-hierarchical graph models which learned peptide representations from the atomic-level and amino acid-level graphs. As shown in Table 4, PepMNet predicted the antimicrobial activity of peptides with a significantly higher accuracy than any of the non-hierarchical models with an AUC–ROC of 0.9619. This demonstrates PepMNet's high reliability for practical applications in identifying AMP peptides, which could be valuable in the research of new antimicrobial peptides. Similar to results from the regression task, for each non-hierarchical model, there was no graph layer that emerged as clearly superior to the others. These results indicate that, for these applications, property prediction is not sensitive to the graph layer choice.| AUC–ROC | ||
|---|---|---|
| Non-HierGraph atomic level | Non-HierGraph amino acid level | |
| TransformerConv | 0.9087 ± 0.0083 | 0.9439 ± 0.0012 |
| SAGEConv | 0.9236 ± 0.0047 | 0.9482 ± 0.0015 |
| GCNConv | 0.9249 ± 0.0090 | 0.9087 ± 0.0428 |
| NNConv | 0.9253 ± 0.0010 | – |
| EGConv | 0.9266 ± 0.0026 | 0.9343 ± 0.0030 |
| GATConv | 0.9329 ± 0.0024 | 0.9375 ± 0.0044 |
| ARMA | 0.9492 ± 0.0008 | 0.9297 ± 0.0133 |
| PepMNet | 0.9619 ± 0.0017 | |
Interestingly, in the testing dataset for antimicrobial peptide classification, we observed that non-hierarchical models based on the amino acid-level graph tended to outperform those trained only on the atomic-level graph (Table 4 and Fig. S2†), while for retention time prediction the reverse was true for the dataset employed in this study (Fig. 4). Retention time is determined by the strength of interactions between the peptide and the stationary phase and the peptide's physical properties, such as hydrophobicity and charge, depend on the distribution and arrangement of atoms at the molecular level. We hypothesize, therefore, that representing the peptide at the atomic level may more accurately capture the contributions of each atom to these interactions, improving RT prediction. On the other hand, antimicrobial activity appears to depend more on the sequence and arrangement of amino acids. The amino acid sequence can determine characteristics like the formation of secondary structures and the peptide's ability to interact with microbial cell membranes.57 In this way, the impact of the representation level (atomic vs. amino acid) may depend on the specific property being predicted and how that property relates to the peptide's structure and function. By implementing a hierarchical model, we ensure to some extent that both types of information are captured by the model for the prediction task.
These results as well as previous studies in the literature1,15,17 suggest that information contained at the amino acid-level is key for accurately predicting anti-microbial activity. To this end, we evaluated whether our model could be further improved by concatenating amino acid features before performing graph convolutions on the amino acid-level graph. These amino acid features consist of a vector containing amino acid molecular weight, aromaticity, GRAVY score, net charge at pH 7, isoelectric point, octanol–water partition coefficient, and number of atoms. We note that all these features except for the GRAVY score can be calculated for any chemical group, not just an amino acid. In this way, only a small modification to our model would be required to introduce non-amino acid components to the molecules. We additionally explored the impact of changing the graph convolutional layer type at the amino acid level to determine whether this has a significant impact on model performance. Our results show that providing the model with amino acid features slightly improves AMP classification and generalizability, with the model achieving a small increase in AUC–ROC values for the test set regardless of graph convolutional layer choice (Fig. 5). Additionally, we observed that the ARMA layer led to a small increase in performance with an AUC–ROC of 0.9619.
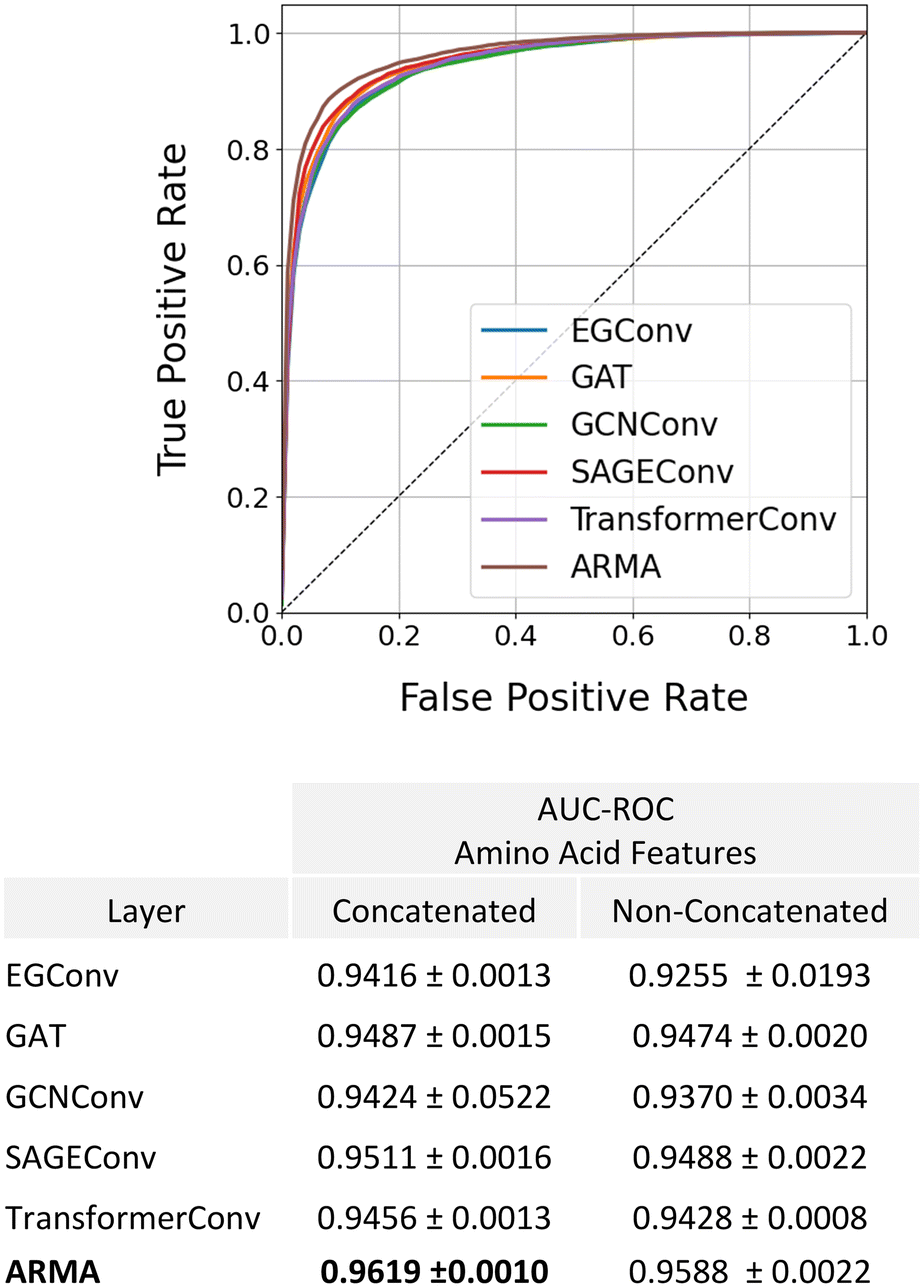 | ||
| Fig. 5 AUC–ROC comparison for AMP classification using different graph convolutional layers at the amino acid level. Incorporating amino acid features slightly improved classification and generalizability, with the ARMA layer yielding the highest. | ||
Finally, to improve model robustness and reduce variability, we trained an ensemble of five separate models using 5-fold cross-validation. Specifically, each model differed in that 1) a different subset of the training set was used for validation and 2) the model weights were initialized randomly. The ensemble prediction was then taken as the average across the five models. Thus, by averaging across multiple models with different training/validation splits and different initialization seeds, we can obtain model predictions that are less sensitive to noise in our model training procedure.58 The loss curves for each fold, as well as the correlation in predictions between folds, are illustrated in Fig. S3 and S4.†
As shown in Fig. 6, the final model achieved an accuracy of 0.9233 using a threshold of 0.5, an average precision of 0.9813, and an AUC–ROC of 0.9775 on the held-out test dataset. These results indicate that the model not only effectively distinguishes between classes but also maintains a high level of precision, minimizing false positives. The high AUC–ROC score further emphasizes the model's ability to discriminate between positive and negative classes across various threshold settings. Together, these metrics suggest that the model is well-suited for peptide classification tasks, demonstrating strong performance across the evaluation metrics employed.
 | ||
| Fig. 6 Performance metrics for peptide classification using the ensembled model on the test dataset. The final model demonstrated strong accuracy, precision, and AUC–ROC, indicating robust performance and effective discrimination between classes across various threshold settings. | ||
3.3 Benchmarking against prior approaches
We tested the multi-scale architecture for predicting the retention time (RT) of peptides across the LC datasets. PepMNet was benchmarked against the framework proposed by Ma et al., which utilizes a capsule network as an alternative to traditional convolutional neural networks. This method involved transfer learning from the eight different datasets.56 For the datasets used in this study, the average R2 value achieved was 0.99, which is on par with the average performance of our model, as shown in Fig. 3.Additionally, we used the test dataset developed by Ruiz et al.33 to compare PepMNet's performance in AMP classification to three publicly available classifications models, two random forest models, AMPEP, and AMPEPpy, and one deep graph network, AMP-Net. Our architecture outperformed the random forest models AMPEP and AMPEPpy in AMP classification, demonstrating higher accuracy, average precision, and AUC–ROC, as depicted in Table 5. Importantly, because it was not necessary to do hyperparameter optimization for the random forest models, these models were trained with 100% of the training dataset, whereas our model was trained with 80% of the dataset and 20% was used for validation. When the random forest models were trained with 80% of the dataset, randomly selected, they achieved an average precision of 0.9709 and 0.9717 for AMPEP and AMPEPpy, respectively, slightly widening the gap between their results and those of PepMNet. Since hyperparameter optimization would normally be performed for random forest training, this scenario is more realistic to compare to. The hierarchical approach also surpassed the model from Ruiz et al., which employed graph representation atomic features. We attribute this improvement to the significance of the amino acid distribution stage, as highlighted by our previous results. By relying solely on atomic composition, the model may overlook important amino acid characteristics of the peptides. Overall, the multi-scale graph neural network proves to be versatile and efficient in handling diverse tasks and predicting various properties compared with the previous state-of-art approaches. This architecture allows for a thorough assessment of the model's generalization capabilities and has emerged as a promising tool for peptide prediction.
| Model | AUC–ROC | Accuracy | Average precision |
|---|---|---|---|
| AMPEP | 0.9674 | 0.9061 | 0.9748 |
| AMPEPpy | 0.9667 | 0.9067 | 0.9740 |
| AMP-net | 0.9444 | 0.8808 | 0.9508 |
| PepMNet | 0.9775 | 0.9233 | 0.9813 |
3.4 Exploring how peptide features impact properties
Both retention time and AMP classification have been viewed in the literature as straightforward functions of the physical characteristics of the peptide.37,56,57 For example, SCX chromatography, which separates peptides based on differences in adsorption to a charged stationary phase, is often thought of as being dictated primarily by the charge of peptide. Similarly, RPLC separates peptides based on adsorption to a hydrophobic stationary phase and is therefore considered to be dictated by peptide hydrophobicity. These two properties also influence the antimicrobial activity: the initial peptide-membrane interaction is driven by their electrostatic attraction and the integration of the peptide into the lipid bilayer is primarily driven by hydrophobic interactions between its hydrophobic residues and the bacteria membrane core.57 This leads to the question: in predicting retention time and antimicrobial activity, is our model simply learning to recognize charged and hydrophobic amino acids? Or is it learning a more complex function of the peptide composition that cannot be reduced to simple physical attributes?To answer these questions, we explored the correlations between charge and hydrophobicity computed using the python package, Biopython,47 and chromatographic retention time. As shown in Fig. 7a, the HILIC retention time dataset was the most strongly correlated with peptide hydrophobicity, with a Pearson-R of 0.536. In contrast, two of the RPLC datasets, HeLa and misc, had lower correlations with hydrophobicity, with Pearson-Rs of 0.256 and 0.271, respectively. HILIC differs from RPLC in that it consists of a nonpolar solvent and a polar stationary phase, while RPLC consists of a nonpolar stationary phase and a polar solvent (typically water). Since both modes of chromatography rely on partitioning between a polar and non-polar phase, it is surprising that a stronger correlation is observed for HILIC compared with RPLC. Finally, we compared retention time on SCX with peptide charge and found that the two quantities were not well correlated (with a Pearson-R of 0.027). This illustrates that the determinant of retention time in SCX is more complex than the formal charge of the peptide.
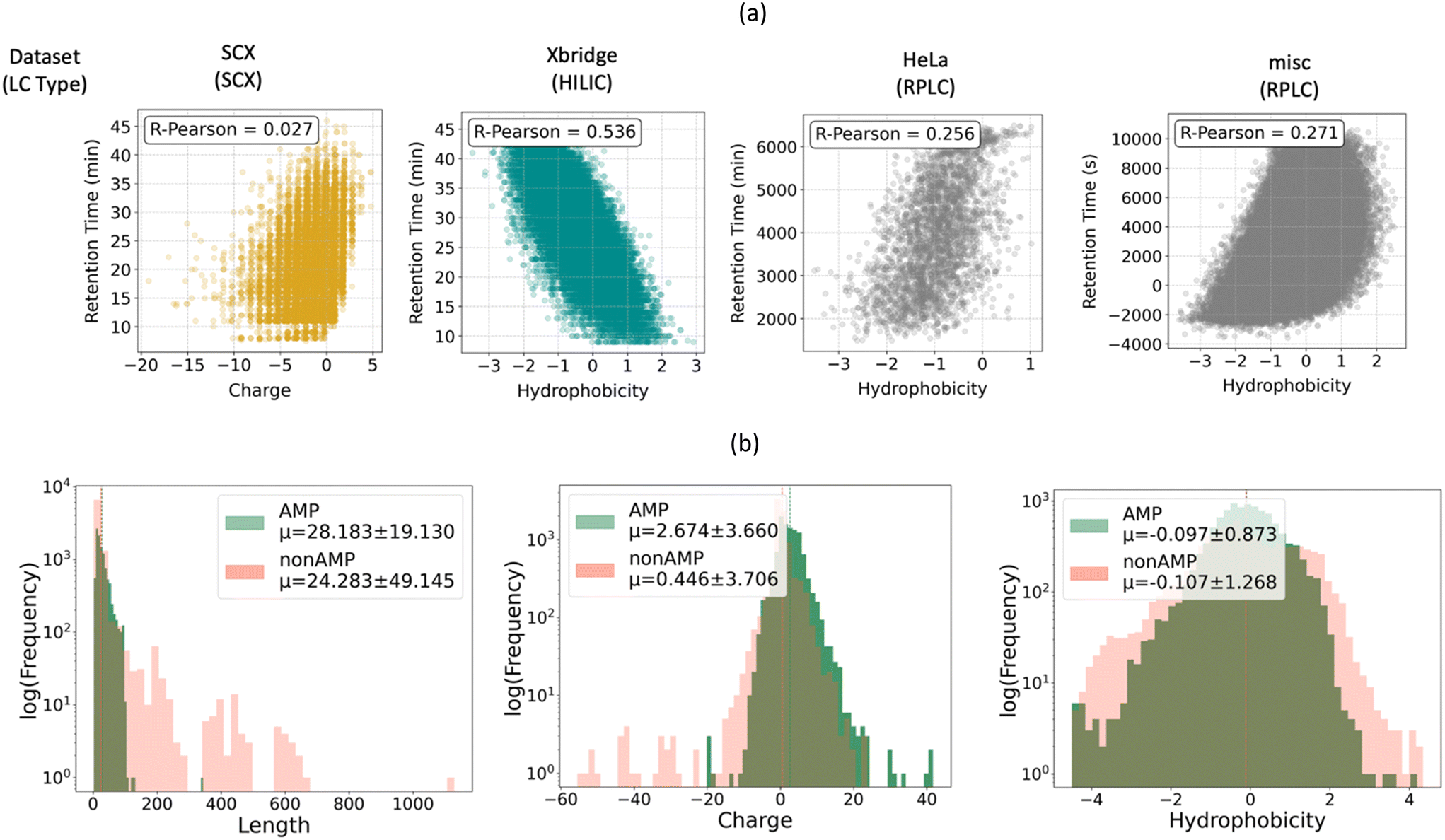 | ||
| Fig. 7 (a) Correlation between peptide properties and RT values. Showing expected trends for different types of LC. In RPLC, RT values increased with hydrophobicity, while an inverse trend was observed in HILIC. (b) Properties Distribution AMP training and validation dataset. The AMP dataset revealed that AMPs are generally more positively charged than non-AMPs, though hydrophobicity did not show a clear distinction. Peptide length was similar between AMP and non-AMPs, with non-AMPs displaying greater variability. | ||
Since antimicrobial activity prediction is represented in our dataset as a binary classification task, it is not possible to correlate antimicrobial activity with peptide properties. In lieu of this, Fig. 7b illustrates the length, charge, and hydrophobicity distributions of peptides in the AMP and non-AMP classes. Overall, the property distributions for both classes of peptides are similar, with non-AMPs having similar properties on average, but with a larger standard deviation than AMPs. Additionally, as expected, non-AMPs were close to neutral on average, while AMPs contained a net positive charge on average.
We can additionally explore the connection between antimicrobial activity and peptide properties by treating each property individually as a predictor of antimicrobial activity and applying different thresholds to obtain a receiver operating curve (ROC). The area under the receiver operating curve (AUC) can then be interpreted as a measure of the strength of the relationship between the two quantities. Fig. 8 illustrates the ROCs for charge, hydrophobicity, and length in predicting antimicrobial activity. Interestingly, charge and length alone are somewhat predictive of antimicrobial activity with AUCs of 0.694 and 0.699, respectively. Because chromatographic retention time and bacterial membrane binding are both adsorption phenomena, we were additionally interested in determining whether retention time was more predictive of antimicrobial activity than the calculated peptide descriptors. To this end, we constructed ROCs for retention times (using our retention time PepMNet model to predict retention times for any peptides that were missing experimental data) and observed a modest increase in AUC with SCX and HILIC both achieving AUCs of 0.704. Overall, this analysis demonstrates that quantifying peptide charge and hydrophobicity individually are not sufficient to predict antimicrobial activity, and a more complex model is required to achieve accurate classification.
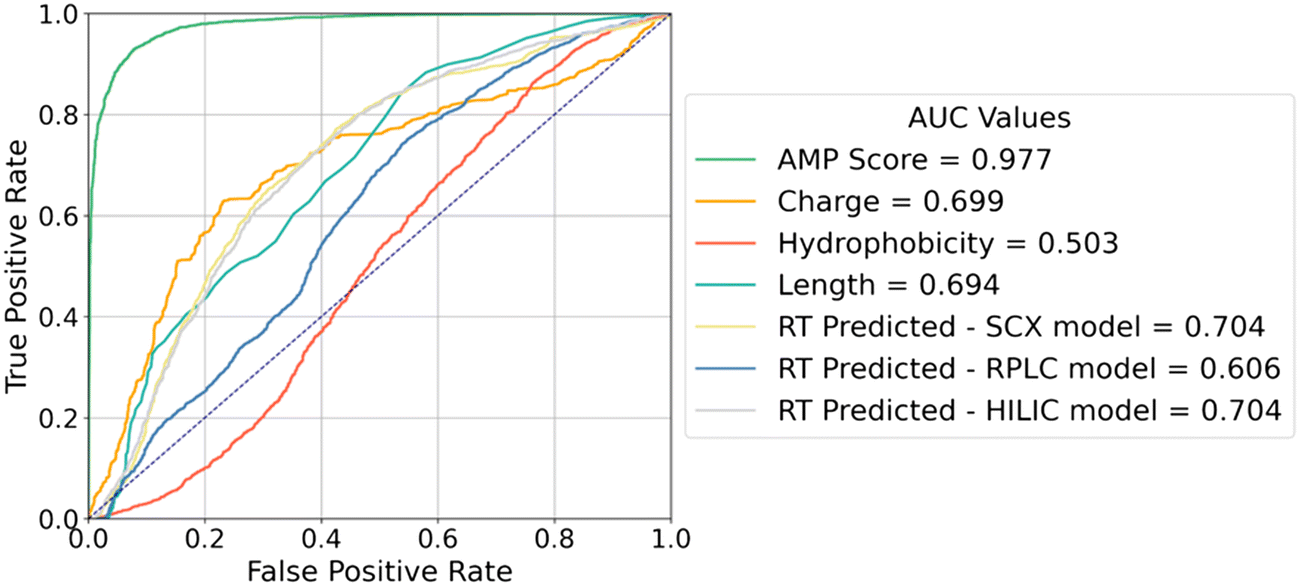 | ||
| Fig. 8 Classification of AMP based on peptide properties and RT values. Peptide charge and length alone provided meaningful insights into AMP classification, Additionally, SCX and HILIC retention times provided information for classifying AMPs, aligning with charge distribution trends. | ||
4. Conclusion
Here we present a hierarchical graph deep learning approach that learns peptide properties directly from atomic and amino acid-level graphs, eliminating the need to rely on human-engineered amino acid descriptors. To test our model, we applied it to two tasks: predicting chromatographic retention time as a regression task and classifying antimicrobial peptides. We found that our model performed on par with previous chromatographic retention time models and better than previous antimicrobial peptide classification models. Additionally, we compared our model with a series of non-hierarchical graph models that relied on different types of graph convolutions and found that regardless of convolutional layer choice, our model outperformed non-hierarchical graph models. Finally, we explored the relationships between peptide properties and prediction tasks and found that while there are some global peptide properties that are correlated with chromatographic retention time or antimicrobial activity (for example charge or hydrophobicity), both properties require a more detailed model to be accurately predicted. Overall, this work provides a new, state-of-the-art neural network for predicting the properties of peptides without relying on human engineered features.It would be valuable in the future to extend this hierarchical approach to include non-natural amino acids and non-linear peptide systems. The atomic graph representation allows for the depiction of complex, nonlinear structures such as stapled and cyclic peptides, providing a more adaptable and less biased alternative. This flexibility is crucial for accurately modeling a broader range of peptide behaviors and functions that are not captured by traditional linear and natural peptide representations. However, a key challenge in translating sequences with non-natural amino acids lies in linking the reading of these sequences with existing libraries for handling chemical compounds. This requires the development of algorithms that can accurately interpret and incorporate non-natural residues. Additionally, future efforts must focus on generating comprehensive experimental datasets with non-natural amino acids, as the current lack of data significantly limits the training and validation of predictive models. Expanding this hierarchical approach to these types of datasets will not only improve the robustness of hierarchical graph models but also pave the way for applying deep learning models to more diverse applications in peptide research.
Finally, because the hierarchical model ensures that peptide information is captured at both atomic and amino acid levels, it is particularly useful when it is unclear whether a property depends more on atomic interactions or on the arrangement of amino acids. In this way, this approach allows for a comprehensive representation of the peptide's characteristics. Further, PepMNet consistently demonstrated a more reliable performance across various datasets of different sizes, highlighting its robustness and adaptability in diverse tasks such as antimicrobial peptide classification. In this context, testing the model's performance in the discovery of novel AMPs would be valuable in the future, especially given the urgent need for innovative therapeutic solutions.2,55 In this work, we have treated AMP classification as a binary problem, however it would be more accurate to classify AMPs into multiple categories such as anti-bacterial, anti-cancer, or anti-fungal peptides. Thus, in the future, it would be beneficial to use PepMNet as a multiclass model to discover peptides that are capable of addressing specific medical problems. This approach could significantly accelerate the discovery pipeline by reducing the reliance on traditional trial-and-error methods in the lab, a highly intensive process that demands substantial time and incurs significant costs.2,31,32 Further, since it is often valuable to know both peptide activity and retention time, it would be valuable to integrate the two models into a single unified workflow. Additionally, beyond AMPs, the implementation of this hierarchical approach could also be extended to other systems, such as polymers or peptide-polymer conjugates, due to its reliance on the atomic graph representation of molecules. This adaptability highlights the broad value of the hierarchical approach, extending its impact beyond peptide prediction to a wider range of molecular systems where atomic-level sequence information is crucial for understanding and predicting material properties.
Data availability
To make it straightforward to reproduce our results and repurpose our models, we have made our code publicly available on GitHub: https://github.com/danielgarzonotero/PepMNet.git. Additionally, the processed datasets used in this project, and the retention time and antimicrobial models, can be found here: https://zenodo.org/communities/pepmnet.Conflicts of interest
The authors declare that there are no conflicts to declare.References
- P. Bhadra, J. Yan, J. Li, S. Fong and S. W. I. Siu, AmPEP: Sequence-Based Prediction of Antimicrobial Peptides Using Distribution Patterns of Amino Acid Properties and Random Forest, Sci. Rep., 2018, 8(1), 1697, DOI:10.1038/s41598-018-19752-w.
- P. Das, T. Sercu, K. Wadhawan, I. Padhi, S. Gehrmann, F. Cipcigan, V. Chenthamarakshan, H. Strobelt, C. Dos Santos, P.-Y. Chen, Y. Y. Yang, J. P. K. Tan, J. Hedrick, J. Crain and A. Mojsilovic, Accelerated Antimicrobial Discovery via Deep Generative Models and Molecular Dynamics Simulations, Nat. Biomed. Eng., 2021, 5(6), 613–623, DOI:10.1038/s41551-021-00689-x.
- W. Chen, E. N. McCool, L. Sun, Y. Zang, X. Ning and X. Liu, Evaluation of Machine Learning Models for Proteoform Retention and Migration Time Prediction in Top-Down Mass Spectrometry, J. Proteome Res., 2022, 21(7), 1736–1747, DOI:10.1021/acs.jproteome.2c00124.
- F. Meier, N. D. Köhler, A.-D. Brunner, J.-M. H. Wanka, E. Voytik, M. T. Strauss, F. J. Theis and M. Mann, Deep Learning the Collisional Cross Sections of the Peptide Universe from a Million Experimental Values, Nat. Commun., 2021, 12(1), 1185, DOI:10.1038/s41467-021-21352-8.
- P. Reiser, M. Neubert, A. Eberhard, L. Torresi, C. Zhou, C. Shao, H. Metni, C. Van Hoesel, H. Schopmans, T. Sommer and P. Friederich, Graph Neural Networks for Materials Science and Chemistry, Commun. Mater., 2022, 3(1), 93, DOI:10.1038/s43246-022-00315-6.
- J. Gilmer, S. Schoenholz, P. F. Riley, O. Vinyals and G. E. Dahl, Neural Message Passing for Quantum Chemistry, arXiv, 2017, preprint, arXiv:1704.01212, DOI:10.48550/arXiv.1704.01212.
- D. Duvenaud, D. Maclaurin, J. Aguilera-Iparraguirre, R. Gómez-Bombarelli, T. Hirzel, A. Aspuru-Guzik and R. P. Adams, Convolutional Networks on Graphs for Learning Molecular Fingerprints, arXiv, November 3, 2015, preprint, arXiv.1509.09292, DOI:10.48550/arXiv.1509.09292.
- X. Xiao, Y.-T. Shao, X. Cheng and B. Stamatovic, iAMP-CA2L: A New CNN-BiLSTM-SVM Classifier Based on Cellular Automata Image for Identifying Antimicrobial Peptides and Their Functional Types, Briefings Bioinf., 2021, 22(6), bbab209, DOI:10.1093/bib/bbab209.
- T.-T. Lin, L.-Y. Yang, I.-H. Lu, W.-C. Cheng, Z.-R. Hsu, S.-H. Chen and C.-Y. Lin, AI4AMP: An Antimicrobial Peptide Predictor Using Physicochemical Property-Based Encoding Method and Deep Learning, mSystems, 2021, 6(6), e00299-21, DOI:10.1128/mSystems.00299-21.
- L. Yu, R. Jing, F. Liu, J. Luo and Y. Li, DeepACP: A Novel Computational Approach for Accurate Identification of Anticancer Peptides by Deep Learning Algorithm, Mol. Ther.--Nucleic Acids, 2020, 22, 862–870, DOI:10.1016/j.omtn.2020.10.005.
- W. Xing, J. Zhang, C. Li, Y. Huo and G. Dong, iAMP-Attenpred: A Novel Antimicrobial Peptide Predictor Based on BERT Feature Extraction Method and CNN-BiLSTM-Attention Combination Model, Briefings Bioinf., 2024, 25(1), 1–9 Search PubMed.
- W.-F. Zeng, X.-X. Zhou, S. Willems, C. Ammar, M. Wahle, I. Bludau, E. Voytik, M. T. Strauss and M. Mann, AlphaPeptDeep: A Modular Deep Learning Framework to Predict Peptide Properties for Proteomics, Nat. Commun., 2022, 13(1), 7238, DOI:10.1038/s41467-022-34904-3.
- C.-R. Chung, T.-R. Kuo, L.-C. Wu, T.-Y. Lee and J.-T. Horng, Characterization and Identification of Antimicrobial Peptides with Different Functional Activities, Briefings Bioinf., 2020, 21(3), 1098–1114, DOI:10.1093/bib/bbz043.
- T. J. Lawrence, D. L. Carper, M. K. Spangler, A. A. Carrell, T. A. Rush, S. J. Minter, D. J. Weston and J. L. Labbé, amPEPpy 1.0: A Portable and Accurate Antimicrobial Peptide Prediction Tool, Bioinformatics, 2021, 37(14), 2058–2060, DOI:10.1093/bioinformatics/btaa917.
- C.-R. Chung, J.-T. Liou, L.-C. Wu, J.-T. Horng and T.-Y. Lee, Multi-Label Classification and Features Investigation of Antimicrobial Peptides with Various Functional Classes, iScience, 2023, 26(12), 108250, DOI:10.1016/j.isci.2023.108250.
- S. A. Pinacho-Castellanos, C. R. García-Jacas, M. K. Gilson and C. A. Brizuela, Alignment-Free Antimicrobial Peptide Predictors: Improving Performance by a Thorough Analysis of the Largest Available Data Set, J. Chem. Inf. Model., 2021, 61(6), 3141–3157, DOI:10.1021/acs.jcim.1c00251.
- P. K. Meher, T. K. Sahu, V. Saini and A. R. Rao, Predicting Antimicrobial Peptides with Improved Accuracy by Incorporating the Compositional, Physico-Chemical and Structural Features into Chou's General PseAAC, Sci. Rep., 2017, 7(1), 42362, DOI:10.1038/srep42362.
- S. Gull, N. Shamim and F. Minhas, AMAP: Hierarchical Multi-Label Prediction of Biologically Active and Antimicrobial Peptides, Comput. Biol. Med., 2019, 107, 172–181, DOI:10.1016/j.compbiomed.2019.02.018.
- D. Veltri, U. Kamath and A. Shehu, Deep Learning Improves Antimicrobial Peptide Recognition, Bioinformatics, 2018, 34(16), 2740–2747, DOI:10.1093/bioinformatics/bty179.
- C. Li, D. Sutherland, S. A. Hammond, C. Yang, F. Taho, L. Bergman, S. Houston, R. L. Warren, T. Wong, L. M. N. Hoang, C. E. Cameron, C. C. Helbing and I. Birol, AMPlify: Attentive Deep Learning Model for Discovery of Novel Antimicrobial Peptides Effective against WHO Priority Pathogens, BMC Genomics, 2022, 23(1), 77, DOI:10.1186/s12864-022-08310-4.
- F. Wan, M. D. T. Torres, J. Peng and C. De La Fuente-Nunez, Deep-Learning-Enabled Antibiotic Discovery through Molecular de-Extinction, Nat. Biomed. Eng., 2024, 8, 854–871, DOI:10.1038/s41551-024-01201-x.
- X. Su, J. Xu, Y. Yin, X. Quan and H. Zhang, Antimicrobial Peptide Identification Using Multi-Scale Convolutional Network, BMC Bioinf., 2019, 20(1), 730, DOI:10.1186/s12859-019-3327-y.
- J. Yan, P. Bhadra, A. Li, P. Sethiya, L. Qin, H. K. Tai, K. H. Wong and S. W. I. Siu, Deep-AmPEP30: Improve Short Antimicrobial Peptides Prediction with Deep Learning, Mol. Ther.--Nucleic Acids, 2020, 20, 882–894, DOI:10.1016/j.omtn.2020.05.006.
- J. Xu, F. Li, C. Li, X. Guo, C. Landersdorfer, H.-H. Shen, A. Y. Peleg, J. Li, S. Imoto, J. Yao, T. Akutsu and J. Song, iAMPCN: A Deep-Learning Approach for Identifying Antimicrobial Peptides and Their Functional Activities, Briefings Bioinf., 2023, 24(4), bbad240, DOI:10.1093/bib/bbad240.
- A. G. B. Grønning, T. Kacprowski and C. Schéele, MultiPep: A Hierarchical Deep Learning Approach for Multi-Label Classification of Peptide Bioactivities, BioMethods, 2021, 6(1), 1–16, DOI:10.1093/biomethods/bpab021.
- I. Dubchak, I. Muchnik, S. R. Holbrook and S. H. Kim, Prediction of Protein Folding Class Using Global Description of Amino Acid Sequence, Proc. Natl. Acad. Sci. U. S. A., 1995, 92(19), 8700–8704, DOI:10.1073/pnas.92.19.8700.
- K.-C. Chou, Pseudo Amino Acid Composition and Its Applications in Bioinformatics, Proteomics and System Biology, Curr. Proteomics, 2009, 6(4), 262–274, DOI:10.2174/157016409789973707.
- C. Ma, Y. Ren, J. Yang, Z. Ren, H. Yang and S. Liu, Improved Peptide Retention Time Prediction in Liquid Chromatography through Deep Learning, Anal. Chem., 2018, 90(18), 10881–10888, DOI:10.1021/acs.analchem.8b02386.
- J. N. Dodds and E. S. Baker, Ion Mobility Spectrometry: Fundamental Concepts, Instrumentation, Applications, and the Road Ahead, J. Am. Soc. Mass Spectrom., 2019, 30(11), 2185–2195, DOI:10.1007/s13361-019-02288-2.
- C. D. Fjell, H. Jenssen, K. Hilpert, W. A. Cheung, N. Panté, R. E. W. Hancock and A. Cherkasov, Identification of Novel Antibacterial Peptides by Chemoinformatics and Machine Learning, J. Med. Chem., 2009, 52(7), 2006–2015, DOI:10.1021/jm8015365.
- J. Xu, F. Li, A. Leier, D. Xiang, H.-H. Shen, T. T. Marquez Lago, J. Li, D.-J. Yu and J. Song, Comprehensive Assessment of Machine Learning-Based Methods for Predicting Antimicrobial Peptides, Briefings Bioinf., 2021, 22(5), bbab083, DOI:10.1093/bib/bbab083.
- R. Gómez-Bombarelli, J. N. Wei, D. Duvenaud, J. M. Hernández-Lobato, B. Sánchez-Lengeling, D. Sheberla, J. Aguilera-Iparraguirre, T. D. Hirzel, R. P. Adams and A. Aspuru-Guzik, Automatic Chemical Design Using a Data-Driven Continuous Representation of Molecules, ACS Cent. Sci., 2018, 4(2), 268–276, DOI:10.1021/acscentsci.7b00572.
- P. Ruiz Puentes, M. C. Henao, J. Cifuentes, C. Muñoz-Camargo, L. H. Reyes, J. C. Cruz and P. Arbeláez, Rational Discovery of Antimicrobial Peptides by Means of Artificial Intelligence, Membranes, 2022, 12(7), 708, DOI:10.3390/membranes12070708.
- K. Sharma, R. C. J. D'Souza, S. Tyanova, C. Schaab, J. R. Wiśniewski, J. Cox and M. Mann, Ultradeep Human Phosphoproteome Reveals a Distinct Regulatory Nature of Tyr and Ser/Thr-Based Signaling, Cell Rep., 2014, 8(5), 1583–1594, DOI:10.1016/j.celrep.2014.07.036.
- N. Nagaraj, N. A. Kulak, J. Cox, N. Neuhauser, K. Mayr, O. Hoerning, O. Vorm and M. Mann, System-Wide Perturbation Analysis with Nearly Complete Coverage of the Yeast Proteome by Single-Shot Ultra HPLC Runs on a Bench Top Orbitrap, Mol. Cell. Proteomics, 2012, 11(3) DOI:10.1074/mcp.M111.013722.
- G. Rosenberger, C. C. Koh, T. Guo, H. L. Röst, P. Kouvonen, B. C. Collins, M. Heusel, Y. Liu, E. Caron, A. Vichalkovski, M. Faini, O. T. Schubert, P. Faridi, H. A. Ebhardt, M. Matondo, H. Lam, S. L. Bader, D. S. Campbell, E. W. Deutsch, R. L. Moritz, S. Tate and R. Aebersold, A Repository of Assays to Quantify 10,000 Human Proteins by SWATH-MS, Sci. Data, 2014, 1(1), 140031, DOI:10.1038/sdata.2014.31.
- D. Gussakovsky, H. Neustaeter, V. Spicer and O. V. Krokhin, Sequence-Specific Model for Peptide Retention Time Prediction in Strong Cation Exchange Chromatography, Anal. Chem., 2017, 89(21), 11795–11802, DOI:10.1021/acs.analchem.7b03436.
- V. Spicer and O. V. Krokhin, Peptide Retention Time Prediction in Hydrophilic Interaction Liquid Chromatography. Comparison of Separation Selectivity between Bare Silica and Bonded Stationary Phases, J. Chromatogr. A, 2018, 1534, 75–84, DOI:10.1016/j.chroma.2017.12.046.
- M. Simonovsky and N. Komodakis, Dynamic Edge-Conditioned Filters in Convolutional Neural Networks on Graphs, arXiv, August 8, 2017, preprint, arXiv.1704.02901, DOI:10.48550/arXiv.1704.02901.
- M. Fey and J. E. Lenssen, Fast Graph Representation Learning with PyTorch Geometric, arXiv, April 25, 2019, preprint, arXiv.1903.02428, DOI:10.48550/arXiv.1903.02428.
- F. M. Bianchi, D. Grattarola, L. Livi and C. Alippi, Graph Neural Networks with Convolutional ARMA Filters, IEEE Trans. Pattern Anal. Mach. Intell., 2021, 1 DOI:10.1109/TPAMI.2021.3054830.
- K. Yang, K. Swanson, W. Jin, C. Coley, P. Eiden, H. Gao, A. Guzman-Perez, T. Hopper, B. Kelley, M. Mathea, A. Palmer, V. Settels, T. Jaakkola, K. Jensen and R. Barzilay, Analyzing Learned Molecular Representations for Property Prediction, J. Chem. Inf. Model., 2019, 59(8), 3370–3388, DOI:10.1021/acs.jcim.9b00237.
- E. Heid, K. P. Greenman, Y. Chung, S.-C. Li, D. E. Graff, F. H. Vermeire, H. Wu, W. H. Green and C. J. McGill, Chemprop: A Machine Learning Package for Chemical Property Prediction, J. Chem. Inf. Model., 2024, 64(1), 9–17, DOI:10.1021/acs.jcim.3c01250.
- J. R. Lobry and C. Gautier, Hydrophobicity, Expressivity and Aromaticity Are the Major Trends of Amino-Acid Usage in 999 Escherichia Coli Chromosome-Encoded Genes, Nucleic Acids Res., 1994, 22(15), 3174–3180, DOI:10.1093/nar/22.15.3174.
- J. Kyte and R. F. Doolittle, A Simple Method for Displaying the Hydropathic Character of a Protein, J. Mol. Biol., 1982, 157(1), 105–132, DOI:10.1016/0022-2836(82)90515-0.
- S. A. Wildman and G. M. Crippen, Prediction of Physicochemical Parameters by Atomic Contributions, J. Chem. Inf. Comput. Sci., 1999, 39(5), 868–873, DOI:10.1021/ci990307l.
- P. J. A. Cock, T. Antao, J. T. Chang, B. A. Chapman, C. J. Cox, A. Dalke, I. Friedberg, T. Hamelryck, F. Kauff, B. Wilczynski and M. J. L. De Hoon, Biopython: Freely Available Python Tools for Computational Molecular Biology and Bioinformatics, Bioinformatics, 2009, 25(11), 1422–1423, DOI:10.1093/bioinformatics/btp163.
- RDKit: Open-Source Cheminformatics, https://www.Rdkit.Org Search PubMed.
- T. Zhang, H. Li, H. Xi, R. V. Stanton and S. H. Rotstein, HELM: A Hierarchical Notation Language for Complex Biomolecule Structure Representation, J. Chem. Inf. Model., 2012, 52(10), 2796–2806, DOI:10.1021/ci3001925.
- W. L. Hamilton, R. Ying and J. Leskovec, Inductive Representation Learning on Large Graphs, arXiv, September 10, 2018, preprint, arXiv.1706.02216, DOI:10.48550/arXiv.1706.02216.
- Y. Shi, Z. Huang, S. Feng, H. Zhong, W. Wang and Y. Sun, Masked Label Prediction: Unified Message Passing Model for Semi-Supervised Classification, arXiv, May 9, 2021, preprint, arXiv.2009.03509, DOI:10.48550/arXiv.2009.03509.
- P. Veličković, G. Cucurull, A. Casanova, A. Romero, P. Liò and Y. Bengio, Graph Attention Networks, arXiv, February 4, 2018, preprint, arXiv.1710.10903, DOI:10.48550/arXiv.1710.10903.
- S. A. Tailor, F. L. Opolka, P. Liò and N. D. Lane, Do We Need Anisotropic Graph Neural Networks?, arXiv, May 9, 2022, preprint, arXiv.2104.01481, DOI:10.48550/arXiv.2104.01481.
- T. Kipf and M. Welling, Semi-Supervised Classification with Graph Convolutional Networks, arXiv, 2016, preprint, arXiv.1609.02907, DOI:10.48550/arXiv.1609.02907.
- M. Magana, M. Pushpanathan, A. L. Santos, L. Leanse, M. Fernandez, A. Ioannidis, M. A. Giulianotti, Y. Apidianakis, S. Bradfute, A. L. Ferguson, A. Cherkasov, M. N. Seleem, C. Pinilla, C. De La Fuente-Nunez, T. Lazaridis, T. Dai, R. A. Houghten, R. E. W. Hancock and G. P. Tegos, The Value of Antimicrobial Peptides in the Age of Resistance, Lancet Infect. Dis., 2020, 20(9), e216–e230, DOI:10.1016/S1473-3099(20)30327-3.
- C. Ma, Y. Ren, J. Yang, Z. Ren, H. Yang and S. Liu, Improved Peptide Retention Time Prediction in Liquid Chromatography through Deep Learning, Anal. Chem., 2018, 90(18), 10881–10888, DOI:10.1021/acs.analchem.8b02386.
- M. R. Yeaman and N. Y. Yount, Mechanisms of Antimicrobial Peptide Action and Resistance, Pharmacol. Rev., 2003, 55(1), 27–55, DOI:10.1124/pr.55.1.2.
- B. Lakshminarayanan, A. Pritzel and C. Blundell, Simple and Scalable Predictive Uncertainty Estimation Using Deep Ensembles, arXiv, 2017, preprint, arXiv.1612.01474, DOI:10.48550/arXiv.1612.01474.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4me00172a |
| ‡ Permanent address: 385 McCormick Road, Charlottesville, VA 22903, USA. |
| This journal is © The Royal Society of Chemistry 2025 |