
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
On the Ostwald ripening of crystalline and amorphous nanoparticles
Manja Annette Behrens†
a,
Alexandra Franzén‡
ab,
Sara Carlertb,
Urban Skantzeb,
Lennart Lindforsb and
Ulf Olsson *a
*a
aDivision of Physical Chemistry, Lund University, Box 124, SE-221 00 Lund, Sweden. E-mail: ulf.olsson@fkem1.lu.se
bAstraZeneca R&D Mölndal, Pharmaceutical Development, Pepperedsleden 1, SE-431 83 Mölndal, Sweden
First published on 25th February 2025
Abstract
Ostwald ripening of crystalline and amorphous nanoparticle dispersions of a model organic compound are compared. While amorphous nanoparticles show a rapid ripening on the timescale of minutes, the crystalline nanoparticles do not ripen within the timescale of weeks. A metastable zone for crystal growth, presumably involving a free energy barrier, is identified, and we propose that this explains the absence of Ostwald ripening in the nanocrystal dispersion. As Ostwald ripening is a process typically occurring near equilibrium, even a small barrier may prevent ripening.
1. Introduction
Finely divided matter, i.e., colloids or nanoparticles, have been studied extensively for more than a century and are still receiving significant attention.1–3 For any kind of application of such materials, in for example catalysis or drug delivery, colloidal stability is generally of major concern, and needs to be addressed. For sufficiently small particles, sedimentation or creaming of dispersions can be avoided, and long-range repulsion, to prevent aggregation, can be achieved by charge or steric stabilization. The small particle size implies a large interfacial area to volume ratio (A/V). For spherical particles of radius R, we have A/V = 3ϕ/R, where ϕ is the particle volume fraction. A large interfacial area is advantageous for applications like catalysis.4 For oral administration of poorly water soluble drugs, nanoparticle formulations may improve bioavailability, due to faster solubilization in the intestine.5,6 However, the large A/V also implies a high interfacial free energy making dispersions prone to coarsening by Ostwald ripening.7,8Ostwald ripening is a coarsening process where larger particles of a dispersion grow, while smaller particles dissolve. The reason is that larger particles have a lower molecular solubility than smaller ones. The interfacial free energy contribution to the chemical potential is size dependent. Assuming spheres of radius R, the chemical potential μ = μ0 + 2γVm/R, where μ0 is the standard chemical potential (the chemical potential in the macroscopic phase), γ is the interfacial tension and Vm is the molecular volume of the compound. Consequently, the molecular solubility, S(R), is size dependent, as given by the Kelvin equation1
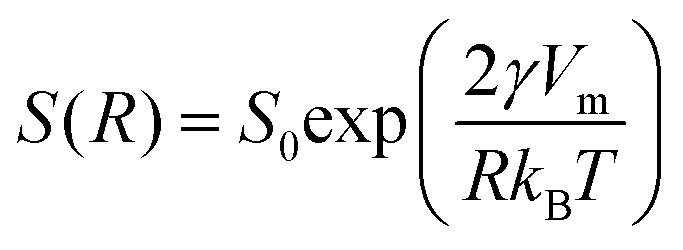 | (1) |
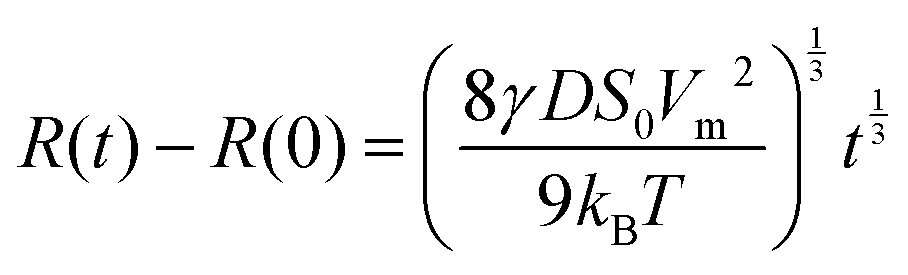 | (2) |
Ostwald ripening has been quantitatively analyzed mainly in liquid emulsion systems,7,11–13 where the predicted t1/3 rate law has been confirmed. A particularly accurate measurement of Ostwald ripening kinetics, and confirmation of the t1/3 rate law, was performed by Stefan Egelhaaf and coworkers,11 using contrast variation small angle neutron scattering to follow the coarsening of a model oil-in-water emulsion.
Ostwald ripening has also been extensively reported for solid particles.14–17 However, here the situation is complicated by the fact that small precipitates can be either amorphous or crystalline,18 and furthermore crystal growth is often reaction limited.8,19,20 In this paper we revisit the question of Ostwald ripening of solid colloidal particles, addressing the criteria for ripening to occur and focus in particular on the potential difference between amorphous and crystalline particles. We have previously shown that amorphous colloidal particles of an organic (model drug) compound, felodipine, undergo Ostwald ripening.21 Here we extend this study to include also crystalline nanoparticles, introducing also a second compound, linaprazan. The chemical structures of felodipine and linaprazan are presented in Fig. 1a.
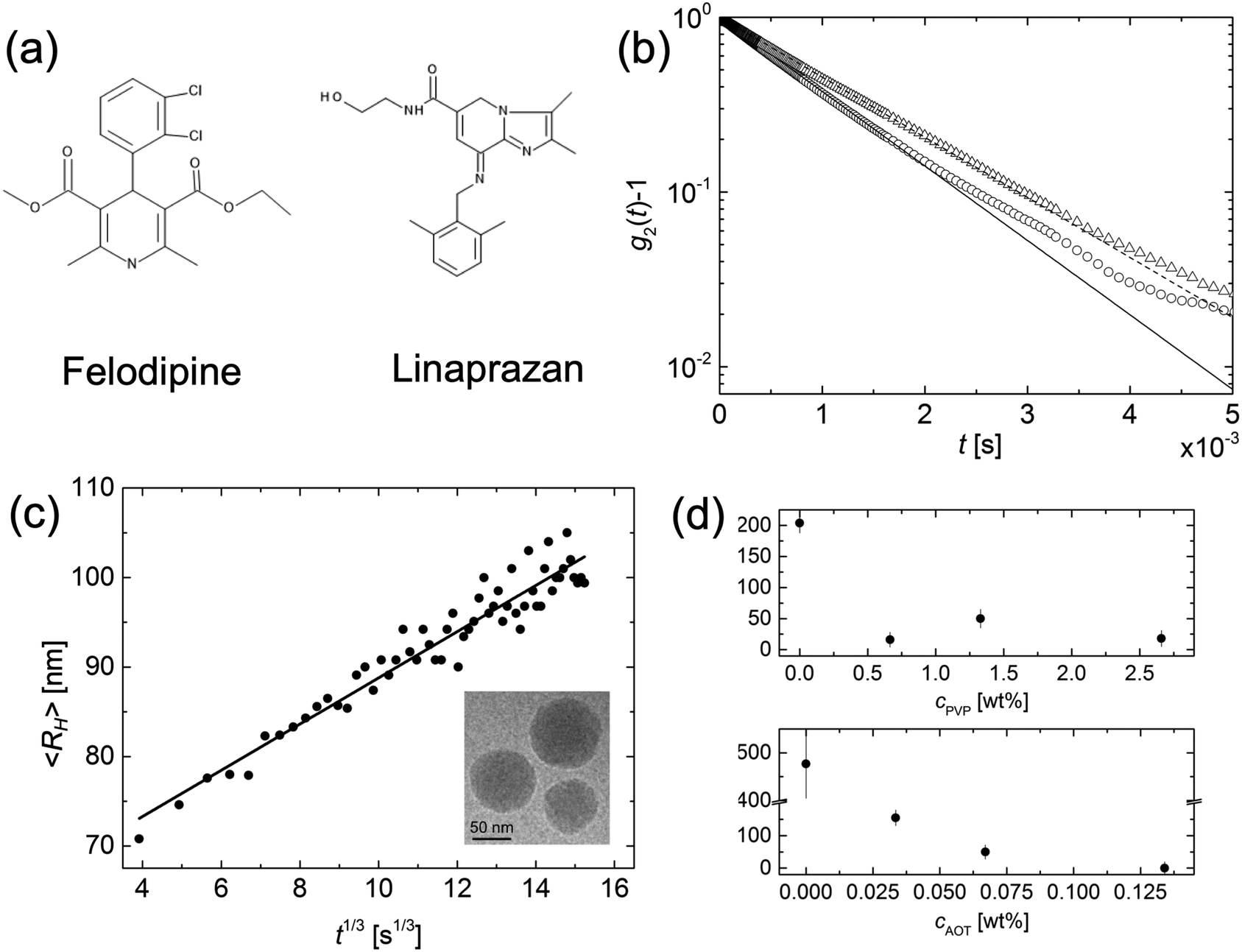 | ||
| Fig. 1 (a) Chemical structures of the two compounds, felodipine and linaprazan, respectively, investigated in this study. (b) Intensity correlation functions, collected from a dispersion of felodipine amorphous nanoparticles one minute (open circles) and 60 minutes (open triangles) after sample preparation. Solid and broken lines are the corresponding fits to a single exponential decay. (c) Growth of felodipine amorphous nanoparticles, stabilized with 0.066 wt% AOT and 1.33 wt% PVP, in time and LSW fit. Inset: Cryo-TEM image of amorphous felodipine nanoparticles obtained approximately 30 minutes after the start of the reaction. (d) The effective interfacial tension of felodipine amorphous nanoparticles as a function of polymer (top) and surfactant concentration (bottom) as obtained from the Ostwald ripening rates. | ||
2. Materials and methods
2.1. Materials
The organic compounds felodipine and linaprazan were provided by AstraZeneca R & D Mölndal, Sweden. Poly(vinylpyrrolidone) (PVP, K30) was obtained from BASF, dioctyl sodium sulfosuccinate (AOT) and N,N-dimethylacetamide (DMA) were obtained from Sigma Aldrich. All chemicals were used as received.2.2. Preparation of amorphous felodipine
Amorphous felodipine was prepared as follows. Felodipine was first solubilized in DMA at a concentration of 100 mM. 10 μl of this solution was then rapidly injected into 990 μl of stabilizer solution placed in an ultrasound bath. The vial remained in the ultrasound bath for approximately 5 seconds. Samples were prepared using stabilizer solutions, with varying polymer (PVP) and surfactant (AOT) content from 0–2.66 wt% and 0–0.134 wt%, respectively. Sample preparations were done at room temperature.2.3. Preparation of crystalline nanoparticles
The crystalline nanoparticles were prepared by milling a suspension of 10 wt% of the compound, felodipine or linaprazan, in a stabilizer solution of 1.33 wt% PVP and 0.066 wt% AOT for 30 minutes. This was repeated four times separated by a resting period of 15 minutes. The milling was performed using the Fritsch Planetary Micromill P7 equipped with 45 mL milling bowls and run at 700 rpm using 0.6–0.8 mm zirconium oxide milling beads. Sample preparations were done at room temperature.2.4. Cryogenic transmission electron microscopy, cryo-TEM
The cryo-TEM images of amorphous and crystalline dispersions were obtained using a Philips CM120 BioTWIN transmission electron microscope, equipped for cryogenic sample environment and with an acceleration voltage of 120 kV. The instrument is situated at the National Center for High Resolution Electron Microscopy (nCHREM) at Lund University, Sweden. The samples are applied as a thin film on a carbon grid, which had been made hydrophilic by glow discharge. The specimens were vitrified in liquid ethane and afterwards transferred to liquid nitrogen.2.5. Dynamic light scattering
Light scattering experiments were performed on a 3D light scattering instrument from LS instruments, which employs a 3D cross-correlation technique with a modulation unit.22,23 The intensity correlation functions displayed in this work were all normalized to one to allow comparison of the shape of the functions.The dynamic light scattering measurements performed on the crystalline nanoparticles were measured at an angle of 90° and with a duration of 60 seconds per measurement. The samples were highly turbid and were diluted 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :200. The high concentration mother dispersion was used to increase the likelihood of observing particle growth, as the rate of Ostwald ripening increases with concentration.24
:200. The high concentration mother dispersion was used to increase the likelihood of observing particle growth, as the rate of Ostwald ripening increases with concentration.24
The dynamic light scattering measurements performed on the amorphous nanoparticles were also measured at an angle of 90° where the time-dependence was investigated by following the sample over an hour in steps of 60 seconds. The samples were not diluted before measuring, as the concentration here, 1 mM, was low and hence the turbidity was low.
An average hydrodynamic radius was obtained by fitting a single exponential decay to the correlation function, g2(t) − 1, obtaining the decay rate, Γ, and hence the diffusion coefficient, D, as these are related by Γ = q2D. Then, by using the Stokes–Einstein relation
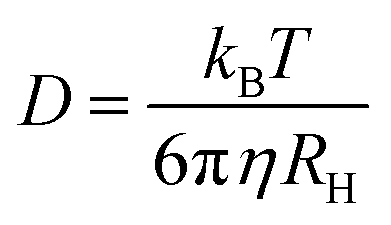 | (3) |
2.6. Fluorescence measurements
Fluorescence measurements were performed as described previously18 on a LS 55 Luminescence Spectrometer from PerkinElmer Instruments, using 2 ml samples in Hellma quarts cuvettes.2.7. Crystal growth experiments
Crystal growth experiments were performed by addition of a small volume of a nanocrystal dispersion to the bottom of a rinsed quartz cuvette, corresponding to 10% crystalline material in the final solution. A solution of a certain supersaturation was then rapidly added, after which the fluorescence measurement was started.3. Results and discussion
Amorphous felodipine nanoparticles nanoparticles are unstable with respect to Ostwald ripening.21 Here we have quantitatively analyzed the ripening using time resolved dynamic light scattering. The felodipine concentration was 1 mM and the concentration of AOT and PVP were 0.066 wt% and 1.33 wt%, respectively. In Fig. 1b the intensity correlation functions, g2(t) − 1, obtained after 1 minute and 60 minutes are compared, and presented together with single exponential fits (solid and broken lines), respectively. After 60 minutes, the decay rate has decreased significantly compared to after 1 minute, consistent with an increase in particle size. Ostwald ripening also leads to a narrow size distribution at the stationary stage of the process.7 A narrowing of the initially broader size distribution was indeed observed in the experimental data. As can be seen, the deviation from a single exponential decay is significantly larger after 1 minute compared to at 60 minutes. Generally, essentially single exponential decays were observed at longer times. In Fig. 1c we present the variation of the average particle hydrodynamic radius, 〈RH〉, plotted as a function of the cubic root of time (t1/3), for the amorphous felodipine particles. t = 0 corresponds to the time of sample preparation. As inset, we present a cryo-TEM image, obtained 30 minutes after particle preparation, showing approximately spherical nanoparticles. From the data in Fig. 1c we see that the particle size distribution is unstable. Significant coarsening occurs within minutes. The slope in the 〈RH〉 vs. t1/3 plot is approximately 2.7 mm s−1/3. For felodipine, the molecular volume, Vm = 0.50 nm3, the amorphous solubility SA = 25 μM, and its diffusion coefficient in water D = 5 × 10−10 m2 s−1.21,25 Using these values and applying eqn (2), an effective interfacial tension γ = 51 μN m−1 is obtained from the slope.The effective γ = 51 μN m−1, obtained from the slope in Fig. 1c, is almost three orders of magnitude lower than a typical water–oil interfacial tension (≈30 mN m−1), and should be considered as an effective, and not a true interfacial tension. The low effective interfacial tension implies that the presence of the AOT–PVP coating of the particles slows down the attachment and detachment of felodipine molecules at the interface. The Ostwald ripening rate, here quantified in terms of this effective interfacial tension, in fact depends on the concentration of AOT and PVP. Two series of experiments have been performed. One at fixed AOT concentration, varying the concentration of PVP, and another at fixed PVP concentration varying the concentration of AOT. The results are presented in Fig. 1c. The effective interfacial tension decreases with increasing concentrations of AOT and PVP.
In the previous study of amorphous felodipine Ostwald ripening, the effective interfacial tension was determined to be approximately 4 mN m−1.21 This study however used a different surfactant (SDS), but probably more important, it was performed with much lower concentrations of both polymer and surfactant.
Having concluded that amorphous nanoparticles of felodipine undergo Ostwald ripening in aqueous dispersions, although the rate depends strongly on the concentration of stabilizer, the focus is now turned to crystalline nanoparticles. A 10 wt% dispersion of crystalline felodipine was milled in the presence of 1.33 wt% PVP and 0.066 wt% AOT, resulting in a particle size of approximately RH = 85 nm. This 10% dispersion has been left to age, and samples from it have been collected at regular intervals. These samples were diluted 1:200, and RH was measured by dynamic light scattering. The dispersion was monitored over a period of approximately two weeks, with samples taken once per day, and the results are presented in Fig. 2.
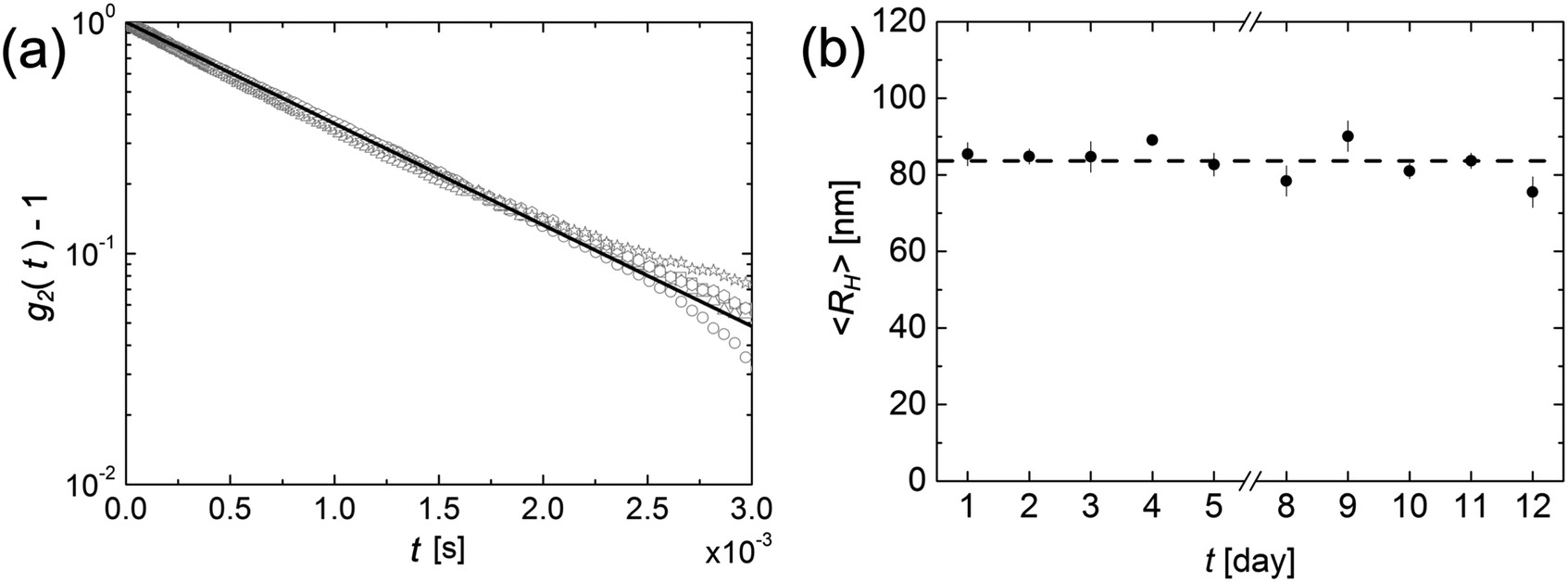 | ||
| Fig. 2 (a) Correlation functions collected for a dispersion of crystalline felodipine nanoparticles at the individual weekdays (day one open square; day two open circle; day three open triangle; day four open hexagon; day five open star) in addition to the single exponential fit (black line) for day one. (b) Hydrodynamic radius of crystalline felodipine nanoparticles, followed over a period of two weeks. The dashed line represents the average value, 84 nm. | ||
In Fig. 2a the intensity correlation functions, recorded during the first five days, are shown. As can be seen, there is essentially no change. The minor deviations observed at longer times are due to uncertainties in the base line. In Fig. 2b the hydrodynamic radius is plotted as a function of time. Over the two-week period, the hydrodynamic radius remains constant at the value 84 ± 6 nm. The crystalline solubility of felodipine, S0 = 2.1 μM, is an order of magnitude lower compared to the amorphous solubility, 25 μM.25 According to eqn (2), a 10 times lower solubility results in a decrease of the Ostwald ripening rate by only a factor of 2. Thus, a lower solubility cannot explain the results of Fig. 2, and we conclude that the crystalline felodipine nanoparticles are kinetically stable, and do not undergo Ostwald ripening.
In a second set of experiments on the stability of crystalline nanoparticles we extended observation time to 10 weeks and also included a second compound, linaprazan. The crystalline solubility of linaprazan is 3.7 μM (AstraZeneca inhouse data). The results are presented in Fig. 3 where the average hydrodynamic radius is plotted as a function of time. As inserts in Fig. 3, representative cryo-TEM images of the crystalline nanoparticles are also shown. The obtained hydrodynamic radii of felodipine, and linaprazan in this experiment are approximately 80, and 65 nm, respectively, and these do not vary over the timescale of 10 weeks.
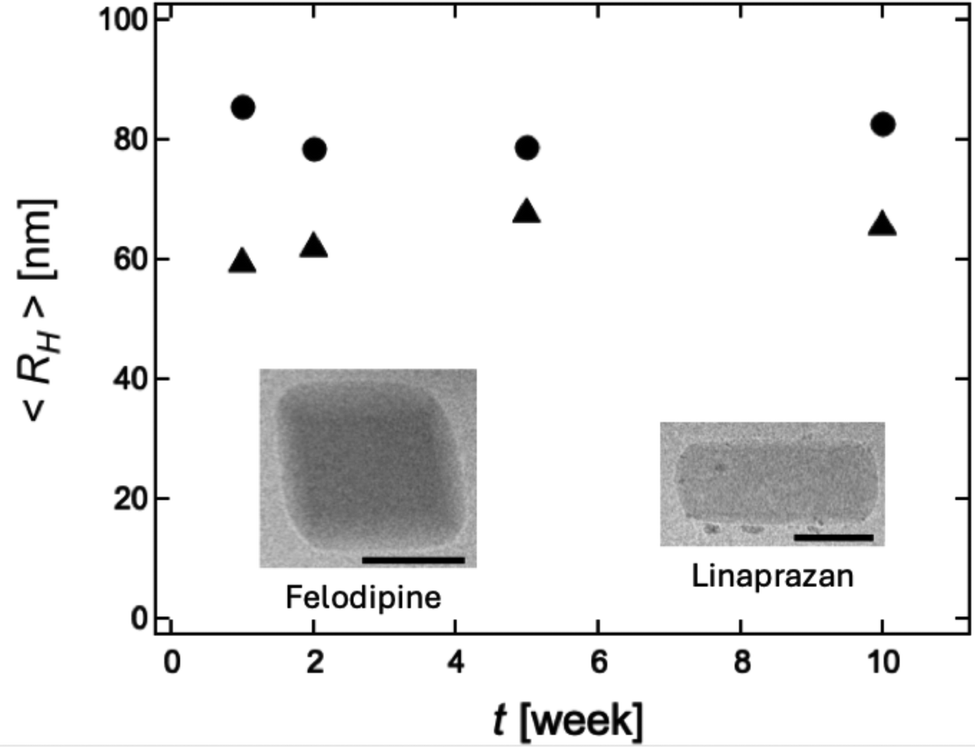 | ||
| Fig. 3 Hydrodynamic radius of felodipine (filled circles), and linaprazan (filled triangles) measured over a period of approximately 10 weeks. As insets we also show representative cryo-TEM images of the respective nanocrystals. Scale bars correspond to 100 nm. | ||
The fact that the crystalline nanoparticles do not undergo Ostwald ripening is indeed striking. To elaborate on this, we consider the two fundamental processes involved in the process, particle growth and particle dissolution, respectively. Focusing on felodipine, we have previously shown that for this compound, crystal dissolution rates are essentially diffusion limited, while the reverse process, crystal growth, is reaction limited.18 This fundamental difference between dissolution and growth is a common observation,8,19 and is also associated with the well-known fact that we can supercool water, but we cannot superheat ice. For macroscopic systems, this apparent absence of microscopic reversibility26 may be explained by the fact that crystalline solids have defects acting as specific sites from which dissolution readily occurs. For a single crystal of colloidal size, the corresponding high energy sites may involve corners and edges.
Thus, a possible explanation for this observation is that the reaction limited crystal growth process involves an activation free energy that requires a certain supersaturation of monomers to occur within a given time. In other words, there is a metastable zone for the crystal growth, analogous to the metastable zone for homogeneous nucleation,20,27,28 and the supersaturation offered by the Ostwald ripening process may not be sufficient to allow the larger crystals to grow.
To test this hypothesis, we performed crystal growth experiments at low supersaturations using time resolved fluorescence experiments to measure the amounts of crystalline felodipine, taking advantage of its intrinsic fluorescence.18,20 Since the quantum yield is approximately two orders of magnitude higher in the crystalline state compared to the monomeric state in solution,18 the fluorescence intensity is to a good approximation proportional to the concentration of crystalline felodipine (unless the monomers are in a large excess). Starting with a known concentration of crystals at time zero, it is straight forward to follow the evolution of the crystalline and monomeric fractions with time. The crystal growth kinetics of felodipine is reaction limited, and at the low concentration of stabilizers used here are not expected to influence the kinetics significantly.18 This is also essentially confirmed here (Fig. 4). In Fig. 4a we present the results from an experiment, performed at a total concentration of 10 μM of felodipine. The initial state at time t = 0 was composed of 1.0 μM of nanocrystalline felodipine in a supersaturated solution of monomers having the concentration 9.0 μM. In Fig. 4a the concentration of monomers, cm, is plotted as a function of time. As can be seen, cm decreases with time and approaches asymptotically a steady state value cm(∞) = 3.6 μM after circa 2 hours. Similar values of cm(∞) were found at total concentrations 15, 20 and 25 μM, respectively. The cm(∞) values are presented in Fig. 4b. The average value of cm(∞) = 3.7 μM is indicated as a broken line. The equilibrium solubility S0 = 2.1 μM is indicated as broken lines in both Fig. 4a and b.
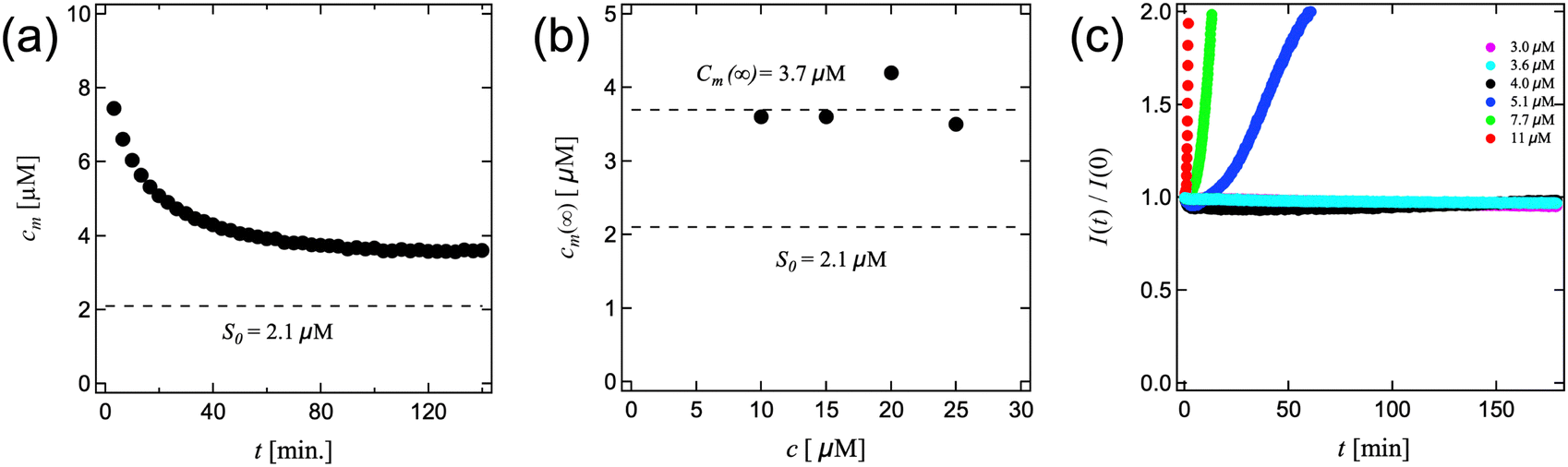 | ||
| Fig. 4 Results from felodipine crystal growth experiments. (a) Variation of the monomer concentration, cm versus time. The total felodipine concentration was 10 μM, with an initial crystalline fraction of 10%. The broken line represents the equilibrium solubility S = 2.1 μM. (b) Steady state monomer concentrations, cm(∞), obtained for different total felodipine concentrations. The average value of cm(∞), 3.7 μM, and the solubility, S0 = 2.1 μM, are both indicated by broken lines. (c) Fluorescence intensity, I(t), normalized by the initial intensity I(0), at t = 0, and plotted versus time for different total initial monomer concentrations. The different colors represent different total concentrations. The initial concentration of crystalline felodipine was 1.5 μM for all monomer concentrations. | ||
We conclude from these experiments that crystal growth terminates when the monomer concentration has reached circa 3.7 μM. This concentration corresponds to a relative supersaturation cm(∞)/S0 = 1.8.
In a second set of crystal growth experiments, we investigated also lower supersaturations, with concentrations ranging from 3.0 to 11 μM. In Fig. 4c we present time resolved fluorescence data, recalling that the fluorescence intensity is essentially proportional to the concentration of crystalline felodipine.18 In these experiments, the initial (t = 0) concentration of crystalline felodipine was 1.5 μM in all experiments. The different supersaturations are indicated by differently colored symbols. As can be seen, no crystal growth is observed within the time window of 3 hours (180 minutes) when the monomer concentration is 4.0 μM or lower. For the higher concentrations, 5.1, 7.6 and 11 μM, crystal growth is clearly observed, with a rate that increases with increasing supersaturation.
Overall, the data in Fig. 4 support the existence of a metastable zone for crystal growth, similar to the rather general metastable zone associated with homogeneous nucleation in the bulk.20,27,28 Crystals often grow layer-by-layer, where each layer is formed by nucleation and layer growth.29 It is most likely that the observed metastable zone is a consequence of a free energy barrier associated with the nucleation of a new molecular layer in the crystal structure.
Ostwald ripening is a relaxation process that typically occurs close to equilibrium. The interfacial free energy contribution to the chemical potential, 2γVm/R, represents only a minor perturbation except for very small R, and hence the relative excess solubilities, S(R)/S0, that we can estimate from the Kelvin equation (eqn (1)) are small. Felodipine has a molecular volume Vm = 0.5 nm3. Assuming further a typical hydrocarbon–water interfacial tension γ = 30 mN m−1 (which is likely an overestimate), and a particle radius of 100 nm, the Kelvin equation predicts at 25 °C S ≈ 1.1S0. This is clearly within the observed metastable zone for felodipine crystal growth that we found to extend to 1.8S0 (Fig. 4b). From this we conclude that because Ostwald ripening is a near equilibrium process, with only minor excess solubilities, a small activation barrier in a fundamental process, such as crystal layer nucleation, is enough to effectively prevent ripening to occur, as demonstrated here for two different crystalline organic compounds. Most likely, the ideas presented here are applicable to other systems where there is a free energy barrier in the particle growth process.
Data availability
Data for this article are available at Open Science Framework at https://osf.io/dxgb2/.Conflicts of interest
There are no conflicts to declare.Acknowledgements
M. A. B. acknowledges financial support from the Villum Kann Rasmussen foundation and U. O. kindly acknowledges the Swedish Research Council for financial support (VR 2020-04633).References
- D. F. Evans and H. Wennerström, The Colloidal Domain, where physics, chemistry and biology meet, Wiley-VCH Wienheim, New York, 2nd edn, 1999 Search PubMed.
- J. Israelachvili, Intermolecular and Surface Forces, Academic Press, Elsevier, San Diego, USA, 3rd edn, 2011 Search PubMed.
- Colloidal Foundations of Nanoscience, ed. D. Berti and G. Palazzo, Elsevier, Amsterdam, 2nd edn, 2022 Search PubMed.
- D. Astruc, Chem. Rev., 2020, 120, 461–463 CrossRef CAS PubMed.
- G. G. Liversidge and K. C. Cundy, Int. J. Pharm., 1995, 125, 91–97 CrossRef CAS.
- D. Horn and J. Rieger, Angew. Chem., Int. Ed., 2001, 40, 4330–4361 CrossRef CAS PubMed.
- A. S. Kabalnov and E. D. Shchukin, Adv. Colloid Interface Sci., 1992, 38, 69–97 CrossRef CAS.
- M. Kahlweit, Adv. Colloid Interface Sci., 1975, 5, 1–35 CrossRef CAS.
- I. M. Lifshitz and V. V. Slyozov, J. Phys. Chem. Solids, 1961, 19, 35–50 CrossRef.
- C. Wagner, Z. Elektrochem., 1961, 65, 581 CrossRef CAS.
- S. Egelhaaf, U. Olsson, P. Schurtenberger, J. Morris and H. Wennerström, Phys. Rev. E, 1999, 60, 5681–5684 CrossRef CAS PubMed.
- A. Kabalnov, Curr. Opin. Colloid Interface Sci., 1998, 3, 270–275 CrossRef CAS.
- P. Taylor, Adv. Colloid Interface Sci., 1998, 75, 107–163 CrossRef CAS.
- P. W. Voorhees, Annu. Rev. Mater. Res., 1992, 22, 197–215 CrossRef CAS.
- Y. Liu, K. Kathan, W. Saad and R. K. Prud’homme, Phys. Rev. Lett., 2007, 98, 036102 CrossRef PubMed.
- R. Viswanatha, P. K. Santra, C. Dasgupta and D. D. Sarma, Phys. Rev. Lett., 2006, 98, 255501 CrossRef PubMed.
- C. L. Kuo and K. C. Hwang, Chem. Mater., 2013, 25, 365–371 CrossRef CAS.
- L. Lindfors, P. Skantze, U. Skantze, J. Westergren and U. Olsson, Langmuir, 2007, 23, 9866–9874 CrossRef CAS PubMed.
- J. W. Mullin, Crystallization, Butterworth-Heinemann, 4th edn, 2001 Search PubMed.
- L. Lindfors, S. Forssén, J. Westergren and U. Olsson, J. Colloid Interface Sci., 2008, 325, 404–413 CrossRef CAS PubMed.
- L. Lindfors, P. Skantze, U. Skantze, M. Rasmusson, A. Zackrisson and U. Olsson, Langmuir, 2006, 22, 906–910 CrossRef CAS PubMed.
- C. Urban and P. Schurtenberger, J. Colloid Interface Sci., 1998, 207, 150–158 CrossRef CAS PubMed.
- I. D. Block and F. Scheffold, Rev. Sci. Instrum., 2010, 81, 123107 CrossRef PubMed.
- P. W. Voorhees, J. Stat. Phys., 1985, 38, 231–252 CrossRef.
- L. Lindfors, S. Forssén, P. Skantze, U. Skantze, A. Zackrisson and U. Olsson, Langmuir, 2006, 22, 911–916 CrossRef CAS PubMed.
- R. C. Tolman, Proc. Natl. Acad. Sci. U. S. A., 1925, 11, 436–439 CrossRef CAS PubMed.
- O. Galkin and P. G. Vekilov, J. Phys. Chem. B, 1999, 103, 10965–10971 CrossRef CAS.
- V. Lattanzi, K. Bernfur, E. Sparr, U. Olsson and S. Linse, JCIS Open, 2021, 4, 100024 CrossRef.
- I. V. Markov, Crystal Growth for Beginners, World Scientific, Singapore, 2nd edn, 2003 Search PubMed.
Footnotes |
| † Current address: Danish Technological Institute, Kongsvang Allé 29, DK-8000, Aarhus, Denmark. |
| ‡ Current address: Södermalmsgatan 14, 507 62 Borås, Sweden. |
| This journal is © The Royal Society of Chemistry 2025 |