
Synthesis of periodic polyolefins based on anionic alternating copolymerization†
Hong
Yan
,
Hongyuan
Bai
,
Xuefei
Wang
,
Haitao
Leng
,
Siwei
Chen
,
Li
Han
 and
Hongwei
Ma
*
and
Hongwei
Ma
*
Department of Polymer Science and Engineering, Dalian University of Technology, China. E-mail: mahw@dlut.edu.cn
First published on 2nd December 2024
Abstract
To avoid sequence bias in the chain growth process of multicomponent periodic polymers, we reported an indirect method for the synthesis of strictly periodic polymers using nonpolar monomers. Using 1,1-diphenylethylene (DPE) as the comonomer, the precise sequence was achieved through living anionic polymerization (LAP) and hydrogenation. Four dienes (isoprene (Ip), 2,3-dimethylbuta-1,3-diene (DMBD), 2,3-diphenylbutadiene-1,3-diene (DPB), and 1-phenyl-1,3-butadiene (1-PB)) and an olefin with cyclic tension 1-cyclobutylvinylbenzene (CBVB) were copolymerized with DPE to prepare the alternating precursors. The periodic sequences of DPE–styrene–propylene (pd-DSP), DPE–propylene–propylene (pd-DPP), DPE–ethylene–styrene (pd-DES), DPE–styrene–styrene (pd-DSS) and DPE–ethylene–styrene–styrene (pd-DESS) were successfully synthesized with hydrogenation of these alternating precursors, and the nonpolar olefin units were successfully introduced into the periodic polymer. The results showed that the modification of the backbone carbon framework structure caused a consistent change in the thermal properties of the polymers. Moreover, the steric hindrance and arrangement density of the side chain substituents could significantly affect the performance of the periodic copolymers. This study provides a feasible method for the synthesis of non-polar periodic copolymers with precise periodic arrangements.
Introduction
The sequence structure of polymers forms the basis for the expression of their properties.1–3 Among various sequences, periodic polymers are composed of multiple monomers arranged in a regular pattern, as well as the side chain groups.4–6 Therefore, compared with other sequences such as random, block, or gradient distributions, the ordered structure of periodic polymers can better guide the folding and assembly of the polymer chains. They exhibit wide applicability in emerging fields such as drug delivery, battery storage and optical components.7–10 Various types of periodic polymers often have defective structures, because it is difficult for multiple types of monomers to undergo strict crossover reactions according to the preset requirements.11 Therefore, only in the polymerization of two monomers, alternating sequences can be obtained by suppressing the homo-propagation reactions.12–14 Constructing alternating sequences is an effective method to obtain precise periodic sequences.Step-growth polymerization employs efficient orthogonal reactions between the functional groups to achieve precise and strict sequences, which can prevent the formation of defective structures; click reactions and multicomponent reactions are widely used in the construction of alternating structures. For example, the Ugi, CuAAC and Passerini reactions have been used to synthesize complex periodic sequences such as [ABCB], [ABCBA] and [ABCDCB].15–18 However, step-growth polymerization can only yield precise sequences of oligomers. Furthermore, in the preparation of high molecular weight periodic copolymers, combining step-growth polymerization and chain-growth polymerization is more promising and challenging. For example, current research has achieved the synthesis of periodic copolymers possessing [ABC], [ABBA], and [ABBCD] repeating structures by the combination of reversible-deactivation radical polymerizations (RDRPs) and coupling reactions.19–22 In addition, radical coupling reactions (RACPs) have also been reported to prepare periodic copolymers with [ABC], [ABAC], and [ABCC] repeating structures.23,24 The above methods are capable of obtaining relatively precise periodic structures that require a combination of multiple mechanisms and rely on the special functional groups of monomers. For non-polar polymers, none of the above methods are applicable. Living ring-opening metathesis polymerization (ROMP) is an extremely efficient synthesis strategy to produce periodic non-polar polymers. Based on the information from periodic sequences, it is necessary to design and construct the required macrocyclic olefins and then obtain periodic sequences through ring opening metathesis polymerization of monomers, such as the periodic sequences of styrene, ethylene, or propylene.25–27 Based on this theoretical foundation, our research group developed a new method for carbon skeleton regulation in previous studies. The styrene derivatives with cyclic substituents underwent anion-migrated ring-opening polymerization, followed by hydrogenation, and we successfully synthesized polymers with pd-SEM and pd-SEE periodic sequence structures.28 This method was able to achieve the goal of obtaining strictly precise periodic sequences through chain polymerization.
Inspired by this work, apart from homopolymers, only alternating polymers can also achieve strict sequence control by chain polymerization. Assuming that multiple nonpolar olefins are directly copolymerized through chain growth to obtain periodic structures, the insertion of monomer units is determined by the reactivity ratio, which is controlled by temperature and solvent;29–32 thus, perfect periodic insertion is impossible. Furthermore, owing to the influence of dynamics, the precision of the sequence sharply decreases during the polymerization process until the sequence structure becomes statistically random. However, in various types of copolymer sequence control, only alternating sequences can be strictly achieved; perfect alternation will occur when both r1 and r2 are equal to zero, or when r1r2 = 0, alternating sequences can also be obtained. Therefore, the problem of statistical randomness can be avoided by synthesizing a strict alternating structure and functionalizing this structure to obtain a periodic structure.33
This process provides an indirect method to obtain strictly periodic structures through chain growth aggregation. Among the various methods for synthesizing alternating copolymers, living anionic polymerization can precisely control the molecular weight of polymers, with no termination or chain transfer during the active chain growth process.34–36 It is difficult to homopolymerize DPE and its derivatives because of steric hindrance. However, owing to the P–π conjugated structure formed between the two benzene rings and the vinyl group in the monomer, DPE can easily copolymerize with other nonpolar olefins. During the process of LAP, the formation of DPE terminal active centers can only lead to alternating cross-growth with comonomers.37,38 Therefore, DPE and various nonpolar olefins can be utilized to synthesize polymers with strictly alternating structures. In this study, four different dienes and a special olefin capable of undergoing a ring-opening reaction were selected for copolymerization with DPE, and five new alternating copolymers were successfully obtained by adjusting the reaction conditions. All copolymers had carbon–carbon double bonds incorporated into their polymer backbones. After simple hydrogenation treatment, five multiblock structured polymers, namely, pd-DSP, pd-DPP, pd-DES, pd-DSS and pd-DESS, were obtained. By comparing the performance of alternating sequence copolymers and periodic sequence copolymers with the 10 different sequence structures mentioned above, the intricate relationship between the polymer structure and performance could be further elucidated. Furthermore, the results from this study indicated that LAP could not only yield fully alternating sequence polymers but also indirectly produce polymers with strictly periodic structures. This method provides a new approach for synthesizing multiperiodic structured polymers with precise sequences. Consequently, developing strategies for synthesizing precisely periodic polymers is highly challenging and valuable; this exploration has the potential to enrich the types of periodic sequences and obtain polyolefin materials with better mechanical properties.
Results and discussion
Polyethylene (PE) and polypropylene (PP) are the most widely used general-purpose plastics in the world, and ethylene-co-propylene also has many potential applications. By utilizing a Ziegler–Natta catalyst to catalyze the trans-1,4-polymerization of Ip and hydrogenation, a precise sequence of ethylene-alt-propylene (alt-EP) can be synthesized.39 This type of alternating polymer was obtained through the complex coordination polymerization of a single monomer and severely compromised the ability to regulate the carbon skeleton of the polymers. Based on the above alt-EP, we introduced other ethylene monomers to copolymerize with Ip to form a fully alternating sequence copolymer; then, a carbon-rich multiperiodic polymer can be obtained in the same manner after hydrogenation. DPE has inherent advantages in the synthesis of alternating copolymers by LAP (rD = 0); thus, periodic polymers of DPE–ethylene–propylene (pd-DEP) can be obtained by controlling the alternation sequence of DPE and Ip. For nonpolar polyolefins, the performance of the copolymer relies more on the arrangement of short branches (methyl) or side groups (phenyl rings) along the main polymer chain when the carbon skeleton structures are similar. Therefore, three types of periodic sequences (DPE–propylene–propylene (DPP), DPE–ethylene–ethylene–St (DEES) and DPE–St–St (DSS)) can be obtained by introducing substituted dienes (DMBD, 1-PB, and DPB). The above four types of periodic polymers have a C6 carbon skeleton. Apart from the arrangement of monomer units, the carbon skeleton length affects the properties of polymers. ROMP is an effective method for preparing long carbon skeletons. Based on this theory, our research group has proposed the AMROP method, which produces a periodic polymer with a C8 backbone structure like DPE–St–ethylene–ethylene (DSEE) (Scheme 1).40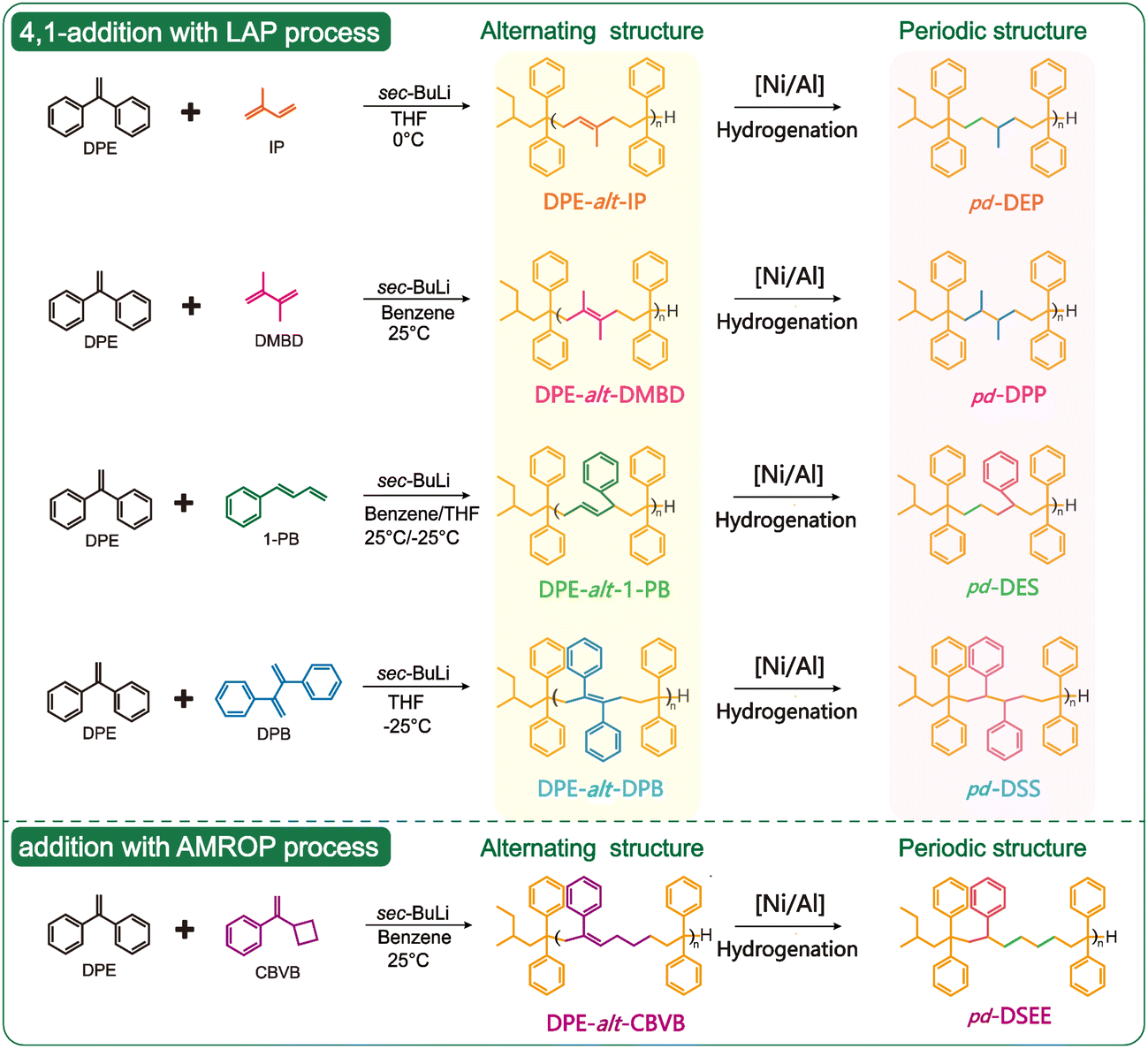 | ||
| Scheme 1 Alternating copolymerization of DPE with dienes. | ||
The strategy of this study is to obtain strict periodic sequences by using strict alternating polymers; thus, the first step is to regulate DPE and various dienes to form strict alternating copolymers. For nonpolar monomers, r1r2 = 0 is a rigid requirement for the synthesis of alternating copolymers; in LAP, the main way of controlling sequences is to alter the reactivity ratios of the monomers. The four main pathways are as follows: (1) by adjusting the feeding strategy, the copolymer sequence structure can be changed. Yuki et al. investigated the reaction characteristics of DPE and St in nonpolar solvents during anionic polymerization, and reactivity ratios could be calculated based on the monomer feed ratio and the proportion of monomer units in the polymer. (2) The addition of a modifier can effectively promote dynamic propagation and regulate the copolymerization behavior of the monomers. For example, alkoxide metal modifiers can form a special four-membered ring transition state structure with an active center, and the two coexisting counterions can isomerize with each other, thereby altering the rate of crossover propagation. (3) The solvation ability and polarity of the solvent directly influence the morphology of the active center, can alter the closeness of the ion pairs and thus affect the reactivity ratios of the monomers. For example, when DPE and Ip are copolymerized in benzene, r2 can reach a maximum of 37, but when they are copolymerized in THF, r2 decreases to 0.11. (4) Temperature affects mainly the reactivity ratios of the monomers by affecting the rate constant of crossover propagation. Generally, anionic polymerization occurs at temperatures ranging from −20 °C to 100 °C.41–46 Based on the various regulation methods mentioned above, DPE can be copolymerized with Ip, DMBD, 1-PB, and DPB to obtain strictly alternating polymers. The specific copolymerization process is described in ESI Table S1.†
The methyl groups of DMBD serve as electron-donating substituents, not only increasing its steric hindrance but also reducing its reactivity in nucleophilic reactions. As described in Yuki's report, even if DMBD (M2) and DPE (M1) were copolymerized in benzene, r2 was only 0.23. For low-reactivity olefins, the polymer structure could be controlled to form alternating sequences by employing a feed strategy. Through the corresponding analysis of proton nuclear magnetic resonance (1H NMR) and size exclusion chromatography (SEC) data (see ESI Fig. S7† and Table 1), the M1/M2 value of DPE and DMBD increased from 1 to 4; this resulted in an increase in the conversion rate of DPE from 69.98% to 88.68% (Covn.M1(%) = Covn.M2(%)/(N2/N1)) and the NDMBD/ND value could reach 1.09 (as calculated via ESI eqn (S7)†). Based on the above results, its structure can be preliminarily identified as an alternating sequence. The obtained polymer had a narrow distribution and a molecular weight close to the predicted value. The conversion rate of DMBD reached 96.66%, indicating that no side reactions occurred during the copolymerization (see Table 1). Matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF-MS) is a direct method for determining the alternating sequence. As shown in Fig. 1(b), using DPE-alt-DMBD as an example, the MALDI-TOF-MS result showed a unique series of proton peaks. The m/z peak at 2967.39 (see ESI Fig. S17†) could be attributed to 10 units of DPE (10 × 180.09 u) + 11 units of DMBD (11 × 68.12 u) + a hydrogen terminal group (1.01 u) + a counterion Ag (106.91 u) + a sec-butyl-terminal group (57.07 u). The difference in the numerical values between each peak consisted of one DPE unit and one DMBD unit.
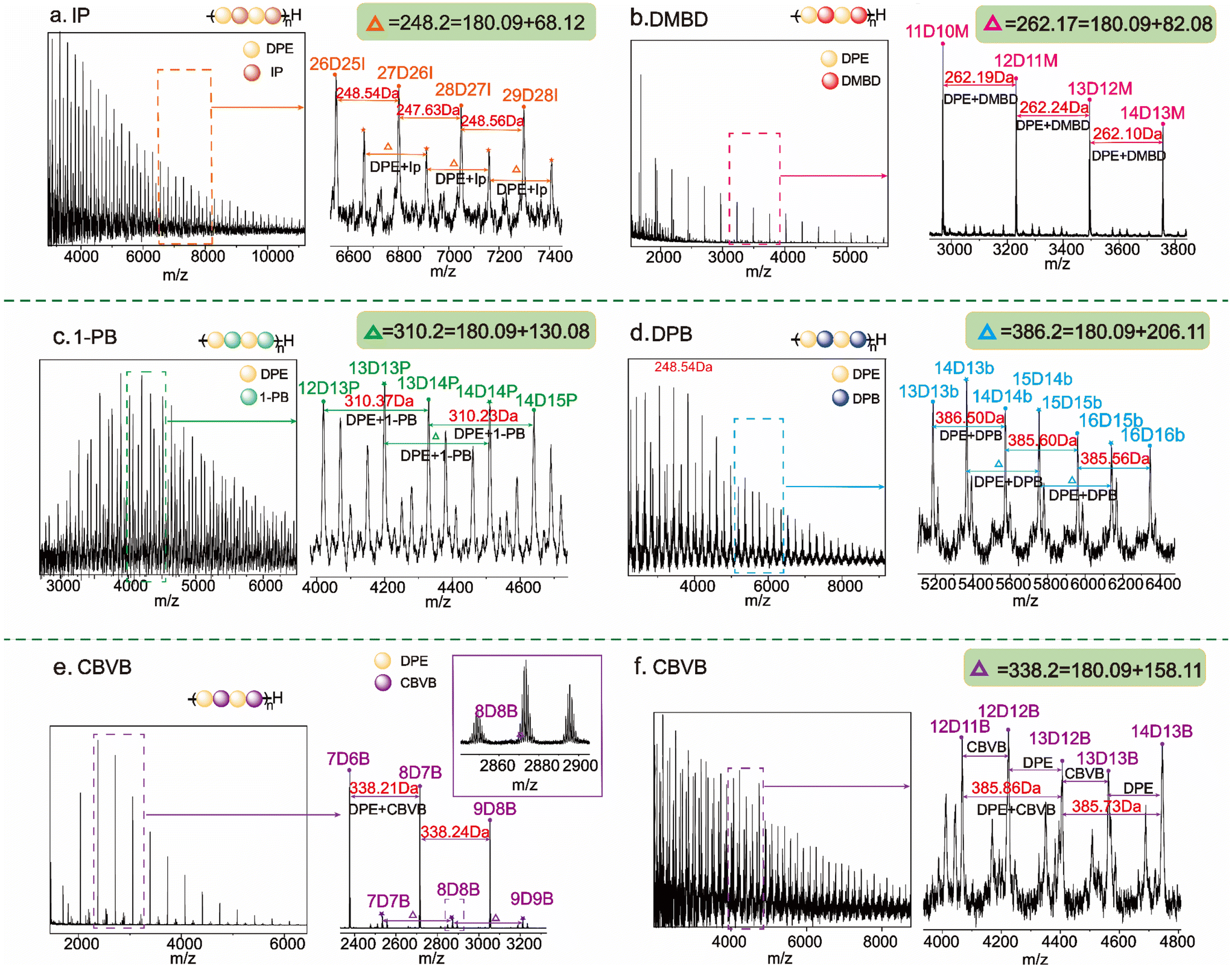 | ||
| Fig. 1 MALDI-TOF-MS of run 1 (a), run 3 (b), run 5 (c), run 6 (d), run 7 (e) and run 8 (f). | ||
| Run | M1/M2 | Eq. | Solvent | Temp./°C | M n [kg mol−1] | PDIa |
N
2/N1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) b b |
N 1 | N 2 | Covn.M2c (%) | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1H NMRb | SECa | ||||||||||
| a M n (SEC) and PDI values for the copolymers were determined using SEC with polystyrene (PS) as a standard. b Ratio of N2/N1 units in the chain calculated by 1H NMR (N1 represents the number of DPE in the chains and N2 represents the number of dienes in the chains). Mn (1H NMR) values for the copolymers calculated through 1H NMR. The calculation basis is shown in ESI Fig. S6–S10† and the calculation formulas are given in ESI eqn (S6)–(S10).† c Covn.M2 represents the conversion rate of dienes (%) and CBVB (%), which was obtained by calculating the ratio of the polymer to the total initial monomer mass. | |||||||||||
| 1 | DPE/IP | 1.5 | THF | 0 | 17.4 | 16.6 | 1.32 | 1.01 | 70 | 71 | 100.0 |
| 2(a) | DPE/DMBD | 1.0 | Benzene | 25 | 12.2 | 13.5 | 1.10 | 1.31 | 43 | 55 | 91.6 |
| 3 | DPE/DMBD | 4.0 | Benzene | 25 | 14.3 | 18.3 | 1.07 | 1.09 | 53 | 58 | 96.7 |
| 4 | DPE/1-PB | 1.5 | Benzene | 25 | 5.6 | 7.1 | 1.21 | 1.79 | 14 | 24 | 41.7 |
| 5 | DPE/1-PB | 1.5 | THF | −25 | 22.5 | 23.7 | 1.87 | 1.06 | 71 | 75 | 100.0 |
| 6 | DPE/DPB | 1.5 | THF | −25 | 17.6 | 15.1 | 1.17 | 1.12 | 43 | 48 | 96.0 |
| 7 | DPE/CBVB | 1.0 | Benzene | 25 | 7.3 | 6.3 | 1.31 | 0.87 | 23 | 20 | 40.0 |
| 8 | DPE/CBVB | 1.0 | Benzene | 25 | 14.0 | 13.5 | 1.25 | 1.03 | 41 | 42 | 84.0 |
However, for highly active dienes, simply increasing the feeding of DPE not only fails to obtain alternating sequences, but also causes waste of resources. The reactivity ratios of DPE and Ip(M2) are strongly influenced by the solvent, with r2 values decreasing from 37 to 0.11 as the polarity of the solvent increases. Therefore, DPE and IP should be copolymerized in THF (run 1), and their corresponding 1H NMR spectra and MALDI-TOF-MS result are presented in ESI Fig. S6 and S16† (calculated using ESI eqn (S6)†). The polymerization conditions of DPE-alt-1-PB and DPE-alt-DPB can be understood by analyzing and comparing the structures of Ip and DMBD, as well as their polymerization conditions. The reaction activity of conjugated dienes is determined by both the conjugation effect and steric hindrance. The 1-position carbon of 1-PB contains a phenyl ring substituent, and the presence of this electron-withdrawing group reduces the electron cloud density on the double bond; this is advantageous for the attack of the carbon anions, and therefore, the reactivity of 1-PB is greater than that of Ip. DPB contains two phenyl ring substituents; these substituents reduce the electron cloud density and increase the steric hindrance. Therefore, its reactivity is greater than that of DMBD but lower than that of Ip. Both DPE-alt-1-PB and DPE-alt-DPB can be obtained in THF (runs 5 and 6). The corresponding analyses of the 1H NMR spectra and the MALDI-TOF-MS result are presented in ESI Fig. S8, S9 and S18, S19,† calculated using ESI eqn (S8) and (S9).†
The sequence regulation in the copolymers obtained by LAP is controlled by their kinetic behavior. In the preliminary stage, our research group has investigated the copolymerization reaction characteristics of DPE and CPVB. At room temperature, the self-growth kinetic constants of both DPE and CPVB are zero (KCP = 0 and KDD = 0 at 25 °C). Therefore, a new polymerization mechanism, AMROP, is proposed to explain the ideal alternating copolymerization process of CPVB and DPE derivatives. The proposed AMROP provides more possibilities for controlling the carbon framework of vinyl monomer polymers; thus, we have shifted our focus to CBVB, which is a styrene derivative with different ring substituents from CPVB. Previously, studies have reported that CBVB can homopolymerize through AMROP; its cyclobutyl can be opened and formed into a C5 skeleton polymer by increasing the temperature; the pd-SEE periodic polymer can be obtained after hydrogenation. In the same way, the self-growth kinetic constants of both DPE and CBVB are zero (KCB = 0 and KDD = 0 at 25 °C). Therefore, DPE and CBVB can likely undergo cross-growth at room temperature by AMROP and yield a quaternary periodic polymer structure of pd-DSEE. Theoretically, CBVB-alt-DPE can be obtained with a 1:1 feed ratio at room temperature. The intramolecular bond angle of the cyclopropyl three-membered ring in CPVB is 60°, and the hydrogen atoms on the ring are in overlapping positions; these hydrogen atoms can easily create tension and therefore facilitate ring-opening reactions. However, the intramolecular angle of the cyclobutyl group in CBVB is 90°, resulting in lower ring tension than that of the ternary ring. Therefore, the polymerization reaction rate of DPE and CBVB is slightly lower. With the same design, the conversion rate of CBVB increased from 46% to 84% by extending the polymerization time from 5 days (run 7) to 10 days (run 8), and the molecular weight of the copolymers also doubled. Based on 1H NMR spectrum characterization (ESI Fig. S10†), regardless of whether the conversion rate of CBVB was high or low, the ND/NB value was close to 1; these results confirmed that DPE-alt-CBVB was successfully synthesized. Here, we tested the MALDI-TOF-MS data of low-molecular-weight (run 7) and high-molecular-weight (run 8) polymers, and several series of proton peaks appeared in the spectra. As shown in Fig. 1(e), the peak with an m/z of 2374.28 corresponded to 7 units of DPE (7 × 180.09 u) repeat units +6 units of CBVB (6 × 158.11 u) repeat units + a hydrogen terminal group (1.01 u) + a counterion Ag (106.91 u) + a sec-butyl-terminal group (57.07 u) (ESI Fig. S20†). Similarly, based on the analysis of the data in ESI Fig. S21,† the polymerization time was prolonged, and DPE and CBVB still grew alternately.
In conjugated dienes, the mobility of the double bond electron cloud is high, and it is easy to induce polarization. When the density of the π electron cloud on the carbon–carbon double bond decreases, the slightly positively charged β-carbon atom is conducive to the attack of the negatively charged anionic active center, causing π-electron heterolysis and initiating anionic polymerization. During the addition reaction of the conjugated dienes with nucleophilic reagents, both 3,4-addition and 1,4-addition can occur. In the case of 1,4-addition, unsaturated double bonds are formed in the polymer skeleton, thereby forming a cis/trans structure. When DPE is copolymerized with Ip, DMBD, 1-PB and DPB, the diolefins need to be copolymerized with DPE by 1,4-addition to obtain long carbon chain structures. In the polymerization of DPE and Ip, DPE is preferentially triggered by sec-BuLi to form DPE-Li; DPE-Li initially attacks the carbon at the 4-position with less steric hindrance, forming an active center at the carbon at the 1-position with the least steric hindrance. Therefore, the next DPE is added to the carbon at the 1-position during the alternating copolymerization. As a result, mainly 1,4-structures are formed, with few 3,4-structures produced and almost no formation of a 1,2-structure. Therefore, the 1,2-structures and 3,4-structures were disregarded in the subsequent description of the polymer structure. In the carbon polymer backbone structure of the DPE-alt-1-PB polymer, 3% 1,2-structures can be clearly observed via1H NMR (ESI Fig. S8,†δ = 5.7–6.2 (ppm)). However, due to its low content, subsequent discussions are focused only on the 1-PB appearing in the polymer with 1,4-structures. Therefore, the polymer backbones can still ensure the rigor of their periodic structure after hydrogenation. Both DMBD and DPB have substituents at the 2-position carbon and 3-position carbon. DMBD contains two methyl groups, which are electron-donating substituents despite their relatively small steric hindrance. On the other hand, DPB has two phenyl rings as substituents, which are electron-withdrawing groups with significant steric hindrance. Together, these factors caused DMBD and DPB to undergo 1,4-addition to the polymer chain. The 1H NMR spectra (ESI Fig. S7 and S9†) also revealed that the unsaturated double bond regions of DPE-alt-DMBD and DPE-alt-DPB had no peaks (δ = 4.0–6.0 (ppm)), showing the absence of a 1,2-structure in the polymer chain. The above results indicated that all five alternating polymers were successfully synthesized according to the proposed structure. A comparison of DPE-alt-Ip and DPE-alt-DMBD revealed that the carbon skeletons of the polymer backbones had the exact same arrangement, but the side chain of DPE-alt-DMBD contained an additional methyl group. When the two polymers had similar molecular weights, the Tg of DPE-alt-DMBD was 16 °C higher. Similarly, DPE-alt-DMBD and DPE-alt-DPB only differed in the size of the side group substitution, and Tg increased by 36 °C. A comparison of the data revealed that the Tg values of DPE-alt-1-PB and DPE-alt-Ip exhibited unexpected trends. Typically, polymers with a benzene ring as the side group had a greater Tg than those with only a single methyl group. However, in this case, the absence of any substituents on the double bonds in the chain of DPE-alt-1-PB enabled increased bond rotation, resulting in a Tg for DPE-alt-Ip that was 36 °C higher than expected. Therefore, the thermal properties of nonpolar olefin copolymers could be controlled by altering the number of substituents, steric hindrance, and position. The polymer backbones of the five alternating structure polymers all contained double bonds. These double bonds could be used for simple hydrogenation modification to obtain the most basic periodic polymers and for more complex substitution and addition reactions to produce polymers with different side groups, thus endowing the polymer with a periodic arrangement of the functional groups (Scheme 2).
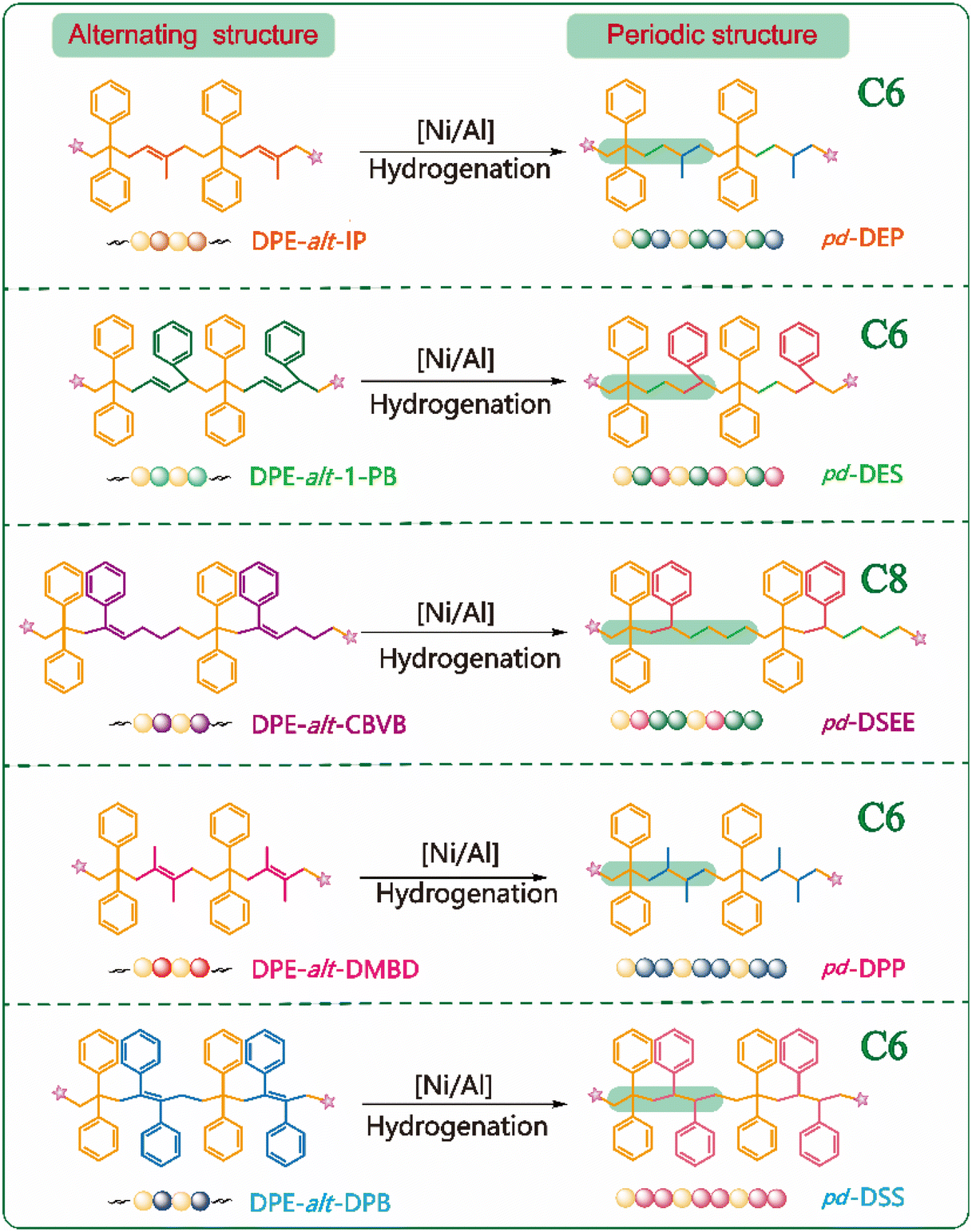 | ||
| Scheme 2 Periodic polymers prepared by the hydrogenation of alternating polymers. | ||
For common nonpolar olefins, controlling the copolymer sequence is often difficult (r1r2 > 0). In our previous work, AMROP was used to develop a new method for regulating the carbon skeleton. After hydrogenation, we successfully obtained strictly periodic polymers of pd-SE, pd-SEM and pd-SEE. Based on the research results, we introduced the special vinyl monomer DPE into the polymer backbone. This not only expanded the carbon skeleton length of the periodic structure and enriched the periodic carbon chain structure, but also increased the richness and feasibility of the design of previous alternating copolymers. The alternating copolymers obtained by LAP in the early stage needed to be hydrogenated to obtain novel periodic polymers. The specific hydrogenation process is described in the ESI.† In this way, five new periodic polymers, pd-DEP, pd-DES, pd-DSEE, pd-DPP and pd-DSS, were obtained.
DPE-alt-IP, DPE-alt-1-PB and DPE-alt-CBVB showed significant changes in the unsaturated region of the 1H NMR spectra before and after hydrogenation because of the double bond of their polymer backbones. Hence, the hydrogenation degree of each hydrogenated product could be directly calculated based on the integral of the hydrogen spectrum (ESI eqn (S13-2), (S14) and (S15)†). The structure of the hydrogenated polymer was characterized by DEPT-135°-13C NMR and 13C NMR (ESI Fig. S13–S15†). The unsaturated carbon peaks of DPE-alt-IP at δ = 130.0–132.0 ppm, DPE-alt-1-PB at δ = 132.0–140.0 ppm and DPE-alt-CBVB at δ = 130.0–137.0 ppm all disappeared; these results further confirmed that the degree of hydrogenation was high. The degree of hydrogenation for the above three structures is shown in Fig. 2. Moreover, it can also explain the acquisition of a strictly periodic structure.
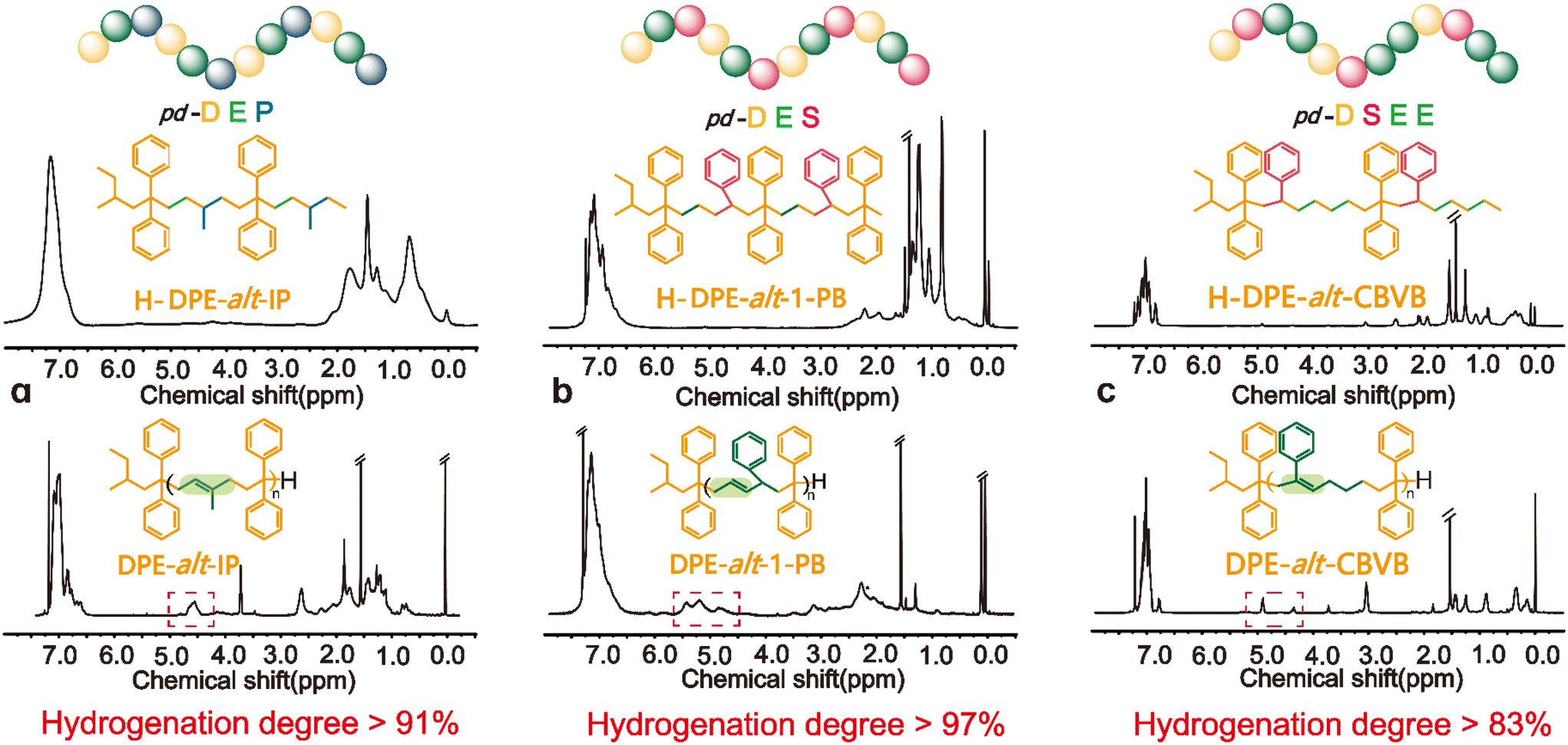 | ||
| Fig. 2 1H NMR spectra of alternating polymers and periodic polymers: (a) for the copolymer of DPE with IP; (b) for the copolymer of DPE with 1-PB; and (c) for the copolymer of DPE with CBVB. | ||
However, no change was observed in the unsaturated hydrogen in the 1H NMR spectra of DPE-alt-DMBD and DPE-alt-DPB before and after hydrogenation. Therefore, the degree of hydrogenation of the polymers could only be analyzed using inverse-gated carbon IG-13C NMR (Fig. 3). Since the NDMBD/NDPE ratio was 1.09, the number of unsaturated seasonal carbons on the DPE structural unit in the polymer chain remained unchanged after hydrogenation (δ = 148.1–148.7 ppm). Using these data as a reference, the degree of hydrogenation could be calculated based on the change in the unsaturated seasonal carbon content of the DMBD structural units. After characterization by IG-13C NMR and the integration of the spectrum, less than 27% of the double bonds in the chain were not hydrogenated. The specific calculations can be found in ESI eqn (S11-1).† The inefficient hydrogenation was caused by the presence of two methyl substituents adjacent to the double bond in the DMBD unit, which affected the hydrogenation efficiency. Similarly, in DPE-alt-DPB, NDPE/NDPB = 1.12. Notably, the number of quaternary carbons on the phenyl rings of DPB remained unchanged after hydrogenation in alternating polymer chains (see Fig. 3, δ = 124.5–129.5 ppm). IG-13C NMR was also used to calculate the unsaturation of the polymer chain. Integral calculations revealed that the degree of hydrogenation of the alternating copolymer was greater than 87% (ESI equation (S12-1)†). Furthermore, the degree of hydrogenation could also be determined by analyzing the ratio of unsaturated carbons on the benzene ring using the DPE unit and the DPB unit. (ESI equation (S12-2)†).
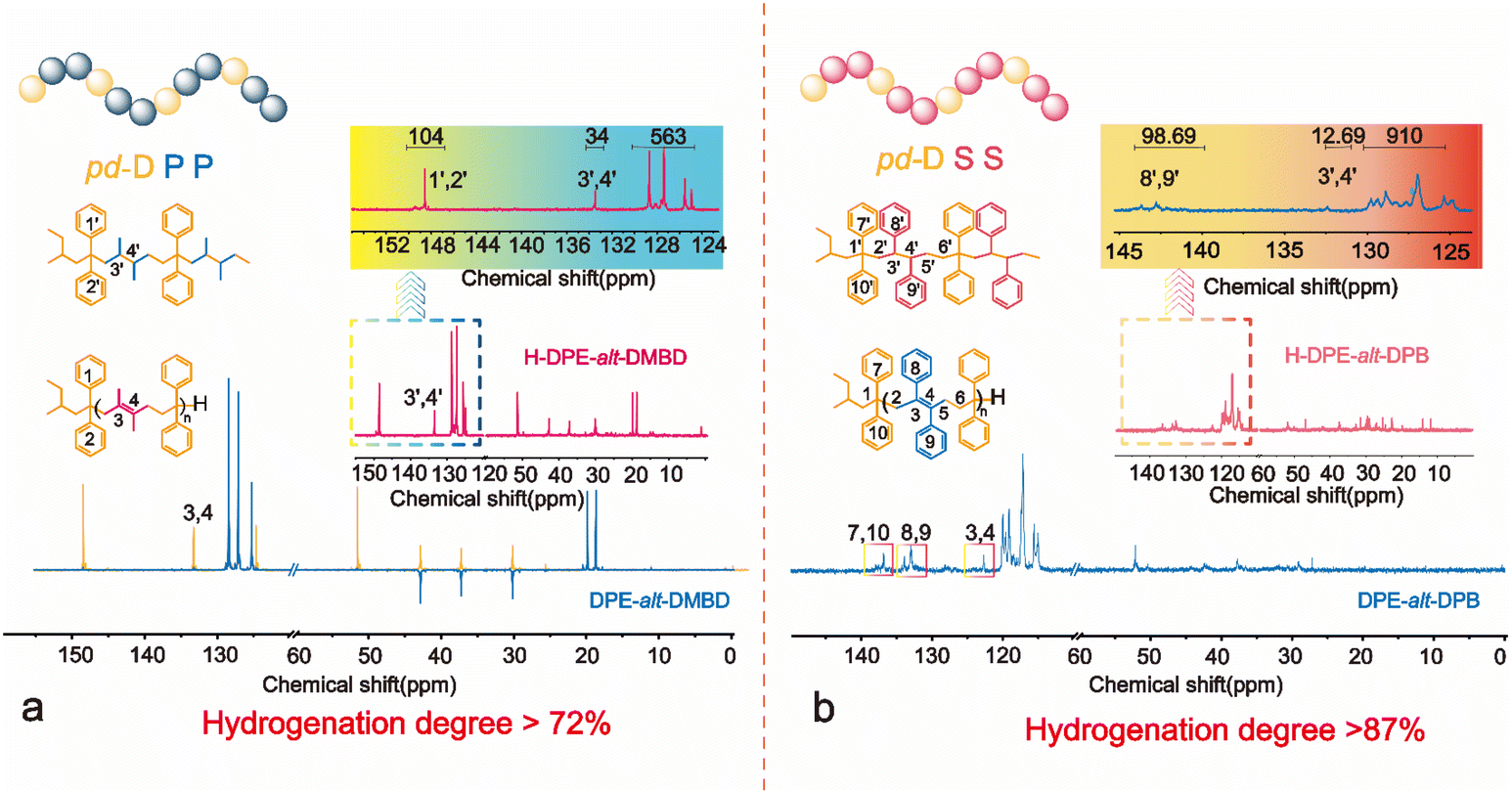 | ||
| Fig. 3 IG-13C NMR spectra of alternating polymers and periodic polymers: (a) for the copolymer of DPE with DMBD and (b) for the copolymer of DPE with DPB. | ||
Therefore, based on the above experimental results, the use of anionic polymerization and simple post-functionalization successfully produced a periodic polymer with a strict sequence arrangement. These periodic sequences contained structures such as ethylene and propylene, which were difficult to obtain using traditional aggregation methods. Nonpolar monomers such as ethylene, propylene, and styrene could not be obtained through LAP with controlled molecular weights and narrow distributions. However, the copolymerization of nonpolar monomers could also yield unique and exceptional properties in the resulting rubber. In this study, DPE-alt-IP and DPE-alt-DMBD polymers were successfully functionalized, and copolymer sequences of ethylene and propylene were easily introduced into the polymer chains. The addition of 1-PB and CBVB introduced copolymer sequences of ethylene and styrene. As the polymer sequence changed from an alternating structure to a periodic structure, Tg significantly decreased compared with that of the non-hydrogenated polymer. Tg is an important factor that indirectly affects the flexibility of polymer chains. Generally, the independent double bonds in the polymer backbone increase the flexibility of polymer chains and reduce the glass transition temperature. However, all five sets of data in this study revealed that saturation of the double bond in the polymer backbone led to a lower glass transition temperature of the polymer (ESI Fig. S22†). This result occurred because the rotation of the carbon–carbon single bond improved the mobility of the polymer chain, which led to a decrease in the glass transition temperature of the polymer (see Scheme 3a and Table 2). Compared with pd-DPP, when the degree of polymerization was similar and when the arrangement density of the methyl substituents in the periodic structure increased (pd-DEP), the friction between the chain ends increased and the Tg of the periodic polymer increased by 26 °C (see Scheme 3b-1). However, compared with pd-DPP, when the methyl substituent in the periodic structure became a benzene ring, the steric resistance between the structural units became greater, resulting in more difficult chain segment movement. Therefore, the Tg of the periodic polymer changed from 117 °C to 133 °C (see Scheme 3b-2). By comparison, pd-DSS and pd-DPP not only had simple periodic arrangement structures but also extended the arrangement of the periodic structure. PDPPDP and SDSSDS are “head–tail” structures. PDPPDP is the equivalent of inserting a second monomer into two propylene monomers. This is a new polymer structure and can be used as a template polymer to provide a reference for the synthesis of more precise positioning sequence polymers. SDSSDS is equivalent to adding a monomer to two styrene monomer units; this structure is difficult to produce using general copolymerization. On comparing SDSSDS with the work of Knoll and coworkers from the BASF on “super-polystyrene”,47 it was a pleasant surprise to find that the Tg of DPE-alt-DPB (161 °C) in this work is very close to that of St-alt-DPE in the aforementioned reference; it was also found that the Tg (133 °C) of pd-DSS is very similar to that of the St/DPE copolymer containing 35% DPE in “super-polystyrene”. The comparison of this data also proves that the relationship between the polymer structure and the thermal properties can be designed and predicted. In addition, the length of the polymer carbon skeleton also affects the thermal performance. pd-DES and pd-DSEE have C6 and C8 skeleton structures, respectively, with the same types of structural units in the polymer backbones. An increase in the length of the periodic sequence enhances the flexibility of the chain, leading to a significant decrease in the Tg (see Scheme 3b-3).
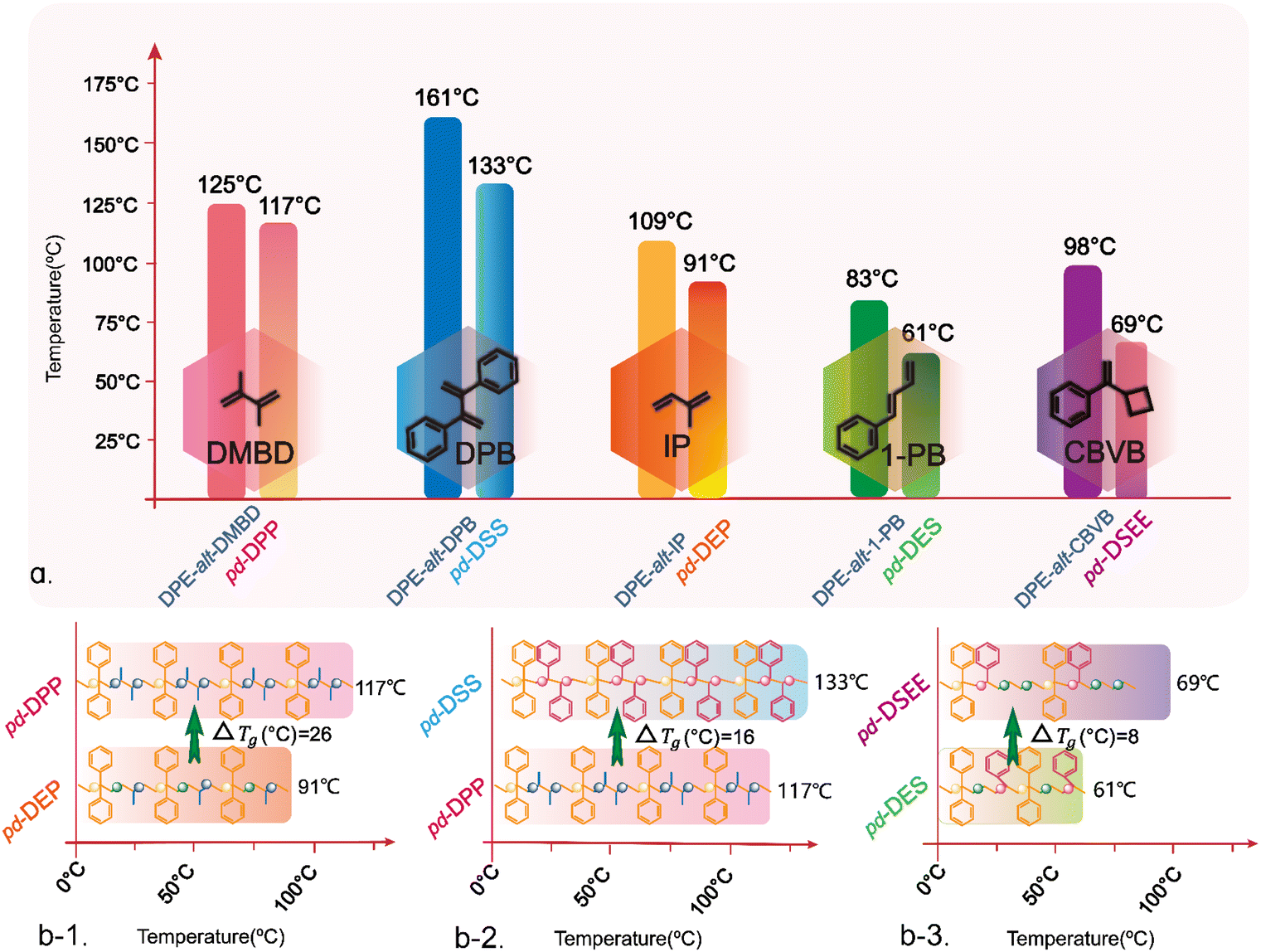 | ||
| Scheme 3 Glass transition temperatures of alternating polymers and periodic polymers. (a) The Tg of base polymers and hydrogenation polymers; (b-1) Run 1 (2#) and run 3 (2#); (b-2) run 3 (2#) and run 6 (2#); and (b-3) run 5 (2#) and run 8 (2#). | ||
| Run | Sample | Before hydrogenation 1# | After hydrogenation 2# | Hydrogenation degreec (%) |
||||
|---|---|---|---|---|---|---|---|---|
| M n [kg mol−1] | PDI |
T
gb (°C) |
M
na [kg mol−1] |
PDI |
T
gb (°C) |
|||
| a Determined by SEC using PS as a standard. b Glass transition temperature. c The degree of hydrogenation was calculated using ESI eqn (S11-1)–(S11-3), (S12-1), (S12-2), (S13-1), (S13-2), (S14) and (S15).† | ||||||||
| 3 | DPE-alt-DMBD | 13.5 | 1.07 | 125 | 14.2 | 1.08 | 117 | 72 |
| 6 | DPE-alt-DPB | 15.1 | 1.17 | 161 | 15.7 | 1.15 | 133 | 87 |
| 1 | DPE-alt-Ip | 16.6 | 1.32 | 109 | 17.5 | 1.21 | 91 | 91 |
| 5 | DPE-alt-1-PB | 23.7 | 1.87 | 83 | 24.0 | 1.74 | 61 | 97 |
| 8 | DPE-alt-CBVB | 13.5 | 1.25 | 98 | 13.5 | 1.22 | 69 | 83 |
Conclusion
The focus of this study was on the synthesis of periodic polymers with precise sequences. Through the synthesis of alternating copolymers, periodic polymers with five new structures, including pd-DEP, pd-DES, pd-DSEE, pd-DPP and pd-DSS, were obtained through hydrogenation. In the synthesis of alternating copolymers, DPE-alt-IP, DPE-alt-DMBD, DPE-alt-1-PB and DPE-alt-DPB were used to change the kinetic behavior of the copolymer by adjusting the polymerization temperature, solvent and feeding ratio through LAP. The sequence was subsequently regulated. DPE-alt-CBVB is a strictly alternating polymer obtained by the AMROP mechanism through common anionic copolymerization and transfer ring-opening polymerization. After the polymeric backbone was hydrogenated and saturated, nonpolar olefins, such as ethylene, propylene and styrene, were successfully inserted into the DPE structural unit according to different arrangement combinations; thus, a periodic polymer with a strict arrangement was obtained. When the polymer backbone was saturated, the Tg of the polymer decreased, indicating that the rotation of the carbon–carbon single bond increased the flexibility of the polymer. The Tg values of the polymers with similar periodic arrangements were different; these results indicated that the size of the polymer substituents and the density of the side group arrangement strongly influenced the polymer properties. Furthermore, when the content of substituents was the same, a change in the carbon skeleton length affected the thermal performance of a polymer. Therefore, controlling the length of the periodic sequence, as well as the position, size, and arrangement density of the substituents, could significantly affect the performance of polymers.Author contributions
The manuscript was written by all the authors and each author has contributed to the experiment. All authors have given approval to the final manuscript.Data availability
The data supporting this article have been included as part of the ESI.†The data that support the findings of this study are available on request from the corresponding author.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (no. 22271041), the China Postdoctoral Science Foundation (2023M740484), the Postdoctoral Fellowship Program of CPSF (GZC20230350) and the Fundamental Research Funds for the Central Universities (2023-BSBA-022).References
- A. J. Destefano, R. A. Segalman and E. C. Davidson, JACS Au, 2021, 1, 1556–1571 CrossRef CAS.
- J. F. Lutz, Eur. Polym. J., 2023, 199, 112465 CrossRef CAS.
- G. Gody, P. B. Zetterlund, S. Perrier and S. Harrisson, Nat. Commun., 2016, 7, 10514 CAS.
- K. Yokota, Prog. Polym. Sci., 1999, 24, 517–563 CAS.
- F. Kubatzki, L. Al-Shok and N. T. Brummelhuis, Polymers, 2017, 9, 166 Search PubMed.
- M. Park, S. J. Hong, S. Lee, N. K. Kim, J. Shin and Y. W. Kim, ACS Sustainable Chem. Eng., 2022, 10, 4538–4550 CAS.
- M. Minoda, T. Otsubo, Y. Yamamoto, J. Zhao, Y. Honda, T. Tanaka and J. Motoyanag, Polymers, 2019, 11(1), 70 Search PubMed.
- X. Deng, B. Huang, Y. Fang, D. Chen, Y. Cheng, S. Chen, J. Zhang, L. Zhang, S. Jeong, F. Wu, J. Liu, L. Chen, C. Yang and Y. Chen, Adv. Funct. Mater., 2024, 34, 23154 Search PubMed.
- S. Jung, Y. Cho, Y. Ji, J. Oh, G. Park, W. Kim, S. Jeong, S. M. Lee, S. Chen, Y. Zhang and C. Yang, Nano Energy, 2023, 106, 108059 CrossRef CAS.
- H. Yin, K. Xing, Y. Zhang, D. M. Aradhana, S. Dissanayake, Z. Lu, H. Zhao, Z. Zeng, J. H. Yun, D. C. Qi and Z. Yin, Chem. Soc. Rev., 2021, 50, 6423–6482 RSC.
- M. Mimura, A. Kanazawa and S. Aoshima, Macromolecules, 2019, 52, 7572–7583 CAS.
- Y. Kametani and M. Ouchi, Polym. Chem., 2020, 11, 6505–6511 CAS.
- H. Kubota and M. Ouchi, Macromolecules, 2022, 55, 4025–4033 CAS.
- L. R. Hutchings, P. P. Brooks, D. Parker, J. A. Mosely and S. Sevinc, Macromolecules, 2015, 48, 610–628 CAS.
- L. Xiang, Z. Li, J. Liu, J. Chen, M. Zhang, Y. Wu and K. Zhang, Polym. Chem., 2018, 9, 4036–4043 CAS.
- Z. Zhang, Y. You and C. Hong, Macromol. Rapid Commun., 2018, 39, 1800362 Search PubMed.
- X. Deng, L. Li, Z. Li, A. Lv, F. Du and Z. Li, ACS Macro Lett., 2012, 1, 1300–1303 CrossRef CAS.
- A. Sehlinger, P. K. Dannecker, O. Kreye and M. A. R. Meier, Macromolecules, 2014, 47, 2774 CrossRef CAS.
- C. Wang, Z. Song, X. Deng, L. Zhang, F. Du and Z. Li, Macromol. Rapid Commun., 2014, 35, 474–478 CrossRef CAS PubMed.
- M. Miyajima, K. Satoh and M. Kamigaito, Polym. Chem., 2021, 12, 423–431 RSC.
- D. A. Shipp, Polym. Rev., 2011, 51, 99–103 CAS.
- C. Boyer, M. Kamigaito, K. Satohc and G. Moad, Prog. Polym. Sci., 2023, 138, 101648 CAS.
- C. Zhang, J. Ling and Q. Wang, Macromolecules, 2011, 44, 8739–8743 CAS.
- K. Satoh, S. Ozawa, M. Mizutani, K. Nagai and M. bKamigaito, Nat. Commun., 2010, 1(1), 6 Search PubMed.
- S. Onbulak and M. A. Hillmyer, Polym. Chem., 2021, 12, 1681–1691 RSC.
- J. Zhang, M. E. Matta and M. A. Hillmyer, ACS Macro Lett., 2012, 1, 1383–1387 CrossRef CAS.
- W. J. Neary and J. G. Kennemur, Macromol. Rapid Commun., 2016, 37, 975–979 CrossRef CAS PubMed.
- H. Bai, L. Han, W. Li, C. Li, S. Zhang, X. Wang, Y. Yin, H. Yan and H. Ma, Macromolecules, 2021, 54, 1183–1191 CrossRef CAS.
- T. Hao, J. Wang, Z. Zhou and D. Yan, Eur. Polym. J., 2024, 214, 113140 CrossRef CAS.
- K. Hayashi, A. Kanazawa and S. Aoshima, Macromolecules, 2022, 55, 1365–1375 CrossRef CAS.
- Y. Sun, Z. Jia, C. Chen, Y. Cong, X. Mao and J. Wu, J. Am. Chem. Soc., 2017, 139, 10723–10732 CrossRef CAS PubMed.
- Y. Ji, L. Zhang, X. Gu, W. Zhang, N. Zhou, Z. Zhang and X. Zhu, Angew. Chem., Int. Ed., 2017, 56, 2328–2333 CrossRef CAS PubMed.
- D. H. S. Ntoukama, H. Mutlub and P. Theatoa, Eur. Polym. J., 2020, 122, 10931 Search PubMed.
- W. Huang, H. Ma, L. Han, P. Liu, L. Yang, H. Shen, X. Hao and Y. Li, Macromolecules, 2018, 51, 3746–3757 CrossRef CAS.
- A. Hirao, S. Loykulnant and T. Ishizone, Prog. Polym. Sci., 2002, 27, 1399–1471 CrossRef CAS.
- Y. Zhang, L. Han, H. Ma, L. Yang, P. Liu, H. Shen, C. Li and Y. Li, Polymer, 2019, 169, 95–105 CrossRef CAS.
- L. Yang, H. Ma, L. Han, P. Liu, H. Shen, C. Li and Y. Li, Macromolecules, 2018, 51, 5891–5903 CAS.
- A. Natalello, J. N. Hall, E. A. L. Eccles, S. M. Kimani and L. R. Hutchings, Macromol. Rapid Commun., 2011, 32, 233–237 CAS.
- G. Ricci, A. C. Boccia, G. Leone, I. Pierro, G. Zanchin, M. Scoti, F. Auriemma and C. D. Rosa, Molecules, 2017, 22, 755 Search PubMed.
- H. Bai, L. Han, C. Li, S. Zhang, X. Wang, Y. Yin, X. Zhang and H. Ma, Macromolecules, 2021, 54, 7269–7281 CAS.
- H. Yuki, J. Hotta, Y. Okamoto and S. Murahashi, Bull. Chem. Soc. Jpn., 1967, 11, 2659–2663 CrossRef.
- P. Liu, H. Ma, W. Huang, L. Han, X. Hao, H. Shen, Y. Bai and Y. Li, Polym. Chem., 2017, 8, 1778–1789 RSC.
- K. Liu, L. Ren, Q. He and W. Xu, Macromol. Rapid Commun., 2016, 37, 752–758 CrossRef CAS PubMed.
- M. Steube, T. Johann, M. Plank, S. Tjaberings, A. H. Gröschel, M. Gallei, H. Frey and A. H. E. Mülle, Macromolecules, 2019, 52, 9299–9310 CAS.
- Q. Ma, L. Han, H. Ma, P. Liu, H. Shen, L. Yang, C. Li, X. Hao and Y. Li, Polymer, 2019, 184, 121907 CAS.
- Z. Wu, Y. Liu, W. Wei, F. Chen, G. Qiua and H. Xiong, Chin. J. Polym. Sci., 2016, 34, 431–438 CAS.
- H. Gausepohl, S. Open, K. Knoll, M. Schenider, G. Mckee and W. Loth, Des. Monomers Polym., 2020, 3, 299–315 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4py00908h |
| This journal is © The Royal Society of Chemistry 2025 |