
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAuCu bimetallic nanocluster-modified titania nanotubes for photoelectrochemical water splitting: composition-dependent atomic arrangement and activity†
Vana
Chinnappa Chinnabathini
 ab,
Karthick Raj
Ag
b,
Thi Hong Trang
Nguyen
a,
Zviadi
Zarkua
a,
Imran
Abbas
a,
Thi Hang
Hoang
a,
Peter
Lievens
a,
Didier
Grandjean
*a,
Sammy W.
Verbruggen
*b and
Ewald
Janssens
*a
ab,
Karthick Raj
Ag
b,
Thi Hong Trang
Nguyen
a,
Zviadi
Zarkua
a,
Imran
Abbas
a,
Thi Hang
Hoang
a,
Peter
Lievens
a,
Didier
Grandjean
*a,
Sammy W.
Verbruggen
*b and
Ewald
Janssens
*a
aQuantum Solid-State Physics, Department of Physics and Astronomy, KU Leuven, Belgium. E-mail: ewald.janssens@kuleuven.be; didier.grandjean@kuleuven.be
bSustainable Energy, Air & Water Technology (DuEL), University of Antwerp, Belgium. E-mail: sammy.verbruggen@uantwerpen.be
First published on 14th November 2024
Abstract
The photoelectrochemical (PEC) water splitting reaction of bimetallic AuxCu1−x (x = 1, 0.75, 0.5, 0.25 and 0) nanocluster-decorated TiO2 nanotube (TNT) photoanodes was investigated using a solar simulator. A strong enhancement in the anodic photocurrent relative to pristine TNTs was found with clear composition-dependent PEC activity, increasing with the Cu content and peaking at Au0.25Cu0.75. Electron microscopy and X-ray absorption fine structure spectra recorded at both Au and Cu edges identified a clear composition-dependent atomic arrangement of the spherical nanoclusters on anatase TNTs, resulting mostly from a time-dependent restructuring of the original metallic nanoalloys in the ambient environment. With time, Cu segregates from the alloy to form a surface oxide layer surrounding a pure gold metallic core in the gold-rich nanoclusters (x = 0.75 and 0.50) or a face centered tetragonal (fct)-intermetallic Au0.5Cu0.5 nanoalloy in copper-rich (x = 0.25) particles. In pure Cu nanoclusters, a metallic Cu core is stabilized by surrounding Cu2O and CuO. The enhanced PEC activity is attributed to a synergy between Au and Cu that upon segregation produces bifunctional catalytic sites consisting of a metallic Au/AuCu alloy and copper oxide at the surface of the nanoclusters. The photoactivity under solar light illumination is boosted by the plasmonic response of the metal. The ordered structure of the fct-AuCu alloy present in the most active Au0.25Cu0.75 may explain its higher stability and photocatalytic performance. Hence, this work provides insight into the relationship between the atomic-level structure of AuxCu1−x nanoalloys on TNTs and their PEC activity.
1. Introduction
The n-type semiconductor titanium dioxide is known for its good stability, high resistance to corrosion, non-toxicity, bio-compatibility, and low production cost. TiO2 nanostructures, including nanoparticles,1 nanowires,2 nanorods,3 nanotubes,4 and nanopores,5 are intensively studied, particularly for photocatalysis. Anodic oxidation of metallic titanium foil allows for straightforward and efficient synthesis of highly ordered titania nanotubes (TNTs), whose length and diameter can be conveniently tuned by the applied voltage, anodizing time, annealing temperature, and the composition of the electrolyte.6–9 Since the pioneering work of Zwilling et al.10 on the anodic oxidation of titanium into highly ordered titania nanotubes, the morphology of TNTs and their potential applications in catalysis have been studied extensively. Self-assembled TNT arrays exhibit exceptional structural, electrical, thermal and optical properties.11–16 They have a highly ordered structure, high mechanical and chemical stability, good corrosion resistance, high specific surface area,17,18 enhanced charge transfer, and a large number of active sites.19,20 These properties make TNTs useful for applications in photocatalysis,21 solar cells, environmental purification, water photolysis, gas sensing, and bio-medicine.22,23 By diminishing dimensions to the nanoscale, the specific surface area increases, while the modified electronic structure of the TNTs diminishes the electron–hole recombination rate. The large effective surface area in direct contact with the electrolyte enables diffusive transport of photogenerated holes to the oxidizable species in the electrolyte of photoelectrochemical (PEC) cells. The effects of annealing temperature, crystallinity, wall thickness, tube length and diameter of the TNTs on the PEC activity have been discussed in several reviews.24–26TiO2 has suitable band-edge positions for electrochemical water splitting with the bottom of the conduction band being more negative than the H2/H2O redox potential (0 V) and the top of the valence band being more positive than the H2O/O2 redox potential (−1.23 V).27,28 A drawback is its relatively large bandgap (≈3.2 eV in the anatase phase), requiring UV light for efficient operation. Various modifications, particularly the incorporation of metal nanoparticles, have been employed to enable visible light driven processes. Cu, Ag, and Au nanoparticles exhibit a strong ability to absorb solar photons due to the localized surface plasmon resonance (LSPR) effect29 and can transfer energetic electrons formed in the LSPR process to a nearby semiconductor.30 LSPR-induced near-field enhancement at the surface of the metallic nanostructures can contribute to the generation of electron–hole pairs. It has been well established that metal nanoparticles on the surface of TiO2 can act as traps for photo-induced electrons, preventing fast recombination of charge carriers,31,32 and thus enhancing the PEC activity of TNT electrodes. Several examples of improved photocurrents in TNTs decorated with monometallic Cu, Ag, or Au nanoparticles can be found in the literature.33–37 For instance, photocurrent densities of about 90 μA cm−2 and 80 μA cm−2 were recorded under 350 nm irradiation at an applied voltage of 1 V versus the saturated calomel electrode, 1 VSCE for TNTs modified with Cu and Ag, respectively.38 The modification of TNTs with plasmonic bimetallic nanoparticles combines the LSPR effect with properties from both metals39–42 and provides highly active metal–support interfaces suitable for PEC reactions.41 For example, TNTs modified with atomically precise AuCu alloy nanoparticles were found to exhibit an improved photocurrent under visible light irradiation in comparison with a blank TNT substrate.43 Also TNTs modified with AuCu nanoparticles that are thermally treated in argon exhibited a higher photocurrent (64 μA cm−2 at −0.2 V) under UV–Vis light as a pristine TNT electrode (32 μA cm−2).44
The modification of TNTs with bimetallic AuCu nanoalloys takes advantage of a synergy between the highly stable and plasmonic active noble metal Au and the less stable but also less expensive and more electrocatalytically active metal Cu. Utilising this promising combination requires a good understanding of the effect of composition on the structural arrangement. Such information is, to the best of our knowledge, lacking.
The aim of the current study is to modify TNTs with well-defined gas-phase prepared AuCu particles with nanometer sizes, hereafter called nanoclusters (NCs) and explore the composition-dependent relationship between their PEC performance for water splitting and the atomic NC arrangement. For this purpose, we have synthesized and deposited small-sized (<5 nm) AuCu NCs at a low metal loading (about 1 μg cm−2) on TNTs with good control over the composition and coverage and tested their PEC water splitting performance. The AuCu-modified TNT photoanodes exhibit excellent photostability and high composition-dependent photoactivity in the visible range of the solar spectrum. Alloy NCs with different compositions were synthesized through the physical cluster beam deposition (CBD) process48 that produced ligand-free and size- and composition-tuneable NCs with a high degree of purity and uniform coverage.
2. Results
2.1. Structural characterization
The gas-phase size distribution of AuxCu1−x clusters, produced using a laser ablation source by ablating alloy targets with x = 1, 0.75, 0.5, 0.25, and 0 followed by inert gas condensation, is recorded using in situ time-of-flight mass spectrometry. Assuming a spherical shape and bulk density for all NC compositions, the diameter distribution of the clusters prior to deposition has a maximum of around 2 nm (see the ESI, Fig. S1†). Because the detection efficiency of larger clusters by the microchannel plate detector is lower, the real average size of the clusters in the gas phase is likely larger. The loading of the NCs is determined by the deposition time and the mass flux measured before and after cluster deposition using a quartz crystal microbalance (QCM). A typical flux value is 0.062 μg per cm2 per minute. A total mass loading of 1.1 μg cm−2 has been deposited for the samples discussed in this work, which can also be expressed in an equivalent height of a hypothetical thin film of 4 atomic monolayers (MLs, i.e. a thickness equivalent to a film of 0.25 nm).X-ray diffraction (XRD) patterns of the annealed TNTs modified with AuCu NCs obtained after PEC measurements are provided in the ESI, Fig. S2.† The characteristic diffraction peaks of TiO2 anatase are observed as well as the underlying metallic titanium substrate of the electrode on which the TNTs were grown. In line with previous studies on TNTs modified with small metal clusters,45,46 no diffraction peaks of the deposited clusters could be detected, likely due to the low loading and their small sizes. The intense (004) diffraction peak observed for all samples indicates a preferred [001]-oriented anatase phase in TNTs with the (004) lattice plane perpendicular to their axis. The variations in the relative intensity of the anatase (004) and (101) peaks reflect variations in the orientation of the TNTs relative to the Ti substrate.47
Fig. 1 shows top-view and cross-sectional scanning electron microscopy (SEM) images of the pristine and NC decorated TNTs. The TNTs have an ordered nano-morphology https://www.sciencedirect.com/topics/materials-science/pore-structure with an average tube diameter of ca. 60 nm, a wall thickness of ca. 15 nm, or an inner diameter of ca. 30 nm, and a tube length of ca. 2.5 μm (Fig. 1a). The metal decorated samples present randomly distributed clusters (i.e. the small bright dots in Fig. 1) on the top surface and the inner walls of the TNTs. Although no apparent sign of cluster agglomeration is observed, it cannot be excluded as the resolution of the SEM (0.8 nm @ 30 keV) is of the order of the size of the clusters. Pure gold NCs appear brighter and slightly larger (Fig. 1b).
 | ||
| Fig. 1 Top-view SEM images of (a) pristine TNTs with a cross-sectional image in the inset and TNTs covered with 4 MLs of (b) Au, (c) Au0.75Cu0.25, (d) Au0.5Cu0.5, (e) Au0.25Cu0.75, and (f) Cu NCs. The NCs are visible in the insets that zoom in on a 140 × 140 nm2 area. | ||
More information about the morphology, size, structure and composition of alloy NCs is obtained by high-angle annular dark-field (HAADF) scanning transmission electron microscopy (STEM) studies. Fig. 2a presents an electron microscopy image of the sample with a loading of 0.67 ML of Au0.25Cu0.75 NCs deposited on a carbon TEM grid. The NCs are uniformly dispersed and have an average diameter of 3.5 ± 0.5 nm. Energy dispersive X-ray (EDX) mapping of the Au0.25Cu0.75 NCs shows that many clusters have an alloyed surface (pale yellow) covered by a significant amount of pure copper patches consisting likely of Cu oxide and a limited amount of pure gold ones (Fig. 2b). EDX elemental mapping over a larger area indicates that the composition of an ensemble of deposited Au0.25Cu0.75 NCs is 22.7% Au and 77.3% Cu, in line with the composition of the ablated alloy target (see the ESI, Fig. S3†).48,49 We assume the same holds for the average compositions of the other AuCu NCs based on our observations for NCs with different Au/Ag ratios that were produced using the same cluster source.40,59
 | ||
| Fig. 2 (a) HAADF-STEM image with the size distribution of NCs obtained from image analysis and (b) EDX elemental mapping of Cu (green) and Au (red) in the Au0.25Cu0.75 NCs. | ||
The slightly larger size distribution observed using TEM compared to the mean diameters obtained by mass spectrometry in the gas-phase may be attributed to the above-mentioned less efficient detection of the larger mass clusters using the microchannel plate detector of the mass spectrometer (particularly for the used acceleration voltage <3 kV).50 Additionally, flattening of the clusters due to cluster–support interaction after deposition and some cluster agglomeration may also contribute to the larger observed sizes.
More information on the composition and the oxidation states of gold and copper in the AuxCu1−x/TiO2 (x = 0.75, 0.50, and 0.25) samples was obtained by X-Ray Photoelectron Spectroscopy (XPS). The XPS survey spectrum (see the ESI, Fig. S4†) shows that O, C, Ti, Au and Cu are present on the surface of all samples. The O 1s contribution is representative of the oxidation of the Cu phase, while the peak intensity of C 1s is relatively consistent for all the samples despite their varying compositions. Au 4f7/2 and 4f5/2 contributions are observed at binding energies of around 84 eV and 88 eV, while the Au 4d5/2 and 4d3/2 core levels are located at around 334 eV and 355 eV. Upon increasing the Cu concentration, a blue shift of the Au 4d5/2 binding energy to 337.8 eV is observed both in Au0.5Cu0.5 and Au0.25Cu0.75, while it is 335.8 eV in Au0.75Cu0.25 (Fig. 3a). The observation that the Au 4d5/2 binding energy in Au0.75Cu0.25 is close to that of bulk gold hints at the presence of pure gold in these NCs. The large blue shift found for Au0.5Cu0.5 and Au0.25Cu0.75 implies the depletion of the Au d-states, which is indicative of the formation of an alloy, as reported for a variety of Au-based alloys, including AuCu.51–55
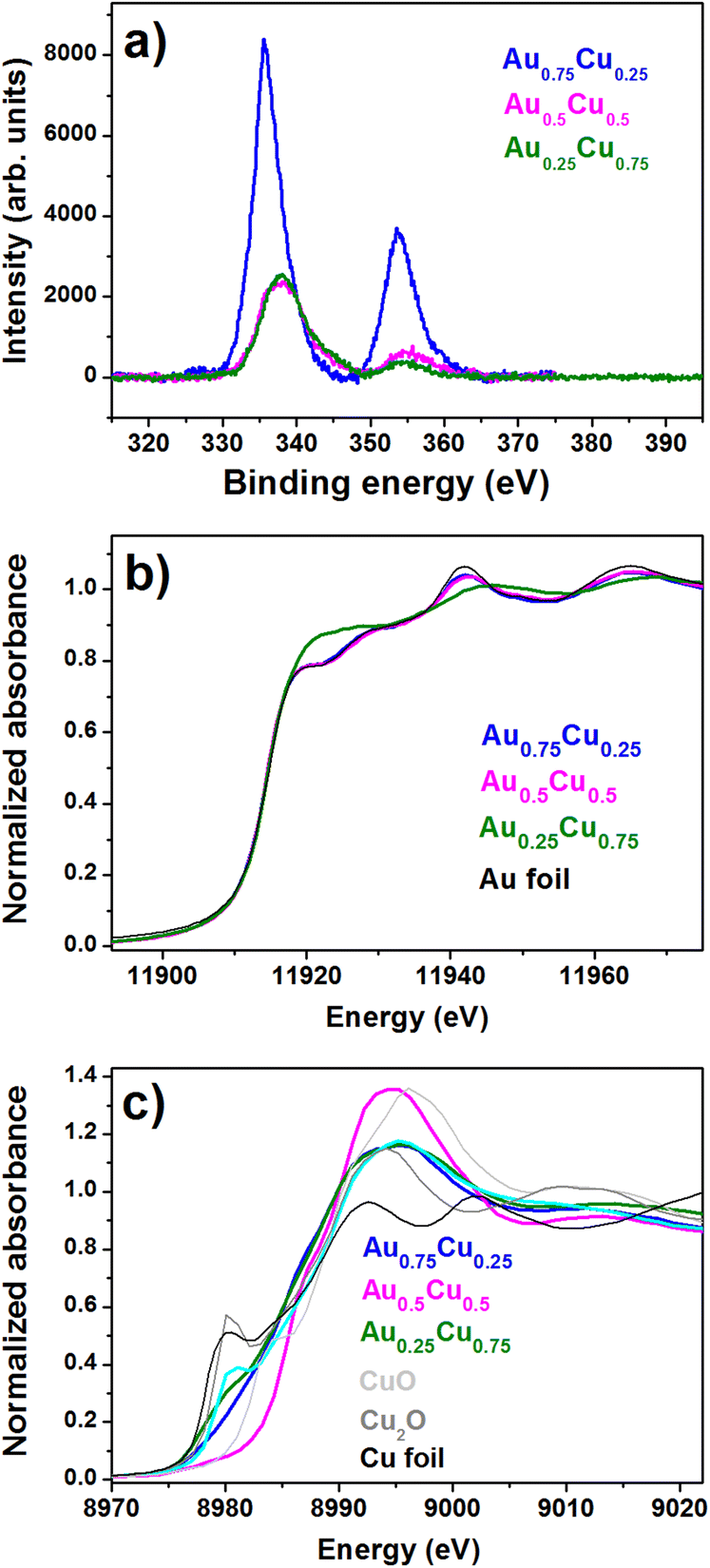 | ||
| Fig. 3 (a) XPS core level Au 4d spectra recorded for AuxCu1−x/TNTs (x = 0.75, 0.5, and 0.25). XANES spectra of (b) Au-L3 edge and (c) Cu K-edge. | ||
The detailed atomic structural arrangement in the AuxCu1−x NCs was further characterized by X-ray absorption spectroscopy, carried out at the Cu and Au edges before performing the PEC studies. X-Ray Absorption Near Edge Structure (XANES) spectra recorded at the Au L3-edge of the alloy samples are presented in Fig. 3b.
The XANES profiles of Au0.75Cu0.25 and Au0.5Cu0.5 are similar to that of the Au foil reference, indicating that mostly pure gold is present in these two alloy samples. On the other hand, in Au0.25Cu0.75 one can see a sharp increase of the absorption near the edge, which is called the white line intensity and reflects for the Au L3-edge the unoccupied density of d states above the Fermi level. This increase indicates a clear charge loss from the Au d states, which is likely due to charge transfer from Au to Cu, occurring upon the formation of an AuCu alloy.56 Similar observations were made for AuAg nanoalloys produced in the same source.48 This is supported by the observation of an increase of the Au 4d binding energy in the XPS data. Unoccupied d states above the Fermi level in Cu, Ag, and Au alloys are generally compensated by a gain of mainly s–p charge from Cu.53,57,58 In Au0.5Cu0.5 a discrepancy is observed between the XPS spectrum recorded a few days after the synthesis, showing the formation of an alloy, and the XANES spectrum recorded a few months later, pointing out an Au rich phase. This could be due to the lower stability of the Au0.5Cu0.5 NCs where Cu tends to segregate from the alloy in the ambient. All alloy AuxCu1−x/TiO2 samples contain a significant amount of copper oxide as shown by the systematic blue shift of the Cu K-edge position, accompanied by the marked increase in the white line intensity (Fig. 3c). Au0.5Cu0.5 contains a large amount of CuO, while a complex mixture of metallic Cu, Cu2O and CuO is found in Au0.75Cu0.25 and Au0.25Cu0.75.
Extended X-ray Absorption Fine Structure (EXAFS) allows for the precise detection of the alloyed structure in nanostructured bimetallic AuCu systems.59,60 The Fourier transform spectra of Au L3-edge k3-weighted EXAFS of Au0.75Cu0.25 and Au0.5Cu0.5 are similar, while that of Au0.25Cu0.75 is distinct (Fig. 4a). Detailed fitting of the Au L3-edge EXAFS data was carried out with a 2-shell structure model based on Au–Au and Au–Cu (see the ESI, Fig. S7 and Table S1†). No Au–O interactions could be detected, indicating the fully metallic state of Au in the clusters. Au–Au atomic bond distances sharply decrease with increasing Cu content from 2.85 Å in gold-rich Au0.75Cu0.25, over 2.84 Å in Au0.5Cu0.5, to 2.81 Å in copper-rich Au0.25Cu0.75. The first two distances are similar to the contracted distance of 2.85 Å (relative to the bulk value of 2.88 Å (ref. 61 and 62)) measured in pure gold clusters,63 indicating the presence of nearly pure gold phase in these materials in line with the XANES analysis. The small Au–Cu contribution of 0.6 Cu with a bond distance of 2.64 Å in Au0.75Cu0.25 implies the presence of a small fraction of a copper-rich alloy in this sample. No Au–Cu contribution could be detected in Au0.5Cu0.5, while in Au0.25Cu0.75 the Au–Cu contribution (5.5 Cu at 2.66 Å) is comparable to the Au–Au signal, indicative of the formation of an ordered intermetallic face centered tetragonal (fct) AuCu alloy. Also, this prediction is consistent with the XANES profile in Fig. 3b.
 | ||
| Fig. 4 Phase-corrected Fourier-transform (a) Au L3-edge and (b) Cu K-edge k3-weighted XAFS spectra of the AuCu NCs/TNT samples. | ||
Fourier transform spectra of Cu K-edge k3-weighted EXAFS in Fig. 4b show distinct profiles for the different samples, but they have a common peak below 2 Å indicative of the presence of a significant fraction of oxides. EXAFS fitting results at the copper edge of bimetallic AuCu and pure Cu clusters (see the ESI, Fig. S8 and Table S2†) show that the first coordination shell consists of 2.27 O at 1.93 Å in pure Cu, 1.8 O at 1.93 Å in Au0.75Cu0.25, 2.9 O at 1.96 Å in Au0.5Cu0.5, and 1.93 O at 1.94 Å in Au0.25Cu0.75. As Cu is four-fold coordinated to O at 1.96 Å in CuO and two-fold coordinated at 1.85 Å in Cu2O, the fraction of copper oxide in each sample, either CuO or Cu2O, can be calculated using a simple combination of both coordination numbers and bond distances. Fractions of ca. 15% (7% of Cu2O and 8% of CuO), 36% of CuOx, 42% (13% of Cu2O and 29% of CuO), and 73% (40% of Cu2O and 33% of CuO) are obtained for Au0.75Cu0.25, Au0.5Cu0.5, Au0.25Cu0.75, and Cu, respectively. Cu–Au contributions of 1.9 Au at 2.63 Å and 4.56 Au at 2.63 Å could be respectively fitted in Au0.75Cu0.25 and Au0.25Cu0.75 confirming the presence of AuCu alloys in these materials. Cu–Cu contributions are 2.3 Cu at 2.57 Å (bulk metal value is 2.56 Å), 1.5 Cu at 2.52 Å, 1.16 Cu at 2.53 Å and 1.37 Cu at 2.56 Å in Au0.75Cu0.25, Au0.5Cu0.5, and Au0.25Cu0.75 and pure Cu, respectively.
Based on the obtained bond lengths, coordination numbers, and the fractions of copper oxide, structural models for the different NCs could be proposed (see Fig. 5). All the Cu containing samples present a significant fraction of copper oxide at the outer shell resulting from the segregation and oxidation of Cu at the surface of the NCs. Pure Cu clusters have the highest fraction of oxides of ca. 73%, as Cu is prone to oxidation under ambient conditions, while Au0.75Cu0.25 has the highest metallic fraction (85%), surrounded by 15% of copper oxide due to the protecting effect of gold. Au0.5Cu0.5 has ca. 50% of Au at the core covered by 14% of AuCu alloy and pure metallic Cu followed by 36% of copper oxide. Interestingly in Au0.25Cu0.75ca. 50% of intermetallic Au0.5Cu0.5 alloy forms at the core covered by 8% Cu metal and 42% copper oxide on the surface. These results highlight the very distinct and complex restructuring processes occurring in the initially alloyed AuxCu1−x NCs as a function of time and composition when they are exposed to ambient conditions. We generally observe the formation of a copper oxide shell consisting of CuO species at the surface and a Cu2O layer between the CuO and the metallic core. The segregation of copper to the surface results in the formation of a mostly pure Au core in the gold-rich samples. However, the high stability of the AuCu intermetallic alloy combined with the thick Cu oxide passivation layer stops Cu segregation in copper-rich nanoalloys when the 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 stoichiometry is reached. In Au0.5Cu0.5 the same AuCu intermetallic alloy is likely formed originally over the whole cluster and remains stable in air for a few days as demonstrated using XPS. However, for longer times the stability of the intermetallic alloy is not sufficient to prevent segregation. Oxidation of copper and destabilisation of the intermetallic structure by departing from the 1:1 stoichiometry and full copper segregation is observed after a few months. This detailed structural characterisation highlights the very distinctive atomic arrangement occurring in the bimetallic NCs as a function of their composition leading to different surface and cores (pure metal, random and/or ordered intermetallic alloys) that are expected to significantly affect their stability and photocatalytic activity. Although Cu oxide is located at the surface of the clusters, it does not form a complete layer but rather grains on the surface of the metallic alloy as shown by the EDX-STEM images of the Au0.25Cu0.75 clusters in Fig. 2. This structural configuration likely results in the formation of a dual structure with both phases accessible for the catalytic reaction.
:1 stoichiometry is reached. In Au0.5Cu0.5 the same AuCu intermetallic alloy is likely formed originally over the whole cluster and remains stable in air for a few days as demonstrated using XPS. However, for longer times the stability of the intermetallic alloy is not sufficient to prevent segregation. Oxidation of copper and destabilisation of the intermetallic structure by departing from the 1:1 stoichiometry and full copper segregation is observed after a few months. This detailed structural characterisation highlights the very distinctive atomic arrangement occurring in the bimetallic NCs as a function of their composition leading to different surface and cores (pure metal, random and/or ordered intermetallic alloys) that are expected to significantly affect their stability and photocatalytic activity. Although Cu oxide is located at the surface of the clusters, it does not form a complete layer but rather grains on the surface of the metallic alloy as shown by the EDX-STEM images of the Au0.25Cu0.75 clusters in Fig. 2. This structural configuration likely results in the formation of a dual structure with both phases accessible for the catalytic reaction.
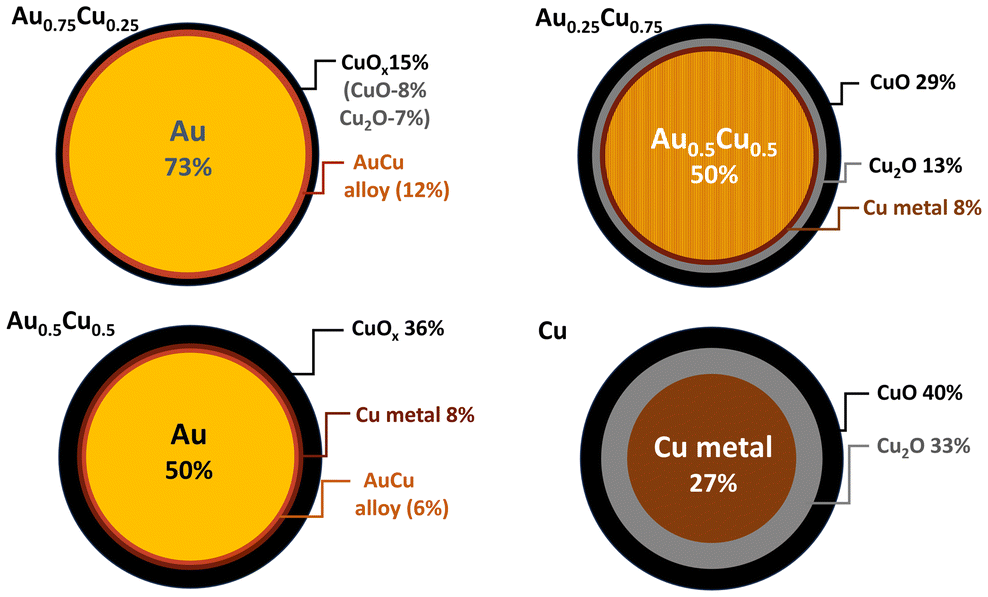 | ||
| Fig. 5 Schematic representation of the predicted NC cross-section as implied from the XAFS data analysis. | ||
As photocatalysis is a complex process, involving plasmonic, catalytic, and charge transfer and separation processes between the TNTs and the nanoalloys, the nature and size of the metallic core of the NCs as well as the nature of their surface are expected to be important for the PEC activity and stability.
2.2 Optical characterization
Fig. 6a presents the UV-Vis absorption spectra of pristine and AuxCu1−x (x = 1, 0.75, 0.5, 0.25 and 0) NC-modified TNTs with a 4 ML loading. All samples exhibit a photoresponse below 400 nm, which can be attributed to the intrinsic band gap of TiO2. This indicates that surface modification of the TNTs with NCs does not affect the inherent absorption of TiO2 in the ultraviolet range. At wavelengths above 400 nm, the absorption significantly changes with the presence of NCs. All cluster-modified samples have a higher absorption in the visible range and the absorption maximum shifts to longer wavelengths compared to bare TNTs. The LSPR intensity of Cu NCs is damped, likely due to their oxidation in air to Cu2O and CuO.64,65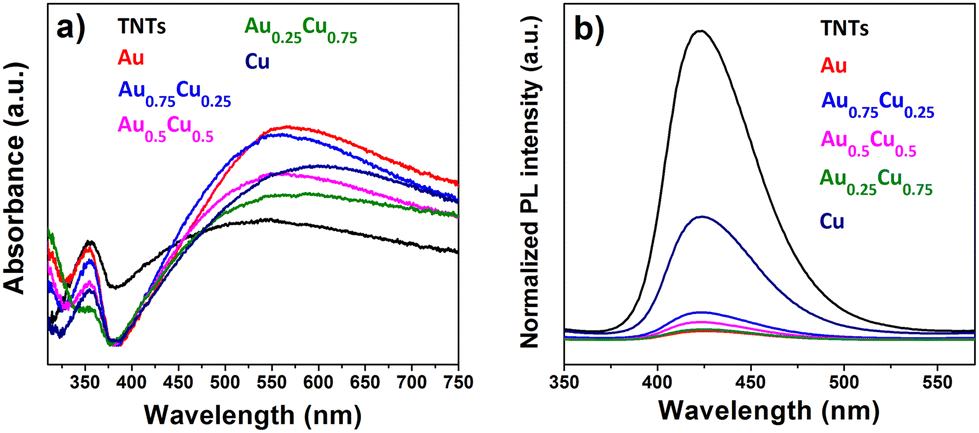 | ||
| Fig. 6 (a) UV-Vis absorbance spectra (300–750 nm) of pristine and AuxCu1−x (x = 1, 0.75, 0.5, 0.25 and 0) cluster-modified TNTs with a loading of 4 MLs. The absorbance spectra are baseline corrected but not normalized. (b) Photoluminescence spectra after excitation of the electrodes at 310 nm. The spectra are normalized for their corresponding optical absorbance at 310 nm. | ||
Although the photoresponse of TiO2 is usually exclusively in the ultraviolet region with wavelengths below 390 nm, the pure TNTs studied in this work show a broad absorption band in the visible range, be it less intense than the one after AuxCu1−x NC deposition. This band can result from light scattering caused by pores and cracks in the nanotubes and/or to oxygen vacancies and Ti3+ states positioned below the TiO2 conduction band.66,67 Finally, the absorption in the visible range may also stem from absorption in the underlying Ti metal related to interband d–d transitions.68 However, incident photon-to-electron conversion efficiency (IPCE) measurements show that no significant photocurrent is generated when the pristine TNTs are illuminated with purely visible light.69 Therefore, the absorption above 500 nm in all the samples could arise not only from the distinctive highly periodic porous structure of TNTs, as previously reported70,71 but also from the LSPR of NCs active in the visible range.
The photoluminescence (PL) spectra of AuxCu1−x (x = 1, 0.75, 0.5, 0.25 and 0) NC-decorated TNT samples, measured at an excitation of 310 nm, are presented in Fig. 6b. Since the radiative recombination of photogenerated carriers and holes takes place close to the surface,72 PL spectra are sensitive to molecular adsorption and chemical reactions at the surface of the semiconductor. The PL intensity quantifies the recombination efficiency of photogenerated charges, which is one of the most important limiting factors for photocatalytic efficiency. The intense PL in the visible range observed for pristine TNTs indicates a rapid recombination. All NC-modified samples exhibited a much lower PL intensity than the bare TNTs, which suggests a significant reduction of the electron–hole recombination.68,73,74 The lowest PL emission is seen for pure Au NCs on TNTs, followed by different alloys Au0.25Cu0.75 < Au0.5Cu0.5 < Au0.75Cu0.25, while the pure Cu NCs reduce the PL of the TNTs the least. When the NCs are deposited on TNTs, migration of excited electrons from the semiconductor to the metal occurs until the two Fermi levels are aligned and a Schottky barrier forms at the AuxCu1−x/TNTs interface, which serves as an efficient electron trap and prevents electron–hole recombination.75 Those trapped electrons are available for redox reactions.
2.3. Photoelectrochemical performance
The photoelectrochemical activity of the pristine and AuxCu1−x (x = 1, 0.75, 0.5, 0.25 and 0) NC-modified TNT electrodes was tested using a standard three electrode PEC cell in 0.5 M Na2SO4 electrolyte (at pH 7.2) under AM 1.5G illumination (100 mW cm−2). When the semiconductor's Fermi level aligns with the redox potential of the electrolyte solution in the PEC cell, band bending takes place and an electric field is generated. This field facilitates the movement of photogenerated charges towards the semiconductor-electrolyte interface. For an n-type semiconductor photoanode, water can oxidize to oxygen.Linear sweep voltammetry (LSV) with the illuminated pristine and AuxCu1−x NC-modified TNT electrodes (Fig. 7a) shows, except for pure Au NCs, an increasing photocurrent density with increasing applied potential. The photocurrent density of the Au/TNTs increases up to 0.6 V versus the reversible hydrogen electrode (RHE) and stabilizes above this value. The pristine TNT electrode shows a linear trend up to 0.3 VRHE, followed by a plateau in photocurrent density until 1.2 VRHE, after which the current increases again. Under illumination TNTs and AuxCu1−x/TNTs electrodes can produce photogenerated electron–hole pairs, whose holes oxidize the water molecules at the anode and electrons reduce protons into molecular hydrogen at the cathode. As summarized in Fig. 7c, the photocurrent density at 1.23 VRHE is strongly enhanced by the presence of the NCs. In particular, the photoanode with Au0.25Cu0.75 NCs produces 244 μA cm−2 at 1.23 VRHE, which is 3.5 times higher than the corresponding value (68 μA cm−2) for pristine TNTs. This Au0.25Cu0.75/TNT result is significantly higher than the values reported (150 μA cm−2 and 64 μA cm−2) in the literature for PEC applications using AuCu nanoparticles on TNTs.43,44 The photoresponse of the electrodes under chopped illumination (switching the light source on and off every 5 seconds) when performing LSV is presented in Fig. 7b. It gives an indication of current under both light and dark conditions exhibited by photoanodes. The current density at a bias of 1.23 VRHE without light is also included in Fig. 7c as a reference for the photocurrent. The dark current probably arises from the electrocatalytic performance of Cu2O. A detailed discussion of the electrocatalytic nature of Cu2O is beyond the scope of this work.
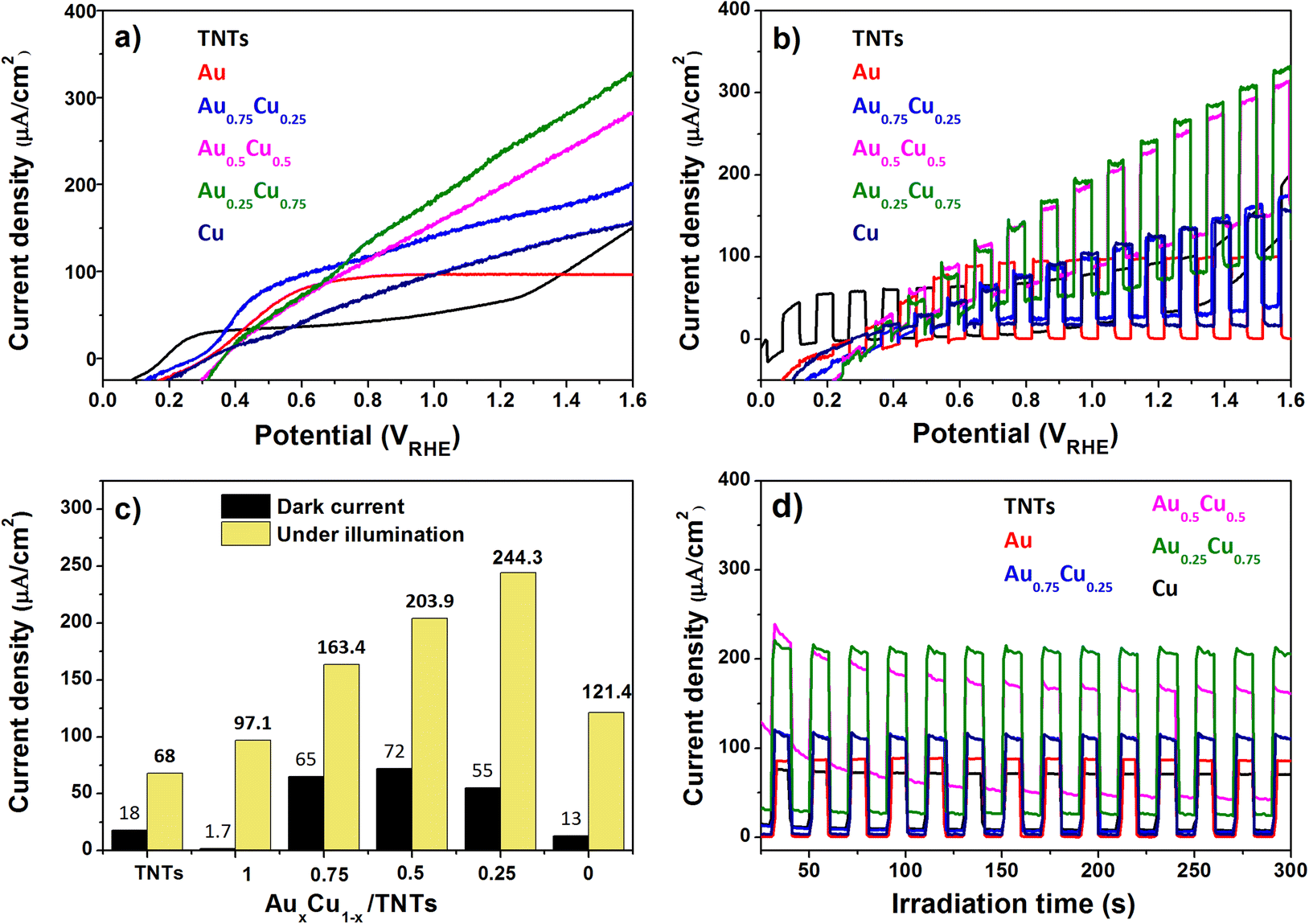 | ||
| Fig. 7 (a) LSV curve of the pristine and AuxCu1−x NC-modified TNT samples under AM 1.5G illumination; (b) LSV curve under chopped illumination; (c) current density of the different photoanodes at 1.23 VRHE, without light and under illumination; and (d) chronoamperometry under chopped illumination at 1.23 VRHE. | ||
Chronoamperometric measurements at 1.23 VRHE (Fig. 7d) show a steady photocurrent for all electrodes and confirm the photocurrent enhancement upon NC modification, particularly with bimetallic AuxCu1−x NCs. The Au0.25Cu0.75 modified TNT photoanode performs the best. It was found to be photostable at 1.23 VRHE for at least 1 hour (see the ESI, Fig. S5†). The small activity drop observed at the beginning of the measurements can be related to the re-oxidation of a fraction of copper in the first seconds (see the assessment of the possible current generated by the oxidation of copper in the ESI†), because the photoanodes were reduced before PEC testing. After a period of 90 days, this electrode retained 90% of its initial activity confirming its resistance to ambient conditions.
The applied bias to photon conversion efficiency (ABPE), as calculated using eqn (1) discussed in section 5.3 from values obtained by LSV under illumination, is depicted in Fig. 8a and b. The pristine TNTs electrode exhibits the lowest ABPE of 0.023% at 0.6 VRHE, while Au0.75Cu0.25/TNTs demonstrates a maximum ABPE of 0.061% at 0.6 VRHE. The Au0.25Cu0.75/TNTs sample which has the highest photocurrent at 1.23 VRHE, shows a slightly lower ABPE of 0.044% at 0.6 VRHE.
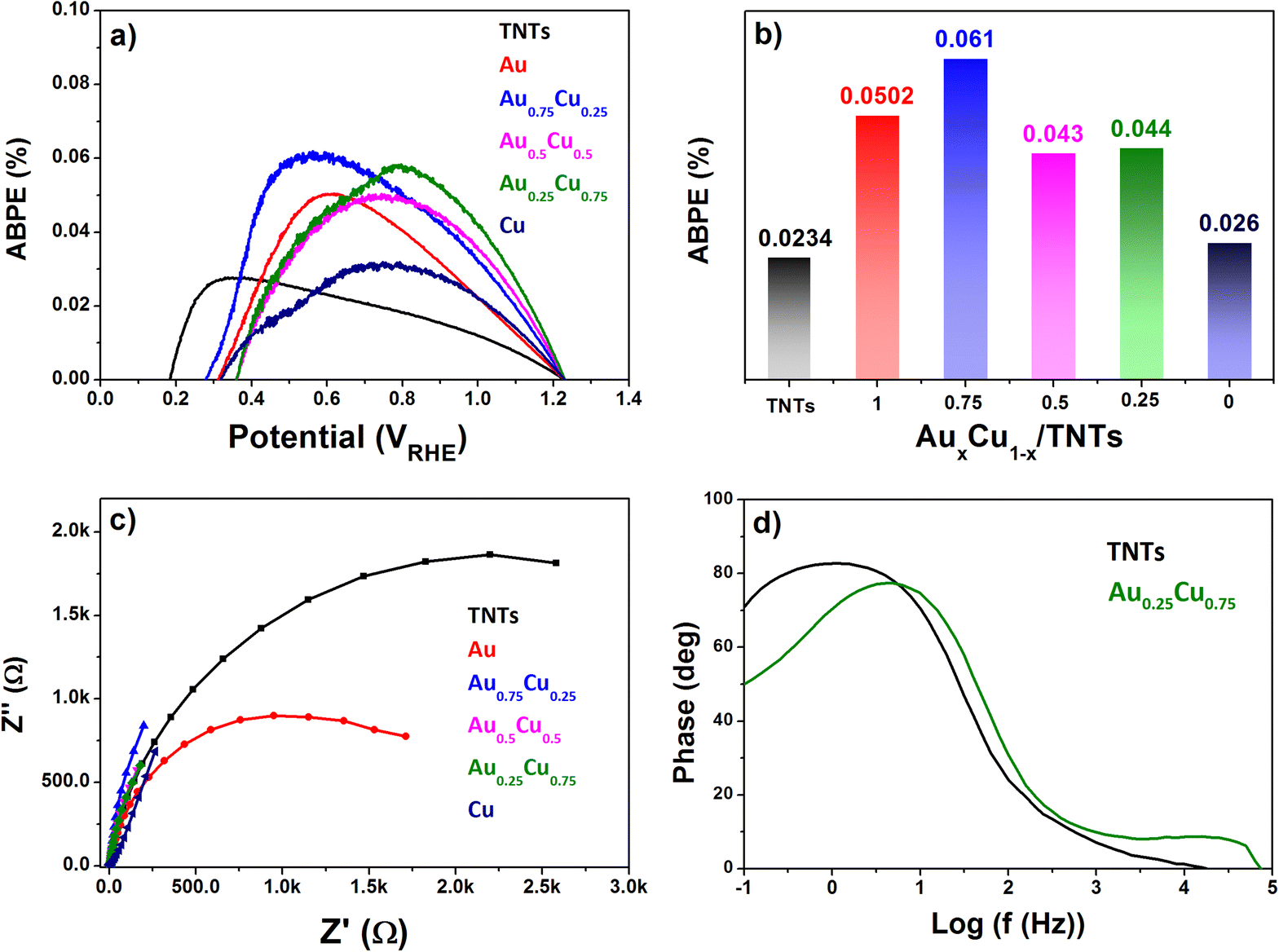 | ||
| Fig. 8 (a) ABPE plot of all electrodes; (b) ABPE values at 0.6 VRHE; (c) EIS spectra of pristine and AuCu1−x/TNT (x = 1, 0.75, 0.5, 0.25 and 0) electrodes measured under AM 1.5G illumination; (d) Bode plot of pristine TNTs and Au0.25Cu0.75/TNTs, measured in 0.5 M Na2SO4 at pH 7.2. | ||
The interfacial charge transfer efficiency of the photoelectrodes at their interface with the electrolyte was evaluated using electrochemical impedance spectroscopy (EIS) measured in 0.5 M Na2SO4 electrolyte at pH 7.2 under AM 1.5G illumination. The Nyquist plot in Fig. 8c shows that Au0.25Cu0.75-modified TNTs exhibit the smallest circle arc followed successively by Au0.5Cu0.5, Au0.75Cu0.25, Cu, and Au NC-modified TNTs, while the pristine TNTs sample exhibits the largest arc. The smaller arc radii in AuCu/TNTs electrodes indicate good electrode–electrolyte interfacial charge transfer with lower charge transfer resistance as compared with pristine TNTs.
The hole relaxation lifetime can be calculated through the Bode phase analysis presented in Fig. 8d. An accelerated lifetime and a minimal hole diffusion length are key factors for a rapid charge transfer process at the interface, resulting in an improved water oxidation performance. Using eqn (2) the hole-relaxation time for Au0.25Cu0.75/TNTs is 40 ms, which is substantially shorter than the corresponding lifetime of bare TNTs of 160 ms. This results, using eqn (3), in a shorter hole diffusion length for Au0.25Cu0.75/TNTs (0.45 mm) than in the bare TNTs (0.91 mm). The dark current densities cannot be ignored since all bimetallic AuCu/TNTs electrodes exhibit a nonzero dark current as seen in LSV under chopped illumination (Fig. 7b and c), originating from the electrocatalytic performance of AuCu in oxidative reactions.76–80 The total current generated by the copper containing electrodes is a combination of electrocatalytic and photocatalytic reactions facilitating a synergistic interaction between the photons and electricity. This synergy contributes to achieving optimal PEC performance.
3. Discussion
CBD technology enabled the production of NCs/TNTs with an optimal 4 ML metal loading which allows the TNT photocatalyst to harvest sunlight effectively and generate more charge carriers, while samples with higher loadings of 6 MLs and 8 MLs exhibit a lower photocurrent density (see the ESI, Fig. S6†). At those higher loadings, the direct exposure of the TNTs to light gradually gets blocked. A similar loading dependence was found in earlier work on photocatalytic stearic acid degradation using titania nanoparticles decorated with pure gold and alloy AuAg NCs.40,81Electrodes modified with AuxCu1−x NCs present composition-dependent atomic arrangements that exhibit very distinctive PEC activity suggesting a direct composition-to-PEC performance relationship. We cannot exclude that small differences in the NC sizes of the different AuxCu1−x NC modified electrodes have an influence on the PEC activity as well, but we expect this influence to be limited since the main contribution of the NCs on the PEC activity is through their plasmonic effect and the plasmonic properties of the NCs vary only slightly with size in the range of a few nanometers.82 Monometallic Au and Cu cluster-modified electrodes exhibit a relatively limited photocurrent enhancement compared to the pristine TNTs. The 40% enhanced PEC activity of Au NCs/TNTs compared to pristine TNTs likely originates from the strong plasmonic properties of the metallic gold clusters, as well as from their low charge carrier recombination, highlighted by the low PL emission intensity. This is supported by the very low dark current of this electrode. The Au/TNT electrode suffers, however, from high charge transfer resistance at the electrode–electrolyte interface as observed by EIS measurements that may limit the PEC activity enhancement. On the other hand, Cu NCs, consisting of a small core of Cu metal surrounded by Cu2O and CuO on the surface, feature a remarkable 80% increase in PEC activity compared to pristine TNTs at 1.23 VRHE. The small dark current of 13 μA cm−2 can originate from the 2 eV band gap of the 33% Cu2O oxide layer, while 89% of activity (108 μA cm−2) is triggered by light. The improvement of the photogenerated charge carrier separation, reflected in the limited attenuation of the PL, as well as its poor stability, may limit the PEC performance of the monometallic Cu/TNT electrode.
All bimetallic AuxCu1−x NCs/TNT electrodes deliver higher photocurrent densities than the monometallic ones. Upon alloying gold with copper, the current density increases by 68%, 91%, and 152% for x = 0.75, 0.5, and 0.25, respectively, compared to that of Au NCs/TNTs. This composition-dependent PEC activity, peaking at Au0.25Cu0.75, can be attributed to a synergy between both metals,82–84 possibly in the form of Au-CuO heterostructures observed by STEM-EDS at the surface of the NPs that consist of two complementary active sites as highlighted previously during CO oxidation where CO adsorbed on Au reacts with a contiguous labile CuO oxygen.77 This synergy is further illustrated by the higher stability of the bimetallic NCs especially Au0.25Cu0.75/TNTs and by the composition-dependent quenching of PL, which is stronger due to the higher copper content in the alloy.
Au-rich Au0.75Cu0.25/TNTs demonstrate the highest ABPE at 0.6 VRHE and show an increase in photocurrent of 68% at 1.23 6 VRHE compared to pure Au NCs. On the other hand, Au0.75Cu0.25/TNTs show a higher recombination of charge carriers compared to its pure Au counterpart as observed by its higher PL intensity but lower charge transfer resistance. It exhibits current values of 65 μA cm−2 in the dark and 98 μA cm−2 under light exposure (Fig. 7c) at 1.23 VRHE. As the structural arrangement of Au0.75Cu0.25 NCs consists of a large pure gold or gold-rich metallic core (around 85%) surrounded by a thin Cu2O/CuO layer at the surface, it features a higher plasmon intensity than the other alloy electrodes. As Au0.75Cu0.25 differs mostly from pure Au by the presence of a thin CuOx layer, this copper oxide may be responsible for its higher activity.
Au0.5Cu0.5/TNTs demonstrate a 25% increase in activity compared to Au0.75Cu0.25 at 1.23 VRHE. On the other hand, it shows lower recombination of charge carriers and charge transfer resistances as observed by its lower PL intensity and EIS, respectively. Au0.5Cu0.5 NCs consist of a pure gold/gold-rich random alloy at the core (56%). They differ from Au0.75Cu0.25 NCs by their smaller metallic core and thicker CuOx (36%) surface layer. This structural arrangement is expected to further reduce the plasmon intensity, while the higher amount of copper oxide at the surface may be at the origin of its higher activity. The discrepancy between XPS and XAFS measurements indicates that NCs with this composition are unstable and their actual in situ structural arrangement may thus be different from the one presented in Fig. 5.
Finally, copper-rich Au0.25Cu0.75/TNTs exhibit the highest PEC activity of all tested electrodes. The main structural difference with Au0.5Cu0.5 lies in the presence of an intermetallic Au0.5Cu0.5 alloyed structure with a small amount (58%) of pure Cu metal in the core. As similar amounts of copper oxide are positioned at the surface of the metallic cores in Au0.25Cu0.75 and Au0.5Cu0.5 NCs, the presence of an intermetallic alloy may be at the origin of the activity increase of Au0.25Cu0.75/TNTs.
All bimetallic AuxCu1−x NCs have core/shell structures in the ambient, resulting from the segregation and oxidation of copper at the surface of the NCs. Similar core/shell structures have been reported for AuCu nanoalloys under oxidative conditions.83,85 This phase segregation leads to the formation of surface CuOx that boosts the CO oxidation catalytic activity,77–80,86 due to a bifunctional mechanism where CO activation takes place on Au atoms, while the O2 species are activated on Cu rich sites.10 A similar mechanism is supported by the present study where we find the significant activity of bimetallic NCs in the absence of light (65 to 72 μA cm−2), as opposed to the negligible activity of their monometallic counterparts. The segregation process depends on the composition and structure of the AuCu alloys as fcc-AuCu and fct-AuCu nanoparticles tend to transform into different Au/CuO and AuCu/CuO core/shell structures, due to distinct Cu diffusion properties, that in turn endow them with distinct catalytic activities.77
The bimetallic AuxCu1−x NCs/TNTs electrodes all present reduced recombination of charge carriers, as indicated by the low PL intensity, implying the generated charge carriers are efficiently separated by the NCs, which act as electron sinks. They also demonstrate good interfacial charge transfer efficiency as evidenced by the EIS results. Besides the photophysical activity of the Cu2O oxide layer at their surface, all bimetallic AuxCu1−x NCs feature a large plasmonic metallic core accounting for 56 to 85% of the cluster atoms that will resonate under visible light illumination. The resulting hot carriers have longer relaxation times and mean free paths and thus become efficient LSPR materials as shown in the literature.87,88 The distinct electronic properties of the fct-AuCu intermetallic alloys may also contribute to the higher activity of Au0.25Cu0.75 as the crystallographic ordering of atoms is indeed expected to significantly affect the band structure of the NPs as this one depends critically on the directional overlap of atomic orbitals.77 Intermetallic compounds show larger hot carrier generation, originating mainly from direct interband and phonon-assisted indirect electronic transitions.87 The resulting hot carriers are energetically higher than those in their monometallic counterparts, which may explain the highest photoactivity of Au0.25Cu0.75, featuring an intermetallic core. The improved stability in time of the intermetallic structure in Au0.25Cu0.75 NCs that, unlike their Au0.5Cu0.5 counterparts, do not segregate after a few days in the ambient as well as under reaction conditions may also explain their higher activity. Hence, the highest activity of Au0.25Cu0.75 results from a combination of factors highlighting the complex and tunable structure–activity relationship in these photocatalysts.
4. Conclusion
In summary, dispersed ligand-free AuxCu1−x (x = 1, 0.75, 0.5, 0.25 and 0) NCs were grown and deposited on TNTs by the CBD method. It was observed that surface modification of TNTs with AuxCu1−x NCs leads to enhanced PEC activity under solar irradiation peaking for the Au0.25Cu0.75 NC composition, which shows a four-fold enhancement in the photocurrent compared to that of pristine TNTs. Reduced recombination of charge carriers as indicated by the lowest PL intensity and the highest interfacial charge transfer efficiency as evidenced by impedance spectroscopy contribute to the superior performance of this electrode. The enhancement is attributed to a synergy between Au and Cu in the NCs that upon segregation produces bifunctional sites consisting of a metallic Au/AuCu alloy and surface copper oxide, whose activity is boosted mostly by the photophysical plasmonic response of the metal under solar light illumination. The distinct fct-AuCu ordered structure of the alloy, present in the most active Au0.25Cu0.75 NCs, may also contribute to the activity as it may improve the NC stability and the photophysical and chemical catalytic performance. Finally, this work highlights the potential of the CBD technique that is used to produce ligand-free NCs of tuneable size and composition with a high degree of purity and high uniformity, and serves as an ideal technology to investigate cluster properties and at the same time to design efficient cluster-based photocatalysts for PEC water oxidation.5. Experimental section
5.1. Preparation of AuCu NC modified samples
A one-step anodization process was used to convert metallic titanium in a porous titanium oxide film with controllable pore size and good uniformity.89 The Ti foil (1 mm thick, 1 × 1 cm2) was first ultrasonically cleaned and rinsed with acetone, ethanol, and distilled water, and then dried under N2 gas flow for a few seconds and finally anodized for 2 h in an electrolyte mixture containing 40 ml of ethylene glycol, 0.5 wt% of ammonium fluoride, and 2 vol% water. Platinum foil (1 × 1 cm2) was used as the cathode and a DC voltage of 30 V was applied (Voltcraft PPS DC power supply). The grown TNTs were cleaned with distilled water and dried under a N2 stream. Subsequently, the samples were subjected to 2 h of heat treatment at 450 °C, aiming to transform the amorphous regions of the nanotube walls into the titania anatase phase.90,91 This step reduces the presence of charge carrier recombination centers. Next, the prepared TNT substrates were modified with AuCu NCs with an equivalent atomic coverage of 4 MLs (Fig. 9).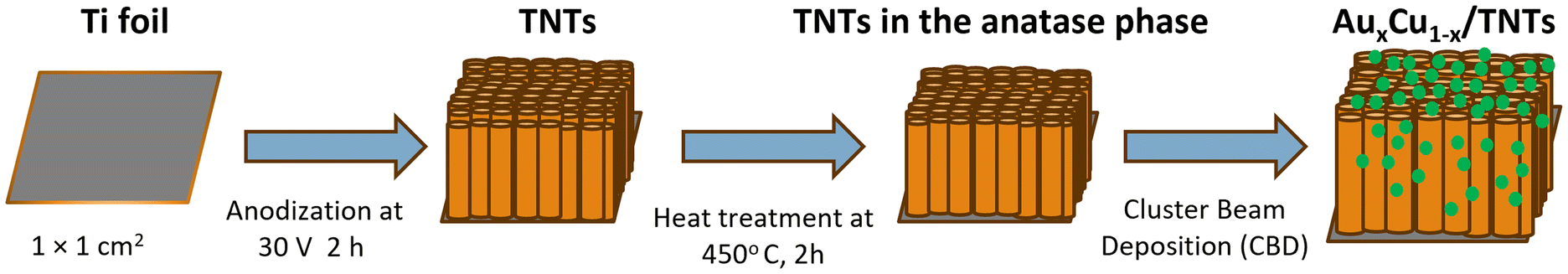 | ||
| Fig. 9 Schematic illustration showing the synthesis of TNTs from titanium foil and the subsequent deposition of AuxCu1−x NCs. | ||
The NCs are produced by pulsed laser (10 Hz, Nd:YAG laser, INDI, Spectra-Physics) ablation of mono and bimetallic AuxCu1−x (x = 1, 0.75, 0.5, 0.25 and 0) plate targets (ACI Alloy, purity 99.995%) and condensation in a helium atmosphere (introduced through a pulsed valve with a backing pressure of 7 bar). Following expansion into a vacuum, a beam of clusters is formed. The cluster size distribution can be tuned by adjusting the laser energy density, the helium pressure, and the time lag between the target ablation and the introduction of the He carrier gas.48,92,93 The AuCu NCs are deposited on the TNT supports by simply placing the supports perpendicular to the molecular beam in a high vacuum deposition chamber (6.8 × 10−8 h Pa). The deposition occurs under soft-landing conditions, given a speed of the cluster beam of about 700 m s−1 (corresponding to a kinetic energy of 0.3 eV per atom).
5.2. Characterization
Different characterization techniques were used to investigate the structural (X-ray diffraction, electron microscopy), optical (UV-Vis and photoluminescence), and photoelectrochemical properties of the TNTs modified with the AuxCu1−x NCs.XRD was performed on all AuxCu1−x NC-modified TNT samples using a Bruker D8 advance diffractometer equipped with a LYNXEYE XE-T detector and Cu Kα source emitting radiation of 1.54 Å. Measurements spanned a 2θ range of 10° to 90°, with a scan rate of 0.5 s per step. The diffractograms were analyzed using Bruker's DIFFRAC.EVA software, with phase identification achieved by comparison with the ICDD database.
The surface morphology of the as-prepared samples was characterized by SEM using an FEI Helios Nanolab G3 CX system (acceleration voltage of 10 kV). The cluster size distributions and compositions were analyzed by Transmission Electron Microscopy (TEM) using an aberration corrected Thermo Fischer Scientific Titan instrument. For the TEM studies, the AuxCu1−x NCs were deposited on molybdenum grids. The TEM instrument was used at an accelerating voltage of 120 kV. Particle size distributions were obtained via ImageJ image analysis software.
Ultraviolet–Visible light absorbance spectra (UV-Vis) of cluster decorated TNT samples were recorded in the 300–700 nm wavelength range using a double beam UV 2600 spectrophotometer (Shimadzu, Kyoto, Japan) equipped with film holders attached to a BaSO4 coated integrating sphere of 60 mm diameter. Steady state photoluminescence (PL) spectra were recorded at room temperature using a Shimadzu RF-6000 fluorescence spectrophotometer equipped with a 150 W xenon lamp and at a resolution of 1 nm. All PL measurements were performed at an excitation wavelength of 310 nm.
XPS measurements were performed at room temperature using a laboratory setup equipped with Mg KαX-rays and an Alpha 110 hemispherical analyzer.
XAFS experiments were performed at the LISA beamline (BM08)94 of the European Synchrotron Radiation Facility (ESRF, Grenoble, France), operating in the 7/8 multi bunch mode with a current of 200 mA. Data were collected at the Au L3-edge (E = 11919 eV) and the Cu K-edge (E = 8979 eV) in grazing incidence (incidence angle of ∼2° with the beam of polarization perpendicular to the surface) and using the fluorescence detection mode. The monochromator was equipped with a pair of Si (111) crystals and run in dynamical focusing mode. The harmonic rejection was carried out by a pair of Si mirrors. The samples were measured in ambient conditions up to a k = 12 Å−1 with typical acquisition times of 40 min. Two spectra were generally averaged to improve the signal to noise ratio. Background subtraction and normalization were carried out by fitting (i) a linear polynomial to the pre-edge region in order to remove instrumental background and (ii) cubic splines simulating the absorption coefficient from an isolated atom to the post-edge region. Phase shifts and backscattering factors were calculated ab initio using Hedin-Lundqvist potentials.
5.3. Photoelectrochemical applications
The photoelectrochemical performance of the pristine and AuCu alloy NC-modified TNTs was studied using a three-electrode configuration in 0.5 M Na2SO4 at pH 7.21. The pristine and NC modified electrodes (1 × 1 cm2), platinum foil and aqueous silver/silver chloride (Ag/AgCl) electrodes were used as working, counter, and reference electrodes, respectively. The performance of each working electrode was evaluated using LSV, chronoamperometry, and EIS. EIS spectra of the prepared photoanodes were measured, in the dark and under illumination, by applying an AC voltage amplitude of 10 mV at open circuit potentials (OCPs) with a frequency ranging from 100 mHz to 100 kHz. All measured potentials were converted to a reversible hydrogen electrode (RHE) scale using the Nernst equation.A Scisun-300 solar simulator from Sciencetech, equipped with a xenon arc lamp and AM 1.5G filter, was used as a light source. The intensity of the light source was adjusted to 100 mW cm−2 by changing the working distance and was measured using an Avantes spectrophotometer. To check and screen the PEC performance, the prepared electrodes were tested in a single PEC cell and the electrodes were positioned as close as possible to the front of the quartz window to minimize the influence of light absorption by water.
The ABPE was calculated from the LSV data as follows:
 | (1) |
where Jph is the photocurrent obtained under the applied potential V and Ptotal is the intensity of the light source. The charge transfer at the interface between the electrode and the electrolyte was studied through Bode phase analysis. The hole-relaxation time or electron lifetime τ and the hole diffusion length Ld are given by:
 | (2) |
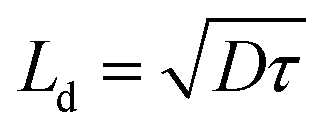 | (3) |
Author contributions
V. C. C.: conceptualization, investigation, formal analysis, visualization, writing original draft, and editing; K. R. A.: investigation, writing, visualization, and formal analysis; T. H. T. N, Z. Z., I. A., and T. H. H.: investigation; P. L.: supervision, editing, and funding acquisition; and D. G., S. V. and E. J.: resources, supervision, writing, editing, validation, and funding acquisition.Data availability
The data of this article are shown in the figures and the ESI.† The raw data behind these figures are available on KU Leuven's institutional research data repository at https://rdr.kuleuven.be/dataverse/rdr with https://doi.org/10.48804/0UXAXQ.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research work was supported by Flanders Innovation & Entrepreneurship (VLAIO) under project No. HBC.2021.0586 (CLUE). V. C. C. was supported by the Jesuit region of the European Low Countries (ELC). T. H. T. Nguyen thanks the Research Foundation Flanders (FWO) for an FWO MSCA SoE postdoctoral fellowship (12ZZI.23N). E. J. acknowledges FWO for a sabbatical bench fee (K8001.24N). The authors thank Thomas Altantzis and Sara Bals (EMAT, University of Antwerp) for providing the HAADF-STEM and EDX data of the Au0.25Cu0.75 clusters. They also thank the ESRF for providing beamtime (HC-5047) and the staff of LISA-BM08 for their assistance.References
- R. K. Keswani, H. Ghodke, D. Sarkar, K. C. Khilar and R. S. Srinivasa, Colloids Surf., A, 2010, 369, 75–81 CrossRef CAS.
- B. Liu, D. Deng, J. Y. Lee and E. S. Aydil, J. Mater. Res., 2010, 25, 1588–1594 CrossRef CAS.
- Z. Zander, R. Yagloski, J. DeCoste, D. Zhang and B. G. DeLacy, Mater. Lett., 2016, 163, 39–42 CrossRef CAS.
- J. M. Macak, H. Hildebrand, U. Marten-Jahns and P. Schmuki, J. Electroanal. Chem., 2008, 621, 254–266 CrossRef CAS.
- G. D. Sulka, J. Kapusta-Kołodziej, A. Brzózka and M. Jaskuła, Electrochim. Acta, 2010, 55, 4359–4367 CrossRef CAS.
- J. M. Macak, H. Tsuchiya, A. Ghicov, K. Yasuda, R. Hahn, S. Bauer and P. Schmuki, Curr. Opin. Solid State Mater. Sci., 2007, 11, 3–18 CrossRef CAS.
- G. D. Sulka, J. Kapusta-Kołodziej, A. Brzózka and M. Jaskuła, Electrochim. Acta, 2013, 104, 526–535 CrossRef CAS.
- J. M. Macak and P. Schmuki, Electrochim. Acta, 2006, 52, 1258 CrossRef CAS.
- S. Bauer, S. Kleber and P. Schmuki, Electrochem. Commun., 2006, 8, 1321–1325 CrossRef CAS.
- V. Zwilling, M. Aucouturier and E. Darque-Ceretti, Electrochim. Acta, 1999, 45, 921–929 CrossRef CAS.
- D. Gong, C. A. Grimes, O. K. Varghese, W. Hu, R. S. Singh, Z. Chen and E. C. Dickey, J. Mater. Res., 2001, 16, 3331–3334 CrossRef CAS.
- H. Brahmi, G. Katwal, M. Khodadadi, S. Chen, M. Paulose, O. K. Varghese and A. Mavrokefalos, Nanoscale, 2015, 7, 19004–19011 RSC.
- X. Chen and S. S. Mao, Chem. Rev., 2007, 107, 2891–2959 CrossRef CAS.
- S. Shen, J. Chen, M. Wang, X. Sheng, X. Chen, X. Feng and S. S. Mao, Prog. Mater. Sci., 2018, 98, 299–385 CrossRef CAS.
- M. Ge, Q. Li, C. Cao, J. Huang, S. Li, S. Zhang, Z. Chen, K. Zhang, S. S. Al-Deyab and Y. Lai, Adv. Sci., 2017, 4, 1600152 CrossRef.
- J. F. De Brito, F. Tavella, C. Genovese, C. Ampelli, M. V. B. Zanoni, G. Centi and S. Perathoner, Appl. Catal., B, 2018, 224, 136–145 CrossRef.
- M. Paulose, K. Shankar, S. Yoriya, H. E. Prakasam, O. K. Varghese, G. K. Mor, T. J. LaTempa, A. Fitzgerald and C. A. Grimes, J. Phys. Chem. B, 2006, 110, 16179–16184 CrossRef CAS PubMed.
- J. M. Macak, M. Zlamal, J. Krysa and P. Schmuki, Small, 2007, 3, 300–304 CrossRef CAS PubMed.
- Y. Jiang, B. Zheng, J. Du, G. Liu, Y. Guo and D. Xiao, Talanta, 2013, 112, 129–135 CrossRef CAS PubMed.
- L. Martínez, L. Soler, I. Angurell and J. Llorca, Appl. Catal., B, 2019, 248, 504–514 CrossRef.
- S. Abela, C. Farrugia, R. Xuereb, F. Lia, E. Zammit, A. Rizzo, P. Refalo and M. Grech, Nanomaterials, 2021, 11, 2823 CrossRef CAS PubMed.
- S. Asadi, B. Mortezagholi, A. Hadizadeh, V. Borisov, M. J. Ansari, H. S. Majdi, A. Nishonova, H. Adelnia, B. Farasati Far and C. Chaiyasut, Pharmaceutics, 2022, 14, 1359 CrossRef CAS PubMed.
- M. Yamaguchi, H. Abe, T. Ma, D. Tadaki, A. Hirano-Iwata, H. Kanetaka, Y. Watanabe and M. Niwano, Langmuir, 2020, 36, 12668–12677 CrossRef CAS PubMed.
- K. Shankar, J. I. Basham, N. K. Allam, O. K. Varghese, G. K. Mor, X. Feng, M. Paulose, J. A. Seabold, K.-S. Choi and C. A. Grimes, J. Phys. Chem. C, 2009, 113, 6327–6359 CrossRef CAS.
- P. Szymanski and M. A. El-Sayed, Theor. Chem. Acc., 2012, 131, 1202 Search PubMed.
- S. A. Batool, M. S. Maqbool, M. A. Javed, A. Niaz and M. A. U. Rehman, Surfaces, 2022, 5, 456–480 Search PubMed.
- M. Grätzel, Nature, 2001, 414, 338–344 CrossRef.
- S. U. M. Khan, M. Al-Shahry and W. B. Ingler, Science, 2002, 297, 2243–2245 CrossRef CAS.
- M. R. Khan, T. W. Chuan, A. Yousuf, M. N. K. Chowdhury and C. K. Cheng, Catal. Sci. Technol., 2015, 5, 2522–2531 RSC.
- S. Linic, P. Christopher and D. B. Ingram, Nat. Mater., 2011, 10, 911–921 CrossRef CAS.
- M. G. Méndez-Medrano, E. Kowalska, A. Lehoux, A. Herissan, B. Ohtani, D. Bahena, V. Briois, C. Colbeau-Justin, J. L. Rodríguez-López and H. Remita, J. Phys. Chem. C, 2016, 120, 5143–5154 CrossRef.
- Z. Hai, N. E. Kolli, D. B. Uribe, P. Beaunier, M. José-Yacaman, J. Vigneron, A. Etcheberry, S. Sorgues, C. Colbeau-Justin, J. Chen and H. Remita, J. Mater. Chem. A, 2013, 1, 10829–10835 RSC.
- P. A. Gross, S. N. Pronkin, T. Cottineau, N. Keller, V. Keller and E. R. Savinova, Catal. Today, 2012, 189, 93–100 CrossRef CAS.
- I. Paramasivam, J. M. Macak and P. Schmuki, Electrochem. Commun., 2008, 10, 71–75 CrossRef CAS.
- S. Zhang, F. Peng, H. Wang, H. Yu, S. Zhang, J. Yang and H. Zhao, Catal. Commun., 2011, 12, 689–693 CrossRef CAS.
- K. Xie, L. Sun, C. Wang, Y. Lai, M. Wang, H. Chen and C. Lin, Electrochim. Acta, 2010, 55, 7211–7218 CrossRef CAS.
- Y. Wang, Z. Li, Y. Tian, W. Zhao, X. Liu and J. Yang, Mater. Lett., 2014, 122, 248–251 CrossRef CAS.
- K. Syrek, J. Grudzień, A. Sennik-Kubiec, A. Brudzisz and G. D. Sulka, J. Nanomater., 2019, 2019, e9208734 Search PubMed.
- R. Ferrando, J. Jellinek and R. L. Johnston, Chem. Rev., 2008, 108, 845–910 CrossRef CAS PubMed.
- V. C. Chinnabathini, F. Dingenen, R. Borah, I. Abbas, J. van der Tol, Z. Zarkua, F. D'Acapito, T. H. T. Nguyen, P. Lievens, D. Grandjean, S. W. Verbruggen and E. Janssens, Nanoscale, 2023, 15, 6696–6708 RSC.
- B. Zhu, L.-Y. Zhang, J.-L. Liu, X.-M. Zhang, X.-S. Li and A.-M. Zhu, J. Hazard. Mater., 2021, 402, 123508 CrossRef CAS.
- Ş. Neaţu, J. A. Maciá-Agulló, P. Concepción and H. Garcia, J. Am. Chem. Soc., 2014, 136, 15969–15976 CrossRef.
- Z.-Y. Li, Y.-H. Chen, J.-R. Zhu, Q. Chen, S.-J. Lu and F.-X. Xiao, Inorg. Chem., 2023, 62, 16965–16973 CrossRef CAS.
- W. Lipińska, K. Grochowska, J. Ryl, J. Karczewski and K. Siuzdak, ACS Appl. Mater. Interfaces, 2021, 13, 52967–52977 CrossRef.
- M. Nischk, P. Mazierski, Z. Wei, K. Siuzdak, N. A. Kouame, E. Kowalska, H. Remita and A. Zaleska-Medynska, Appl. Surf. Sci., 2016, 387, 89–102 CrossRef CAS PubMed.
- Q.-L. Mo, X.-C. Dai and F.-X. Xiao, Small, 2023, 19, 2302372 CrossRef CAS.
- Y. Pang, G. Xu, Q. Feng, J. Lv, Y. Qin, Y. Zhang, Z. Zheng and Y. Wu, Phys. Lett. A, 2018, 382, 2759–2762 CrossRef CAS.
- B. Pauwels, G. Van Tendeloo, E. Zhurkin, M. Hou, G. Verschoren, L. Theil Kuhn, W. Bouwen and P. Lievens, Phys. Rev. B: Condens. Matter Mater. Phys., 2001, 63, 165406 Search PubMed.
- P. Milani and S. Iannotta, Cluster Beam Synthesis of Nanostructured Materials, Springer Science & Business Media, 2012 Search PubMed.
- R. Liu, Q. Li and L. M. Smith, J. Am. Soc. Mass Spectrom., 2014, 25, 1374–1383 CAS.
- D. Hong, A. Sharma, D. Jiang, E. Stellino, T. Ishiyama, P. Postorino, E. Placidi, Y. Kon and K. Koga, ACS Omega, 2022, 7, 31260–31270 CrossRef CAS.
- T. K. Sham, Y. M. Yiu, M. Kuhn and K. H. Tan, Phys. Rev. B: Condens. Matter Mater. Phys., 1990, 41, 11881–11886 CrossRef CAS PubMed.
- R. E. Watson, J. Hudis and M. L. Perlman, Phys. Rev. B: Solid State, 1971, 4, 4139–4144 CrossRef.
- W. Eberhardt, S. C. Wu, R. Garrett, D. Sondericker and F. Jona, Phys. Rev. B: Condens. Matter Mater. Phys., 1985, 31, 8285–8287 CrossRef CAS PubMed.
- T. K. Sham, M. L. Perlman and R. E. Watson, Phys. Rev. B: Condens. Matter Mater. Phys., 1979, 19, 539–545 CrossRef CAS.
- M. Kuhn and T. K. Sham, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 49, 1647–1661 CrossRef CAS.
- P. Weinberger, A. M. Boring, R. C. Albers and W. M. Temmerman, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 38, 5357–5362 CrossRef CAS PubMed.
- J. E. Müller, O. Jepsen and J. W. Wilkins, Solid State Commun., 1982, 42, 365–368 CrossRef.
- T.-W. Liao, A. Yadav, K.-J. Hu, J. van der Tol, S. Cosentino, F. D'Acapito, R. E. Palmer, C. Lenardi, R. Ferrando, D. Grandjean and P. Lievens, Nanoscale, 2018, 10, 6684–6694 RSC.
- I. J. Godfrey, A. J. Dent, I. P. Parkin, S. Maenosono and G. Sankar, J. Phys. Chem. C, 2017, 121, 1957–1963 CrossRef CAS.
- J. T. Miller, A. J. Kropf, Y. Zha, J. R. Regalbuto, L. Delannoy, C. Louis, E. Bus and J. A. van Bokhoven, J. Catal., 2006, 240, 222–234 CrossRef CAS.
- R. E. Benfield, D. Grandjean, M. Kröll, R. Pugin, T. Sawitowski and G. Schmid, J. Phys. Chem. B, 2001, 105, 1961–1970 CrossRef CAS.
- A. Yadav, Y. Li, T.-W. Liao, K.-J. Hu, J. E. Scheerder, O. V. Safonova, T. Höltzl, E. Janssens, D. Grandjean and P. Lievens, Small, 2021, 17, 2170139 CrossRef CAS.
- S. Zhao, Z. Cheng, S. Wang, H. Hao and Y. Fang, Appl. Phys. A, 2021, 127, 930 CrossRef CAS.
- D. Albinsson, S. Nilsson, T. J. Antosiewicz, V. P. Zhdanov and C. Langhammer, J. Phys. Chem. C, 2019, 123, 6284–6293 CrossRef CAS.
- Z. Guo, O. V. Prezhdo, T. Hou, X. Chen, S.-T. Lee and Y. Li, J. Phys. Chem. Lett., 2014, 5, 1642–1647 Search PubMed.
- S. Kurian, H. Seo and H. Jeon, J. Phys. Chem. C, 2013, 117, 16811–16819 CrossRef CAS.
- F.-X. Xiao, S.-F. Hung, J. Miao, H.-Y. Wang, H. Yang and B. Liu, Small, 2015, 11, 554–567 Search PubMed.
- M. Szkoda, K. Siuzdak, A. Lisowska-Oleksiak, J. Karczewski and J. Ryl, Electrochem. Commun., 2015, 60, 212–215 CrossRef CAS.
- Z. Zhang and P. Wang, Energy Environ. Sci., 2012, 5, 6506–6512 RSC.
- Z. Zhang, L. Zhang, M. N. Hedhili, H. Zhang and P. Wang, Nano Lett., 2013, 13, 14–20 CrossRef CAS.
- T. H. Gfroerer, in Encyclopedia of Analytical Chemistry, John Wiley & Sons, Ltd, 2006 Search PubMed.
- B.-H. Lee, S. Park, M. Kim, A. K. Sinha, S. C. Lee, E. Jung, W. J. Chang, K.-S. Lee, J. H. Kim, S.-P. Cho, H. Kim, K. T. Nam and T. Hyeon, Nat. Mater., 2019, 18, 620–626 CrossRef CAS.
- T. Kiyonaga, M. Fujii, T. Akita, H. Kobayashi and H. Tada, Phys. Chem. Chem. Phys., 2008, 10, 6553–6561 RSC.
- A. L. Linsebigler, G. Lu and J. T. Yates Jr., Chem. Rev., 1995, 95, 735–758 CrossRef CAS.
- X. Liu, S. Cui, Z. Sun, Y. Ren, X. Zhang and P. Du, J. Phys. Chem. C, 2016, 120, 831–840 CrossRef CAS.
- W. Zhan, J. Wang, H. Wang, J. Zhang, X. Liu, P. Zhang, M. Chi, Y. Guo, Y. Guo, G. Lu, S. Sun, S. Dai and H. Zhu, J. Am. Chem. Soc., 2017, 139, 8846–8854 CrossRef CAS PubMed.
- T. E. R. Fiuza and D. Zanchet, ACS Appl. Nano Mater., 2020, 3, 923–934 CrossRef CAS.
- Z. Xu, E. Lai, Y. Shao-Horn and K. Hamad-Schifferli, Chem. Commun., 2012, 48, 5626–5628 RSC.
- Y. Yu, X. Dong, P. Chen, Q. Geng, H. Wang, J. Li, Y. Zhou and F. Dong, ACS Nano, 2021, 15, 14453–14464 CrossRef CAS PubMed.
- T. W. Liao, S. W. Verbruggen, N. Claes, A. Yadav, D. Grandjean, S. Bals and P. Lievens, Nanomaterials, 2018, 8, 30 CrossRef PubMed.
- S. W. Verbruggen, M. Keulemans, J. A. Martens and S. Lenaerts, J. Phys. Chem. C, 2013, 117, 19142–19145 CrossRef CAS.
- A. Shah, M. Akhtar, S. Aftab, A. H. Shah and H.-B. Kraatz, Electrochim. Acta, 2017, 241, 281–290 CrossRef CAS.
- U. Latif-ur-Rahman, A. Shah, R. Qureshi, S. B. Khan, A. M. Asiri, A.-H. A. Shah, M. Ishaq, M. S. Khan, S. K. Lunsford and M. A. Zia, Adv. Mater. Sci. Eng., 2015, 2015, e638629 Search PubMed.
- T.-S. Kim, H. Choi, D. Kim, H. C. Song, Y. Oh, B. Jeong, J. Lee, K.-J. Kim, J. W. Shin, H. R. Byon, R. Ryoo, H. Y. Kim and J. Y. Park, Appl. Catal., B, 2023, 331, 122704 CrossRef CAS.
- X. Liu, A. Wang, X. Wang, C.-Y. Mou and T. Zhang, Chem. Commun., 2008, 3187 RSC.
- C. Jian, J. Zhang, W. He and X. Ma, Nano Energy, 2021, 82, 105763 CrossRef CAS.
- C. Jian, J. Zhang and X. Ma, RSC Adv., 2020, 10, 13277–13285 RSC.
- D. Gong, C. A. Grimes, O. K. Varghese, W. Hu, R. S. Singh, Z. Chen and E. C. Dickey, J. Mater. Res., 2001, 16, 3331–3334 CrossRef CAS.
- Z. Hua, Z. Dai, X. Bai, Z. Ye, P. Wang, H. Gu and X. Huang, Chem. Eng. J., 2016, 283, 514–523 CrossRef CAS.
- K. Grochowska, N. Nedyalkov, J. Karczewski, Ł. Haryński, G. Śliwiński and K. Siuzdak, Sci. Rep., 2020, 10, 20506 CrossRef CAS PubMed.
- P. Ferrari, J. Vanbuel, Y. Li, T.-W. Liao, E. Janssens and P. Lievens, in Gas-Phase Synthesis of Nanoparticles, John Wiley & Sons, Ltd, 2017, pp. 59–78 Search PubMed.
- T. W. Liao, A. Yadav, P. Ferrari, Y. Niu, X. K. Wei, J. Vernieres, K. J. Hu, M. Heggen, R. E. Dunin-Borkowski, R. E. Palmer, K. Laasonen, D. Grandjean, E. Janssens and P. Lievens, Chem. Mater., 2019, 31, 10040–10048 CrossRef CAS.
- F. D'Acapito, G. Lepore, A. Puri, A. Laloni, F. Manna, E. Dettona, A. Luisa and A. Martin, J. Synchrotron Radiat., 2019, 26, 551–558 CrossRef.
- G. Xing, N. Mathews, S. Sun, S. S. Lim, Y. M. Lam, M. Grätzel, S. Mhaisalkar and T. C. Sum, Science, 2013, 342, 344–347 CrossRef CAS PubMed.
- S. D. Stranks, G. E. Eperon, G. Grancini, C. Menelaou, M. J. P. Alcocer, T. Leijtens, L. M. Herz, A. Petrozza and H. J. Snaith, Science, 2013, 342, 341–344 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Additional sample characterisation data and assessment of the potential current generated by the oxidation of copper. See DOI: https://doi.org/10.1039/d4nr03219e |
| This journal is © The Royal Society of Chemistry 2025 |