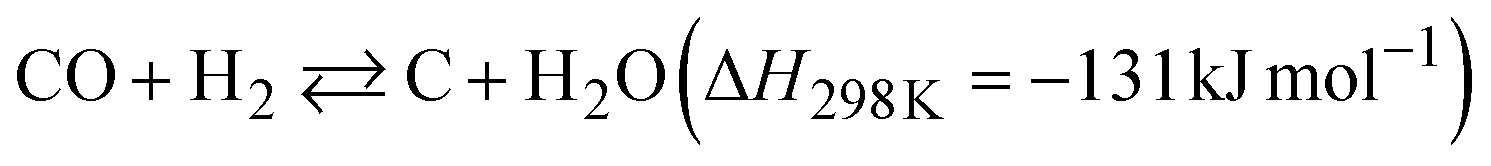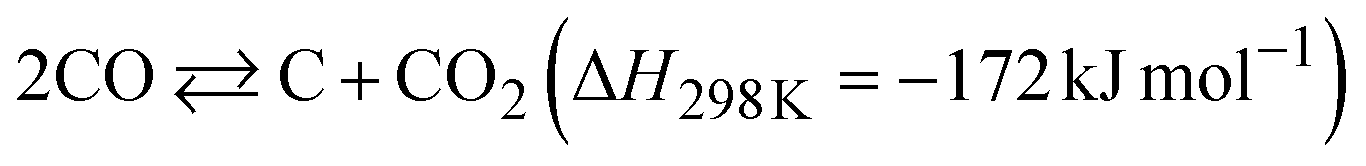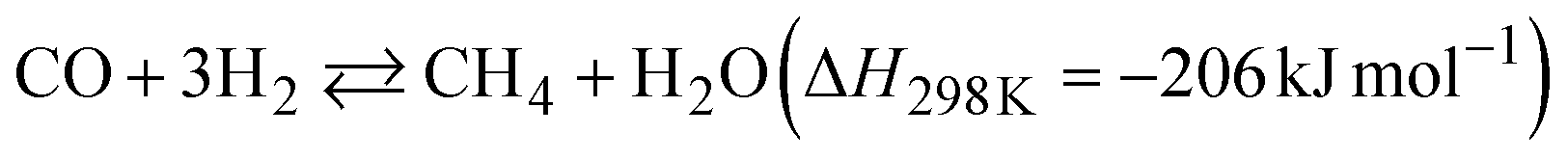DOI:
10.1039/D4NA00740A
(Paper)
Nanoscale Adv., 2025,
7, 346-353
Carbon nanotube growth catalyzed by metal nanoparticles formed via the seed effect of metal clusters†
Received
5th September 2024
, Accepted 2nd November 2024
First published on 2nd December 2024
Abstract
Carbon nanotubes (CNTs) are useful nanomaterials owing to their distinct functions that depend on their structure and diameter; therefore, CNTs have recently attracted much attention. Catalytic chemical vapor deposition using metal nanoparticles as catalysts is one of the most useful methods for synthesizing CNTs. However, fine control of the CNT diameter has been technically difficult at the 1 nanometer level owing to the size distribution of metal nanoparticles and the difficulty in suppressing their aggregation during the CNT growth reaction. In this study, we developed a method for simultaneously conducting nanoparticle formation and CNT growth through the in situ preparation of nanoparticles using highly dispersed metal clusters on a support substrate as seeds. In this method, the migration and aggregation behavior of seed metal clusters on a substrate upon heating uniformly induces nanoparticle formation until nanoparticles are enlarged to suitable size for CNT growth, and then CNTs grow with a diameter corresponding to that of the nanoparticles. The nanoparticle size does not change afterward by spatial separation of the nanoparticles from the substrate due to the tip-growth mechanism of CNTs. Detailed analysis focusing on cobalt as a catalyst demonstrated that the diameters of the multi-walled CNTs could be simply tuned solely by modifying the reaction temperature and time. This result reveals that the synthetic concept using the seed clusters enables the control of the CNT size to a certain extent using only temperature control (thermal energy), without conventional separation processes for synthesizing nanoparticles and determining conditions to retain nanoparticle size.
Introduction
Based on its unique and diverse properties, nanosized carbon is one of the key substances used to create highly functional materials. One key feature of nano-carbon is its variety of allotropes and dimensions, such as nanodiamonds, graphene, fullerenes, and carbon nanotubes (CNTs). CNTs have outstanding functions and are expected to be beneficial in a wide range of industrial fields. For instance, CNTs have a distinctively high mechanical strength (approximately 20 times that of steel), high electrical conductivity (approximately 1000 times that of copper), and high thermal conductivity (approximately 10 times that of copper), despite their lightness and low density (approximately half that of aluminum).1–3 These properties can be further modified by controlling the structural configurations of CNTs, such as the number of walls (single-walled or multi-walled) and the thickness of the tubes (diameter). Since CNTs were first discovered as a byproduct in the synthesis of fullerenes in 1991,1,4 researchers have studied the synthesis of CNTs using various methods such as arc discharge evaporation,1,4,5 laser ablation,6,7 and catalytic chemical vapor deposition (CCVD).8–10 The CCVD method, which affords CNTs with high purity and yield compared to other methods, is an efficient synthetic approach. In this method, metal nanoparticles (NPs) supported on substrates are primarily used as catalysts for CNT growth. They catalytically decompose gaseous carbon sources (e.g., CO, CH4, and CH3OH) and enable the subsequent growth of CNTs originating from the NP surface, at high temperatures.11,12 In this mechanism, fine control of the size of the metal NPs is important for precise synthesis of the CNTs because the diameter of the CNTs is directly correlated with that of the NPs.13,14 In previous studies, researchers attempted to control the diameter of CNTs by adjusting various parameters such as the reactant, metal species of the catalysts, compositional ratio, reaction pressure, and reaction temperature.15–18 However, synthesizing CNTs of various diameters with a 1 nanometer precision is generally difficult due to the challenges in precisely synthesizing metal NPs with controlled diameters with small distribution (particularly those up to ∼10 nm) and in suppressing NPs' enlargement by their aggregation behavior during the CNT growth reaction.19–22 The properties of CNTs depend on their structural configurations such as the number of walls and the diameter. Therefore, the selective fabrication of CNTs with a uniform diameter remains one of the most fundamental issues in CNT chemistry.
To overcome this obstacle, in this study, we propose the concept of the ‘seed method,’ in which the diameter of CNTs prepared via CCVD is controlled through the in situ preparation of metal NPs using metal clusters as seeds (seed clusters are denoted as SCs). Clusters are ultrasmall particles with a diameter of ∼1 nm composed of a few dozen metal atoms, and their properties and reactivity differ significantly from those of conventional NPs;23–25 for example, they do not catalyze CNT growth. In this method, SCs that are highly dispersed on a substrate are initially prepared as NP precursors. Their air-oxidized surfaces are reduced to the metal state by H2 (and/or CO) and the metal particles subsequently aggregate on a substrate under the action of thermal energy during CO-CCVD at high temperatures, automatically forming NPs with uniform diameters. Simultaneously, the NPs grow to a suitable size for CCVD and immediately serve as the catalyst for the CNT growth reaction, leading to fine control of the diameter of the CNTs. Therefore, this approach involving simultaneous NP formation and CNT growth using SCs is expected to be beneficial for the mass synthesis of CNTs. The shape, diameter, and yield of the CNTs, depending on the properties of the SCs, reaction temperature, and reaction conditions, are evaluated to demonstrate this synthetic concept.
Results and discussion
Preparation of Co SCs dispersed on a silica substrate
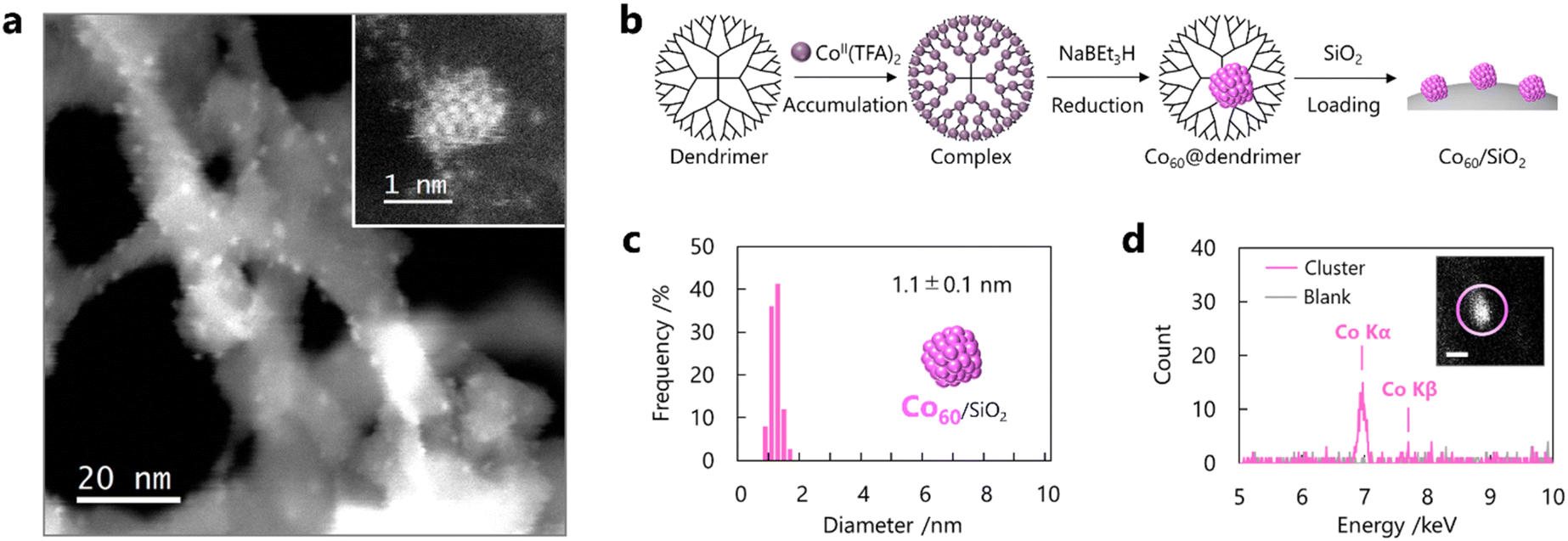
Cobalt (Co) was selected as a model catalyst for CNT growth because it has been widely applied in CCVD using carbon monoxide (CO-CCVD) owing to its high reactivity with CO molecules and high reductivity.15,16 Co SCs were prepared by the template method for the synthesis of metal clusters using a macromolecular capsule in an organic solvent (Fig. S1†).26–28 In this method, a dendrimer containing 60 Lewis basic imine units was adopted as a capsule, and 60 Lewis acidic cobalt salts were accumulated in the nanospace through monodentate complexation. Following the formation of a multinuclear complex with 60 Co atoms, atomicity-defined Co SCs (Co60) were synthesized by chemical reduction (Fig. 1a and S2†). These SCs were loaded onto silica, and other impurities, including dendrimers and reductants, were removed from the sample by filtration with a chloroform and methanol rinse. High-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) observations indicated that the SCs with a diameter of 1.1 ± 0.1 nm were uniformly dispersed on silica (Fig. 1b and c). This average diameter is well consistent with the Co oxide cluster. XPS and XAFS measurements revealed that Co SCs were automatically oxidized by air exposure (Fig. S3 and S4†). The size distribution mainly originates from frequent structural change of clusters to non-spherical shapes such as elliptical or semicircular structure associated with a support on silica. Atomic-resolution observations using a graphene support indicated that each particle consisted of an appropriate number of atoms (Fig. 1b). Elemental analysis using energy-dispersive X-ray spectroscopy (EDS) revealed that the particles were composed of Co (Fig. 1d and S5†). These results indicate the successful synthesis of Co SCs with finely controlled sizes using the template method.
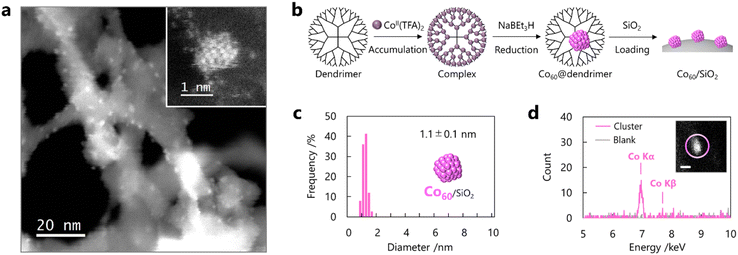 |
| | Fig. 1 Preparation of Co SCs by the template method and identification using STEM/EDS. (a) STEM images of Co SCs supported on silica at low magnification and (inset) on graphene at atomic resolution. (b) Preparation scheme of Co SCs on silica using a dendrimer. Cobalt trifluoroacetate tetrahydrate as a precursor of SCs is abbreviated to Co TFA. (c) Histogram of the diameter of Co SCs. (d) EDS profiles of a Co SC on silica obtained from (inset) the circled area in the STEM image (scale bar: 1 nm). Prepared Co SCs were automatically oxidized in air. | |
CNT growth from Co NPs formed via Co SCs
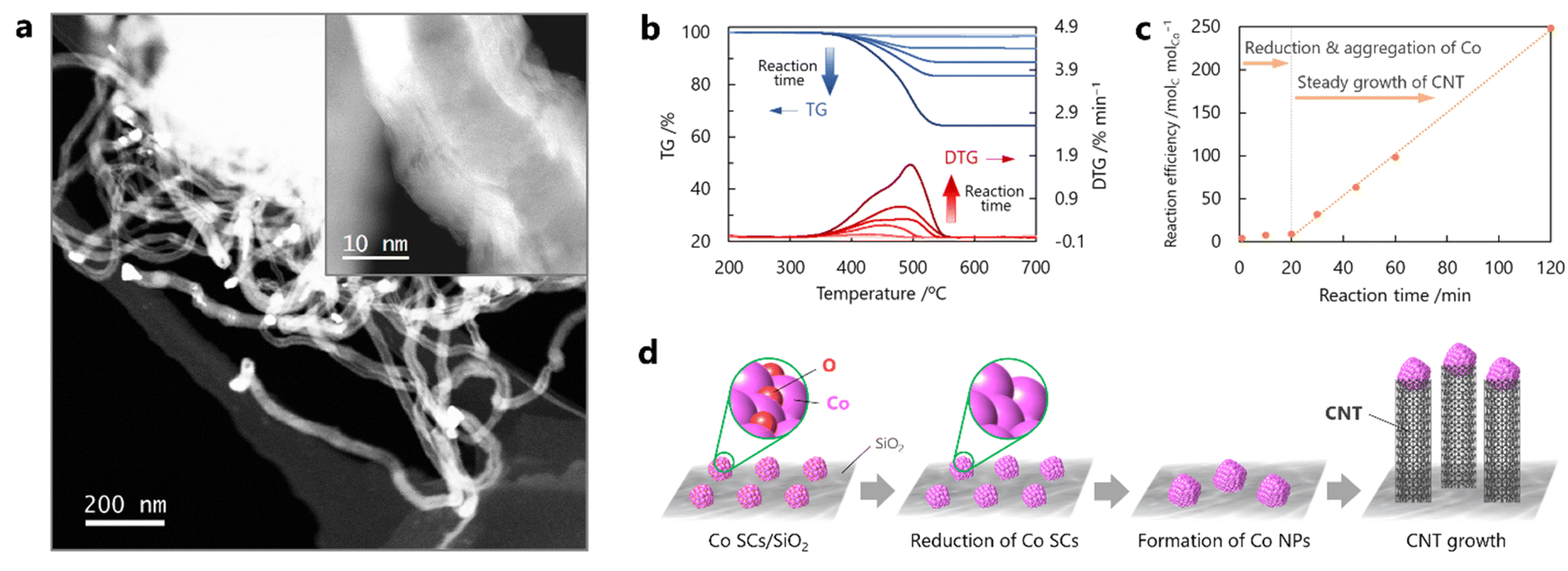
The Co SCs dispersed on silica were used as catalysts for subsequent CNT growth. This study utilized the CCVD technique with CO as the carbon source under a simultaneous flow of H2 for chemical reduction of the Co catalysts and preventing re-poisoning by oxygen during the reaction. Initial attempts were made to grow the CNTs under milder conditions (1 atm and 600 °C) than those employed in previous reports.16,29 Many CNTs grown from the Co NPs were observed by STEM after the CO-CCVD reaction at 600 °C for 60 min, and the Co SCs disappeared (Fig. 2a). The produced carbon would have tube-type structures, judging from its hollow structure, which is confirmed in the STEM image. Note that it is difficult to observe the hollow structure of CNTs in the defocused area of STEM images. Because the individual Co NPs were at the tip of each CNT, it was suggested that CNT growth proceeded via the tip-growth mechanism rather than the base-growth mechanism.30–32 High-magnification STEM observations indicated that the synthesized CNTs were multi-walled. The amount of obtained CNTs was quantified by thermogravimetric (TG) analysis under an air atmosphere, where a decrease in the weight of the sample due to the combustion of carbon materials by oxygen was observed at 400–550 °C.33,34 As a result, an increase in the generation of CNTs with an increase in the reaction time was observed, although almost no weight change was observed in the TG analysis of the sample after a shorter reaction time (1–20 min) (Fig. 2b, c and S6†). It has been reported that CNT growth is catalyzed on pure (non-oxidized) metal surfaces rather than on the oxidized surfaces.12,34 Aggregation of the Co SCs was confirmed, whereas almost no CNTs were formed in the reaction for 10 min (Fig. S7†). Based on these results and the fact that the Co SCs were oxidized in air before the reaction (Fig. S4 and S5†), it was deduced that CNT growth proceeded by a stepwise scheme as follows: [1] air-oxidized Co SCs were first reduced by H2 (and/or CO); [2] the Co SCs aggregated and subsequently formed Co NPs on silica, driven by thermal energy; and [3] CNTs finally grew from the Co NPs in an elevator-like fashion (Fig. 2d). The TG results suggest that the Co SCs required approximately 20 min for the first two steps ([1] and [2]) (Fig. 2c). This mechanism involved moderate interaction between the reduced Co SCs and silica, in comparison with other supports. When alumina was used as the support substrate, the main product was amorphous carbon with no CNTs because the Co NPs underwent excessive enlargement (Fig. S8†).
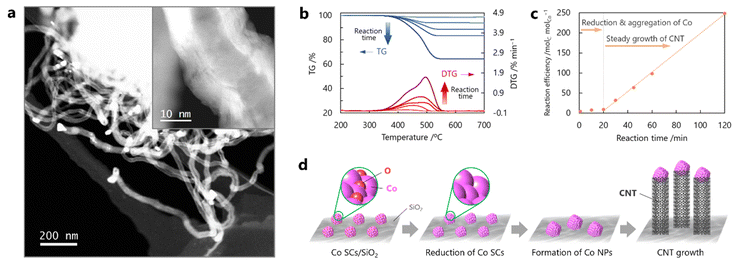 |
| | Fig. 2 Dependence of CNT growth on reaction time for the seed method using Co SCs. (a) High-magnification and low-magnification (inset) STEM images of CNTs from Co NPs after reaction using a mixture of CO/H2 = 1 at 600 °C for 60 min. (b) TG profiles of the sample acquired under an air atmosphere after the CO-CCVD reaction using a mixture of CO/H2 = 1 at 600 °C for different durations of time (1, 5, 10, 30, 60, and 120 min). (c) TG measurements showing the effect of reaction time on the weight loss of the sample prepared by the CO-CCVD reaction. (d) Proposed scheme for CNT growth by the seed method using Co SCs. | |
Temperature-induced control of the diameter of CNTs
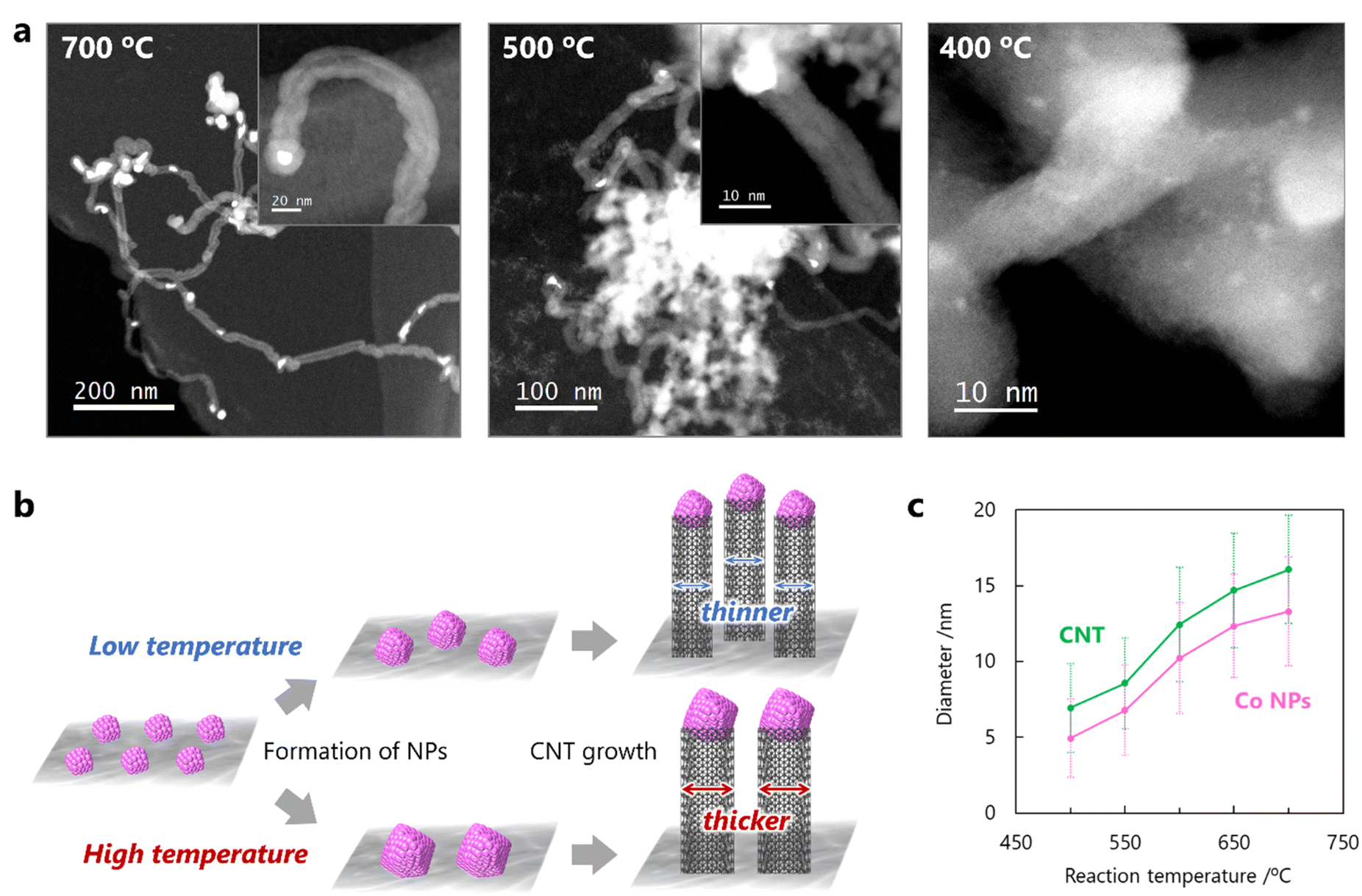
The influence of the reaction temperature on the diameter of the CNTs was investigated in the CO-CCVD reaction for 60 min at several temperatures from 400 to 800 °C. Large Co NPs with diameters of 10–100 nm covered with carbon were observed at 800 °C (Fig. S9†). The shape of the obtained carbon materials was distorted and CNTs could not be formed, indicating that this temperature was not suitable for CNT growth using this method. On the other hand, CNT growth from Co NPs was observed below 700 °C, and even at 500 °C (Fig. 3a). At 400 °C, no CNTs were observed, and the Co SCs remained dispersed on the silica without aggregation because this temperature was not high enough to reduce air-oxidized Co SCs. This speculation was also supported by the temperature-programmed reduction test using H2 (H2-TPR), which indicated that air-oxidized Co SCs were reduced by H2 at approximately 500 °C or higher (Fig. S10†). These results suggest that CNT growth via this mechanism depends significantly on the reaction temperature (Fig. 3b). In particular, the migration behavior of the reduced Co SCs flexibly combined with silica plays an important role in the tunable formation of the Co NPs, leading to the growth of CNTs with finely controlled diameters. Consequently, successful control of the diameters of the CNTs and corresponding Co NPs was achieved by optimizing the reaction temperature (Fig. 3c and S11†).
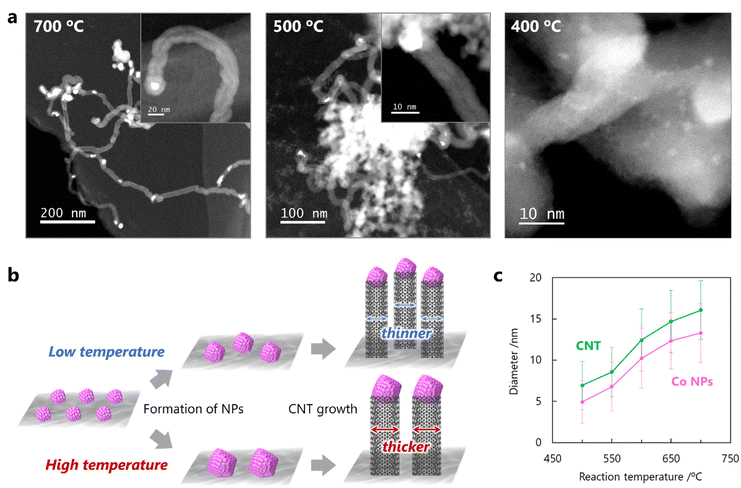 |
| | Fig. 3 Effect of reaction temperature on CNT growth by the seed method using Co SCs. (a) High-magnification and low-magnification (inset) STEM images of CNTs grown from Co NPs using a mixture of CO/H2 = 1 at 700, 500, and 400 °C for 60 min. (b) Proposed scheme of CNT growth by the seed method using Co SCs depending on the reaction temperature. (c) Temperature-dependence of the diameters of Co NPs and grown CNTs. | |
In the base-growth mechanism, continuous aggregation of Co NPs on the support has been observed even during CNT growth.30–32 On the other hand, the CNT growth in this work proceeded via the tip-growth mechanism, in which the Co NPs were detached from the silica substrate and the isolated NPs were carried upward in the CNTs. In this study, it was revealed that the spatial separation of the NPs from the support played an important role in inhibiting further excessive aggregation of Co NPs and realized fine control of their diameters. In the reaction without CO as a carbon source, the average diameter of the Co NPs increased, and their distribution broadened at each reaction temperature because CNT growth reaction did not occur (Fig. S12†). Consequently, the control of diameters of CNTs by the reaction temperature would be achieved by the change in the balance between the rate of cluster reduction, the rate of NP formation by cluster migration/aggregation, and the rate of CNT growth with the separation of NPs from the support substrate.
The influence of the concentration ratio of CO/H2 was also investigated. After the CO-CCVD reaction using a mixture of CO/H2 = 2 at 700 °C, severely aggregated Co NPs surrounded by carbon materials were observed with a small amount of tube-like carbon (Fig. S13†). The main product was layered carbon or amorphous carbon rather than CNTs, whereas CNTs were the main products of the reaction using a mixture of CO/H2 = 2 at 600 °C. H2 gas prevents re-poisoning by oxygen and the selective etching of amorphous carbon.35,36 Consequently, CO-rich conditions are not appropriate for CNT growth in this method because the generation of amorphous carbon is predominant over H2-etching.
A seeding method using other metal SCs (Ni and Fe) was investigated (Fig. S14–S17†). XPS measurements also demonstrated that these SCs were automatically oxidized in air in the same manner as the Co SCs (Fig. S18 and S19†). Similar to the Co NPs, Ni NPs were formed via Ni SCs (Ni60) and provided CNTs using a CO/H2 = 1 mixture at 500 and 700 °C (Fig. S20 and S21†). Interestingly, at 500 °C, the Ni NPs provided many CNTs with very short lengths, and distorted and fractured structures. It is speculated that a suitable balance between the reduction of Ni NPs and re-poisoning by oxygen partially restricted CNT growth, leading to the fabrication of short CNTs. On the other hand, the Fe SCs (Fe60) did not act as a catalyst even at 700 °C, and the diameter was maintained at ∼1 nm. Ostensibly, this temperature may be insufficient for chemical reduction of the oxidized Fe SCs.
Effect of reaction temperature on the yield of CNTs
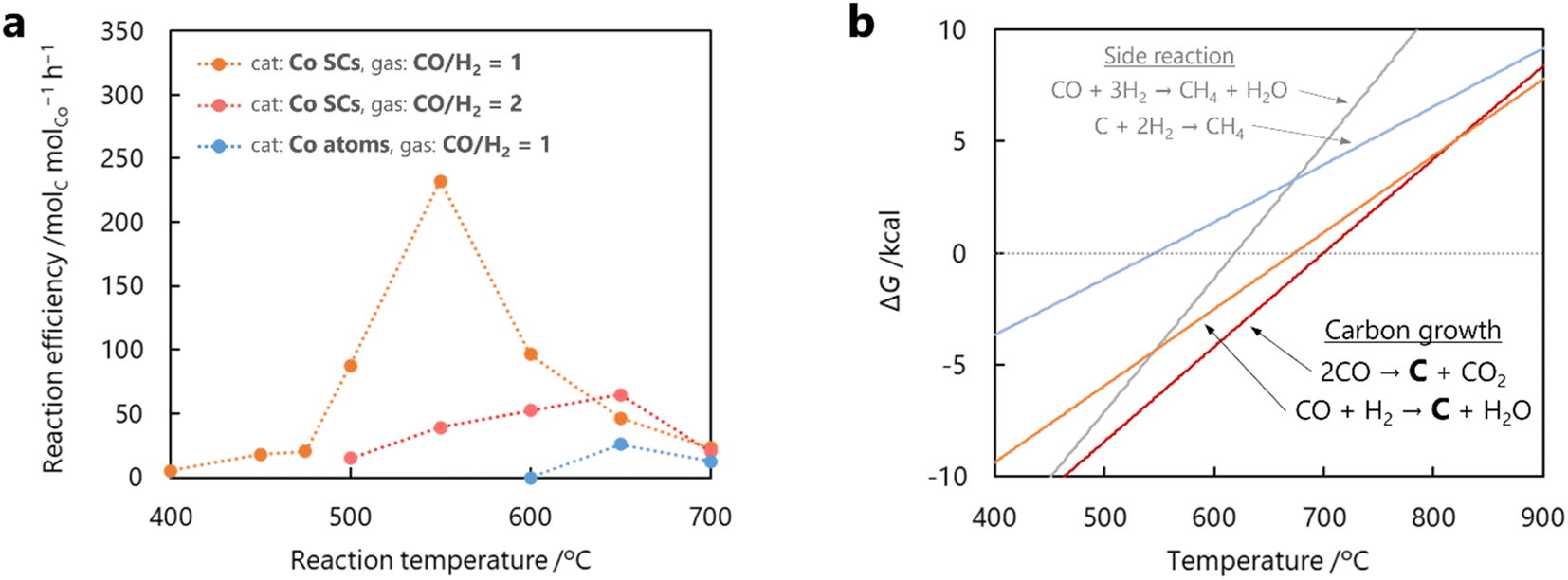
The dependence of the CNT yield on the reaction temperature was evaluated by TG analysis after 60 min of CO-CCVD (Fig. 4a and S22†). Almost no weight loss of the Co-silica catalyst due to carbon combustion was detected for the reaction at temperatures below 475 °C. This is because the chemical reduction of Co SCs at this temperature was insufficient for the formation of Co NPs, which required migration and aggregation; thus, the CNT growth reaction was not catalyzed (Fig. S10†). On the other hand, in the case of CO-CCVD at above 500 °C, the sample underwent weight loss due to the combustion of carbon, and the weight loss was maximal at 550 °C. This result is supported by the thermodynamic profiles of the CO-CCVD process shown below. In the CO-CCVD method, carbon is typically generated by the Fischer–Tropsch (FT) (eqn (1)) and Boudouard (eqn (2)) reactions.37| | 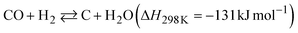 | (1) |
| | 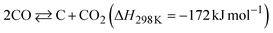 | (2) |
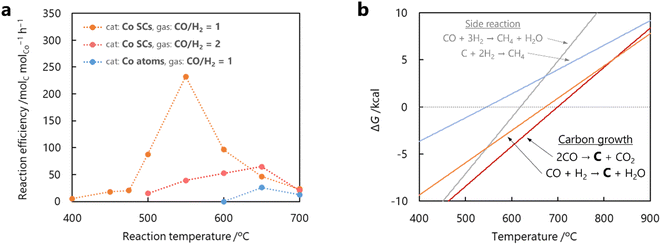 |
| | Fig. 4 Effect of reaction temperature on reaction efficiency for CO-CCVD by the seed method using Co SCs. (a) Reaction efficiency of Co catalysts used in the CNT growth reaction at several temperatures in the reaction for 60 min, determined by TG measurements under an air atmosphere. The amount of Co on silica was set to 2 wt% for all catalyst samples. (b) Gibbs free energy of the CO-CCVD reaction, simulated by thermodynamic equilibrium calculation (Gibbs free energy minimization algorithm). | |
The two reactions are expected to proceed as side reactions that inhibit CNT growth (eqn (3) and (4)).
| | 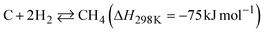 | (3) |
| | 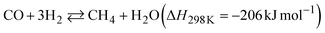 | (4) |
The Gibbs free energy (ΔG) in these four reactions depending on the temperature was simulated by thermodynamic equilibrium calculation (Fig. 4b). The graph indicates that both the FT reaction and Boudouard reaction are involved in carbon generation, and predominate below 700 °C. Therefore, the decline in the yield of carbon generated around 700 °C is due to the disadvantageous thermodynamic equilibrium (ΔG > 0). Furthermore, the carbon yield decreased with a change in the reactant gas ratio from CO/H2 = 1 to CO/H2 = 2 (Fig. 4a and S23†). The dependence of the carbon yield on the amounts of both CO and H2 indicates that the main CO-CCVD reaction in this study is the FT reaction. H2O (generated by the FT reaction) was detected in much larger quantities than CO2 (generated by the Boudouard reaction) (Fig. S24†). However, a decrease in the CO/H2 ratio was expected to lead to side reactions (eqn (3) and (4)).38 Based on these results, it is suggested that CO/H2 = 1 is the optimum ratio for CO-CCVD.
A single-atom catalyst, in which Co atoms were dispersed on silica with the same loading amount as in Co SCs (2 wt% Co atoms on silica), was also evaluated for the CO-CCVD reaction using a CO/H2 = 1 mixture (Fig. S25 and S26†). In this case, no progress in CNT growth was observed at 600 °C over the course of 60 min, and CNTs were generated in low yields even at higher reaction temperatures. The H2-TPR profiles shown in Fig. S10† indicate that higher temperatures (≥620 °C) were required for chemical reduction of air-oxidized Co atoms and their nucleation on silica. This is due to strong adsorption and stabilization of Co single atoms to O atoms on the silica surface compared to Co SCs. Therefore, a single-atom catalyst in this ‘seed method’ cannot grow CNTs at temperatures below at least 620 °C. On the other hand, Co SCs can be reduced even at as low as 500 °C, leading to formation of NPs and growth of CNTs at lower temperatures. Consequently, it was demonstrated that sub-nanosized clusters, with higher reducibility than single atoms and without CO-activating ability like in the case of NPs, serve as superior precursors in realizing the seed method for obtaining diameter-controlled CNTs under mild conditions.
Conclusion
Co SCs highly dispersed on silica served as seeds for the in situ formation of size-defined Co NP catalysts. The diameters and yields of multi-walled CNTs were adequately tuned by modifying the reaction temperature and time, in contrast with conventional methods.29,39 The CO-CCVD system successfully enabled CNT growth with a finely controlled diameter at a 1 nanometer precision with a narrow distribution (≤30%) and high reaction efficiency (348 molC molCo−1 h−1) at several reaction temperatures, in contrast with other techniques.14,34,40 This result was achieved by a process whereby the migration and aggregation behavior of seed metal clusters uniformly induces NP formation before CNT growth begins, and then spatial separation of the NPs from the support substrate based on the tip-growth mechanism of CNTs suppresses excessive enlargement of NPs. In contrast with the seed clusters, Co single atoms on silica did not catalyze CNT growth because they could not be activated by H2 at the temperature required for CO-CCVD because of their strong interaction with silica. Therefore, metal clusters offer many advantages for the seed method, such as higher reducibility than that of single atoms, suitable mobility on the silica substrate, and inertness to CO. These properties are unique to clusters with moderate particle sizes that belong neither to atoms nor to NPs; thus, this synthetic approach exploiting the advantages of clusters will lead to the efficient and selective production of highly functional nanomaterials containing CNTs.
Experimental
Materials
The dendrimer was synthesized according to a previous report.41 Dichloromethane, acetonitrile, and methanol were purchased from Kanto Chemical Co., Inc. Trifluoroacetic acid was purchased from Wako Pure Chemical Industries, Ltd. All the solvents were of anhydrous grade. Sodium triethylborohydride (NaBEt3H) (1.0 M, in toluene) and iron nanopowder (25 nm) were supplied by Sigma-Aldrich Co., LLC. Nickel trifluoroacetate tetrahydrate (NiII(TFA)2) was purchased from Hydrus Chemical, Inc. Cobalt carbonate (CoCO3) was purchased from Wako Pure Chemical Industries, Ltd. Silica (Aerosil 300) and alumina (Aeroxide Alu C) were supplied by Nippon Aerosil Co., Ltd. High-surface-area graphene nanopowder (diameter: 1.6 nm, surface area: 400–800 m2 g−1) was purchased from Alliance Biosystems, Inc. Carbon monoxide (G1 grade, >99.95%) and hydrogen (G1 grade, >99.99999%) were obtained from Kayama Oxygen. Cobalt trifluoroacetate tetrahydrate (CoII(TFA)2(aq)4) and iron trifluoroacetate tetrahydrate (FeII(TFA)2(aq)4) were prepared using the methods described in the ESI.†
Instruments
UV-vis absorption spectra were recorded using a UV-3150 spectrometer (Shimadzu Corp). STEM measurements were performed using a JEM-ARM200F (JEOL, Ltd) system equipped with an energy-dispersive X-ray spectroscopy (EDS) analyzer (acceleration voltage: 80 kV). STEM samples were prepared by dropping the SNP solution onto molybdenum TEM grids with a carbon-coated microgrid film (Nisshin EM Co., Ltd). XPS analysis was performed using a PHI VersaProbe 3 instrument (ULVAC-PHI, Inc). XAFS data were acquired at BL9C of the PF (proposal no. 2023G054, KEK-IMSS-PF, Tsukuba, Japan). CO-CCVD and H2-TPR measurements were performed using a BELCAT II instrument (MicrotracBEL Corp). Qualitative analysis of the products using H2-TPR was conducted using a BELMass (Microtrac-BEL Corp.) mass spectrometer connected to a BELCAT II instrument. TG analysis was conducted using a TG8120 instrument (Rigaku Corp). A theoretical thermodynamic equilibrium simulation was conducted using the Gibbs free energy minimization algorithm in HSC Chemistry 10 software (Outotec Research Oy).
Titration method using UV-vis absorption spectra
A 3.0 × 10−6 M dendrimer solution (dichloromethane, 3 mL, 20 °C) was prepared in a 1 cm optical quartz cell. A 3.0 × 10−3 M CoII(TFA)2 solution (dichloromethane/acetonitrile = 50) was added to 3 mL of the dendrimer solution in a stepwise fashion (0, 1, 2, 3, 4, 6, 8, 10, 12, 16, 20, 24, 28, 36, 44, 52, and 60 equiv.) under an inert gas. The dendrimer used in this study had 60 Lewis basic imine units in its structure and could accumulate Lewis acidic metal salts through complexation (Fig. S1†). Additionally, the intramolecular negative potential gradient of the dendrimer originating from the branched π-conjugation system governs the order of basicity of the imines, conferring stronger basicity to imines at the inner layers. Therefore, the Co salts preferentially accumulated in the inner imines without random complexation (Fig. S2†). This complexation behavior was confirmed by titration using UV-vis absorption because the absorption peaks of the complexes of the four types of imines differed.
Preparation of Co SCs and STEM observations
The metal-salt/dendrimer complex was prepared under the same conditions as those used in the titration method. NaBEt3H solution (10 equiv. of 1.0 M in methanol on the accumulated metal salts) was added to the complex solution with stirring, and Co SCs were then formed by chemical reduction of CoII. This SC solution was quickly added to a suspension of silica or alumina (50 mg in 100 mL of dichloromethane) and stirred for 1 min to immobilize 2 wt% Co SCs on the silica or alumina. The Co SCs supported on silica or alumina were collected by filtration and subsequently rinsed with chloroform and methanol to remove the dendrimers and NaBEt3H from the sample. All processes were performed under N2 in a glove-box. For low-magnification STEM observations, 5 μL of a dispersion of SCs on silica or alumina in dichloromethane was dropped onto a molybdenum TEM grid and dried overnight under vacuum. For STEM observations with atomic-level resolution, the SC solution was added to a dispersion of graphene in dichloromethane (1 mg in 2 mL of dichloromethane) and stirred for 1 s to immobilize 2 wt% Co SCs on graphene. This suspension (5 μL) was placed on a molybdenum TEM grid and dried overnight under vacuum.
XPS measurements
XPS data were recorded using a PHI VersaProbe 3 (ULVAC-PHI, Inc.) instrument with Mg-Kα radiation (50 W, 15 kV). All XPS samples (powder state) were loaded onto carbon tape for analysis. The binding energies were standardized relative to the Si 2p peak of the silica substrate at 103.5 eV.
XAFS measurements
XAFS data were acquired in transmission mode at the BL9C of PF (proposal no. 2023G054, KEK-IMSS-PF, Tsukuba, Japan). The electron-storage ring was operated at 2.5 GeV and 45 mA. Synchrotron radiation from the storage ring was monochromatized using Si (111) channel-cut crystals. Ionization chambers filled with N2 gas and an 85% N2 + 15% Ar gas mixture were used as detectors to monitor the incident (I0) and transmitted X-rays (I), respectively. The angle of the monochromator was calibrated using Co foil, with the inflection point at the edge set to 7712.5 eV. All XAFS samples (powder state) were molded into pellets.
General procedure for CO-CCVD
CO-CCVD was performed using a BELCAT II catalyst analyzer (MicrotracBEL Corp). Each sample (15 mg) was placed in a quartz tube. As a pre-treatment, the sample was calcined at 200 °C under Ar at a flow rate of 30 cm3 min−1 for 10 h to remove H2O and air. Subsequently, CO-CCVD was conducted under a mixture of CO/H2 = 1 or 2 gas at a flow rate of 20 cm3 min−1 and Ar at a flow rate of 30 cm3 min−1 at constant temperature (400, 450, 475, 500, 550, 600, 650, and 700 °C) for various durations (1, 10, 20, 30, 60, and 120 min).
General procedure for H2-TPR
H2-TPR measurements were performed using a BELCAT II catalyst analyzer (MicrotracBEL Corp). Each sample (100 mg) was loaded into a quartz tube. As a pre-treatment, the sample was calcined at 200 °C under Ar at a flow rate of 30 cm3 min−1 for 10 h to remove H2O and air. Subsequently, H2-TPR measurement was conducted under an Ar/H2 = 4 mixture gas at a flow rate of 50 cm3 min−1 with temperature ramping at a rate of +5.0 °C min−1 in the temperature range of 100–900 °C. The generation of H2O was monitored based on the mass intensity of the signal at m/z = 18 using a BELMass (MicrotracBEL Corp.) mass spectrometer, by applying the following equation:
TG measurements
Thermogravimetric (TG) measurements were performed using a differential thermal analyzer (TG8120, Rigaku Corp.) under an air atmosphere. The sample (10 mg) was loaded into an alumina sample pan and calcined at 120 °C for 10 h to remove H2O. Subsequently, the temperature was ramped to 700 °C at a rate of +5.0 °C min−1. The change in the TG curve of the sample due to carbon combustion by oxygen in air was monitored.
Data availability
The data supporting this article have been included as part of the ESI.†
Author contributions
T. M. and T. T. initiated this study under the supervision of K. Y. and T. I. T. M. and T. T. carried out the accumulation of the metal salts in the dendrimer, synthesis and identification of particles, and CO-CCVD reactions. T. T. synthesized the metal salts for cluster preparation. T. M. conducted the STEM observations and TG measurements of the prepared CNTs. T. M., T. T., and K. Y. wrote the manuscript through discussions with all the authors.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This study was supported by grants from JST ERATO (Grant Number JPMJER1503 (K. Y.)), JST PRESTO (Grant Number JPMJPR20AA (T. T.)), JSPS KAKENHI (Grant Numbers JP19K15583 (T. T.), JP22H01904 (T. T.), JP23K19268 (T. M.), and JP24K21707 (T. T.)), a research grant from the Advanced Technology Institute (T. T.), the Yoshinori Ohsumi Fund for Fundamental Research (T. T.), the JGC-Saneyoshi Scholarship Foundation (T. T.), the Toshiaki Ogasawara Memorial Foundation (T. T.), the Iwatani Naoji Foundation (T. T.), the Inamori Foundation (T. T.), the Iketani Science and Technology Foundation (T. T.), the Mizuho Foundation for the Promotion of Sciences (T.T.), the Japan Association for Chemical Innovation (T. T.), the Special Fund of the Institute of Industrial Science, University of Tokyo (T. T.),and a grant from the University of Tokyo Excellent Young Researcher (T. T.).
References
- S. Iijima, Nature, 1991, 352, 56–58 CrossRef.
- X. Peng and S. S. Wong, Adv. Mater., 2009, 21, 625–642 Search PubMed.
- S. Bellucci, Phys. Status Solidi C, 2005, 2, 34–47 Search PubMed.
- S. Iijima and T. Ichihara, Nature, 1993, 363, 603–605 CrossRef CAS.
- Y. Yan, J. Miao, Z. Yang, F.-X. Xiao, H. B. Yang, B. Liu and Y. Yang, Chem. Soc. Rev., 2015, 44, 3295–3346 RSC.
- A. Thess, R. Lee, P. Nikolaev, H. Dai, P. Petit, J. Robert, C. Xu, Y. aH. Lee, S. G. Kim, A. G. Rinzler, D. T. Colbert, G. E. Scuseria, D. Tománek, J. E. Fischer and R. E. Smalley, Science, 1996, 273, 483 Search PubMed.
- F. Kazemizadeh, S. M. Bellah and R. Malekfar, J. Laser Appl., 2017, 29, 042004 CrossRef.
- R. Vajtai, B. Wei, Y. J. Jung, A. Cao, S. K. Biswas, G. Ramanath and P. M. Ajayan, IEEE Trans. Nanotechnol., 2003, 2, 355–361 Search PubMed.
- S. Maruyama, R. Kojima, Y. Miyauchi, S. Chiashi and M. Kohno, Chem. Phys. Lett., 2002, 360, 229–234 Search PubMed.
- M. A. Cassel, A. J. Raymakers, J. Kong and H. Dai, J. Phys. Chem. B, 1999, 103, 6484–6492 CrossRef.
- P. B. Amama, M. R. Maschmann, T. S. Fisher and T. D. Sands, J. Phys. Chem. B, 2006, 110, 10636–10644 Search PubMed.
- M. V. Singh, A. K. Tiwari and R. Gupta, ChemistrySelect, 2023, 8, e20220471 Search PubMed.
- S. Sato, A. Kawabata, D. kondo, M. Nihei and Y. Awano, J. Aerosol Res., 2005, 20, 108–115 CAS.
- C. L. Cheung, A. Kurtz, H. Park and C. M. Lieber, J. Phys. Chem. B, 2002, 106, 2429–2433 Search PubMed.
- L. J. E. Hofer, E. Sterling and J. T. McCartney, J. Phys. Chem., 1955, 59, 1153–1155 CrossRef CAS.
- R. K. Rana, Y. Koltypin and A. Gedanken, Chem. Phys. Lett., 2001, 344, 256 CrossRef CAS.
- K. Cui, A. Kumamoto, R. Xiang, H. An, B. Wang, T. Inoue, S. Chiashi, Y. Ikuhara and S. Maruyama, Nanoscale, 2016, 8, 1608–1617 RSC.
- K. Otsuka, R. Ishimaru, A. Kobayashi, T. Inoue, R. Xiang, S. Chiashi, Y. K. Kato and S. Maruyama, ACS Nano, 2022, 16, 5627–5635 CrossRef CAS PubMed.
- K. Bromann, C. Flix, H. Brune, W. Harbich, R. Monot, J. Buttet and K. Kern, Science, 1996, 274, 956–958 Search PubMed.
- N. K. Chaki, Y. Negishi, H. Tsunoyama, Y. Shichibu and T. Tsukuda, J. Am. Chem. Soc., 2008, 130, 8608–8610 CrossRef CAS PubMed.
- D. Kçnig, K. Richter, A. Siegel, A.-V. Mudring and A. Ludwig, Adv. Funct. Mater., 2014, 24, 2049–2056 CrossRef.
- M. Yamamoto, Y. Kashiwagi and M. Nakamoto, Langmuir, 2006, 22, 8581–8586 Search PubMed.
- T. Moriai, T. Tsukamoto, M. Tanabe, T. Kambe and K. Yamamoto, Angew. Chem., Int. Ed., 2020, 59, 23051–23055 CrossRef CAS PubMed.
- K. Yamamoto, T. Imaoka, W.-J. Chun, O. Enoki, H. Katoh, M. Takenaga and A. Sonoi, Nat. Chem., 2009, 1, 397–402 CrossRef CAS PubMed.
- M. Huda, K. Minamisawa, T. Tsukamoto, M. Tanabe and K. Yamamoto, Angew. Chem., Int. Ed., 2019, 58, 1002–1006 CrossRef CAS.
- T. Tsukamoto, T. Kambe, A. Nakao, T. Imaoka and K. Yamamoto, Nat. Commun., 2018, 9, 3873 CrossRef PubMed.
- K. Yamamoto and T. Imaoka, Bull. Chem. Soc. Jpn., 2006, 79, 511–526 Search PubMed.
- K. Yamamoto and T. Imaoka, Acc. Chem. Res., 2014, 47, 1127–1136 CrossRef CAS PubMed.
- P. Nikolaev, M. J. Bronikowski, R. K. Bradley, F. Rohmund, D. T. Colbert, K. A. Smith and R. E. Smalley, Chem. Phys. Lett., 1999, 313, 91–97 Search PubMed.
-
M. Kumer and S. Yellampalli, Carbon Nanotubes, 2010 Search PubMed.
- A. Gohier, C. P. Ewels, T. M. Minea and M. A. Djouadi, Carbon, 2008, 46, 1331–1338 CrossRef CAS.
- S. Sakurai, T. Tsuji, J. He, M. Yamada and D. N. Futaba, ACS Appl. Nano Mater., 2024, 7, 12745–12751 Search PubMed.
- W. E. Alvarez, B. Kitiyanan, A. Borgna and D. E. Resasco, Carbon, 2001, 39, 547–558 Search PubMed.
- Y. Murakami, Y. Miyauchi, S. Chiashi and S. Maruyama, Chem. Phys. Lett., 2003, 374, 53–58 CrossRef CAS.
- V. Gaikwad, T. K. Rout, D. V. Plas, R. V. Dennis, S. Banerjee, S. P. Benito and L. Lefferts, Carbon, 2012, 50, 4722–4731 CrossRef.
- S. B. Pacheco and L. Lefferts, Carbon, 2010, 48, 2862–2872 CrossRef.
- B. Panek, H. Kierzkowska-Pawlak, P. Uznański, S. Nagy, V. Nagy-Trembošová and J. Tyczkowski, Catalysts, 2023, 13, 1302 Search PubMed.
- W. E. Alvarez, B. Kitiyanan, A. Borgna and D. E. Resasco, Carbon, 2001, 39, 547–558 Search PubMed.
- M. J. Bronikowski, P. A. Willis, D. T. Colbert, K. A. Smith and R. E. Smalley, J. Vac. Sci. Technol., A, 2001, 19, 1800 Search PubMed.
- J. Ma, J. N. Wang and X. X. Wang, J. Mater. Chem., 2009, 19, 3033–3041 Search PubMed.
- O. Enoki, H. Katoha and K. Yamamoto, Org. Lett., 2006, 8, 569–571 Search PubMed.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *cde,
Kaori
Fukuhara
a,
Takane
Imaoka
*cde,
Kaori
Fukuhara
a,
Takane
Imaoka