
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectrification of fertilizer production via plasma-based nitrogen fixation: a tutorial on fundamentals
Mikhail
Gromov
 *ab,
Yury
Gorbanev
cd,
Elise
Vervloessem
c,
Rino
Morent
a,
Rony
Snyders
ef,
Nathalie
De Geyter
a,
Annemie
Bogaerts
cd and
Anton
Nikiforov
a
*ab,
Yury
Gorbanev
cd,
Elise
Vervloessem
c,
Rino
Morent
a,
Rony
Snyders
ef,
Nathalie
De Geyter
a,
Annemie
Bogaerts
cd and
Anton
Nikiforov
a
aResearch Unit Plasma Technology (RUPT), Department of Applied Physics, Ghent University, 9000 Ghent, Belgium. E-mail: mikhail.gromov@ugent.be
bLeibniz Institute for Plasma Science and Technology (INP), 17489 Greifswald, Germany
cResearch Group PLASMANT, Department of Chemistry, University of Antwerp, 2610 Wilrijk, Belgium
dElectrification Institute, University of Antwerp, 2020 Antwerp, Belgium
eChimie des Interactions Plasma-Surface (ChIPS), CIRMAP, University of Mons, 7000 Mons, Belgium
fMateria Nova Research Centre, Parc Initialis, 7000 Mons, Belgium
First published on 9th January 2025
Abstract
Nitrogen-containing fertilizers are key chemicals for our population, ensuring the constantly growing demands in food production. Fertilizers promote vegetative growth, specifically through the formation of amino acids, the building blocks of proteins. However, the current synthesis method relies on the Haber–Bosch process for ammonia synthesis, one of the largest-volume chemicals made globally, having a significant environmental impact. The need for a sustainable and green industry with low CO2 emission triggers the demand to reconsider the current fertilizer production approach. In this context, electrified, local, small-scale production emerges as a promising option to address current environmental and economic challenges. This approach allows production to be consumer-oriented while adhering to environmental regulations. In light of this, non-equilibrium plasma technology has gained a wave of attention. Plasma-based nitrogen fixation has a long history, starting more than a century ago. It was one of the first nitrogen fixation methods invented and later replaced by more energy-efficient technologies. In the current paradigm, this approach can fulfill all industrial and social demands: it perfectly aligns with non-stable renewable energy, is carbon-neutral, relatively simple to maintain, and can provide a valuable source of fixed nitrogen on a small-scale, on-farm production with complete control over land processing. The plethora of existing publications on plasma-based nitrogen fixation addresses the concept of synthesizing nitrogen-containing fertilizers. However, despite significant advancements in the field and the availability of numerous reviews, they tend to focus on specific aspects, such as plasma physics (e.g., the role of vibration excitation), plasma-initiated chemistry (e.g., nitrogen oxidation or reduction), or reactor design. This tutorial review aims to bridge these gaps by presenting an integrated and accessible explanation of the interconnections between different aspects affecting plasma-based nitrogen fixation. It is designed both for newcomers to the field and those who want to broaden their knowledge, highlighting the current state-of-the-art and offering insights into future research directions and implementations.
Sustainability spotlightPlasma technology presents an attractive alternative for converting N2 into nitrogen-based fertilizers in a manner that aligns with current sustainability goals. However, this necessitates a reconsideration of the existing soil fertilization paradigm: a pivot from large-scale centralized production to on-site direct synthesis. Although challenging, this can bring immense collective benefits. In this approach, valuable nitrogen species (NO3− and/or NH4+) do not require separation or recycling, thereby reducing associated energy costs. Instead, they can be synthesized directly from inexpensive feedstock (air) using plasma oxidation or reduction processes, and applied shortly thereafter. Our work emphasizes the importance of the following UN sustainable development goals: zero hunger (SDG 2); industry, innovation and infrastructure (SDG 9), climate action (SDG 13). |
1 Introduction
The challenge of sustainable nitrogen fixation has garnered increasing attention in the context of mitigating climate change and achieving global food security. Plasma-based nitrogen fixation (NF), leveraging non-thermal plasma technologies, offers a promising alternative to conventional processes like the Haber–Bosch process. Plasma NF has the potential to decentralize fertilizer production, reduce greenhouse gas emissions, and utilize renewable energy sources.This tutorial is designed to serve as a learning resource for those who are new to the field of plasma-based nitrogen fixation or wish to broaden their understanding of its fundamentals. It aims to provide a comprehensive overview of the key process aspects, including the basics of plasma phenomena and plasma-initiated chemistry in different feedstock atmospheres, and, more importantly, to illustrate the interconnections between various process parameters. By synthesizing knowledge from various plasma-related disciplines with state-of-the-art examples, this review seeks to create a cohesive narrative explaining how plasma technologies can contribute to the nitrogen fixation and fertilizers industry.
The primary audience for this review includes:
• Graduate students and early-career researchers entering the field of plasma-based technologies or green chemistry.
• Industry professionals and engineers exploring sustainable nitrogen fixation alternatives for local or small-scale applications.
• Policymakers and educators interested in understanding the potential of plasma-based systems for environmental and economic benefits.
Structure of the tutorial.
To achieve its objectives, the tutorial is organized into the following sections:
(1) Nitrogen fixation and electrification of the chemical industry: a brief overview of nitrogen fixation methods, from the Haber–Bosch process to modern plasma-based approaches.
(2) Fundamentals of plasma and plasma-based nitrogen fixation: this section explains the basics of plasma-initiated kinetics, the generation of reactive species, and the vibrational ladder-climbing phenomenon that makes non-thermal plasma so chemically attractive.
(3) Plasma nitrogen fixation: chemistry: an overview of the state-of-the-art N2 oxidation and N2 reduction processes, focusing on underlying chemical mechanisms in different plasma systems.
(4) From plasma nitrogen fixation to NH4NO3 fertilizer: based on insights into nitrogen fixation fundamentals, this section proposes and discusses a conceptual process to convert atmospheric N2 into ready-to-use fertilizer.
(5) Conclusion and outlook: a summary of progress in plasma-based nitrogen fixation to date, with possible future research directions outlined.
Through this structured approach, the review aims to bridge existing knowledge gaps and inspire further innovation in plasma-based nitrogen fixation.
2 Nitrogen fixation and electrification of the chemical industry
2.1 Nitrogen fixation in nature
Nitrogen (N), phosphorus (P), and potassium (K) are essential elements for plant growth and the foundation of modern soil fertilization. Nitrogen, a vital component of all living organisms, forms the structural basis of life.1 For plants, it supports chlorophyll formation, which is crucial for photosynthesis, and plays a key role in amino acid and protein synthesis. Nitrogen deficiency slows growth and destroys chlorophyll, while excess weakens plants' resistance to diseases. Phosphorus drives energy storage, photosynthesis, and respiration, while potassium regulates water balance, strengthens cell walls, and enhances stress resistance. Together, phosphorus and potassium stimulate the synthesis of nitrogen-based biopolymers and improve water retention, optimizing resource use. These elements collectively ensure energy balance, cellular stability, and plant productivity, boosting resilience to adverse conditions. Therefore, nitrogen, phosphorus, and potassium are all equally important for efficient crop growth.2 Potassium is mined as potassium chloride, and phosphate is derived from phosphate rock, both of which undergo refinement and manufacturing. In contrast, nitrogen, comprising about 78% of the atmosphere, is converted into fertilizer through fixation via reduction or oxidation.The only source of N for plants is from molecular nitrogen in air that is first required to be activated through:
• N2 oxidation into (H)NOx
• N2 reduction into NH3.3
To some extent, this process of nitrogen fixation (NF) occurs in nature via abiotic (lightning) and biotic processes (fixation by aquatic and non-aquatic microorganisms), thus providing the basis for the growth of crops as a food source.4–6
2.2 The current landscape of artificial nitrogen fixation
The increasing food demands due to the ever-growing human population require more fixed N2 than nature supplies. For this reason, the soil has to be supplemented with additional nitrogen, originating initially from natural sources (e.g., organic waste or planting N2 fixing legumes7) and further from synthetic fertilizers.3 Currently, synthetic fertilizers used to grow crops as a direct feedstock, as well as food for sustaining livestock, are estimated to sustain half of the planet's population.7The thermo-catalytic Haber–Bosch (HB) process for nitrogen fixation into ammonia (NH3), commercialized in 1913, is one of the main industrial chemical processes that has been frequently described as the major reason for the rapidly growing human population during the last century.8,9 NH3 is synthesized from N2 and H2 gases (R1.1) in the presence of a catalyst (Fe, Ru). Natural gas is typically used as the H2 source for NH3 and as the source of energy to create the high pressure (200–300 atm) and temperature (>700 K) required for the synthesis. In the next step, the produced gas is cooled down, compressed, and condensed, yielding liquid NH3 as the final product.10 Although the diverse NH3 applications span from textile and plastics production to pharmaceutics and automotive industry, up to 80% of the globally produced NH3via the HB process is used for the production of fertilizers.3
NH3 itself is not a direct agricultural fertilizer but serves as a vital component in the production of various fertilizers, including ammonium salts. Ammonium nitrate (NH4NO3) stands out as the most widely used. Nitric acid (HNO3), also necessary to synthesize NH4NO3, ranks among the world's 15 largest commodity chemicals.11 It is manufactured through the Ostwald process, which involves the catalytic oxidation of NH3, prepared via the HB process, and further steps involving NOx, as summarized in global reactions (R1.2)–(R1.6).12
| 3H2 + N2 → 2NH3 | (R1.1) |
| 4NH3 + 5O2 → 4NO + 6H2O | (R1.2) |
| 2NO + O2 → 2NO2 | (R1.3) |
| 3NO2 + H2O → 2HNO3 + NO | (R1.4) |
| 4NO2 + 2H2O + O2 → 4HNO3 | (R1.5) |
| NH3 + HNO3 → NH4NO3 | (R1.6) |
Therefore, the production of HNO3 is directly limited by NH3 synthesis through the HB process.
2.3 Drivers of change in the nitrogen fixation industry
In the current stage, almost 50% of food production and consumption depends on a single chemical process, which is HB. The tremendous production volume triggers many harmful consequences. The process requires nearly 2% of the total energy produced worldwide, 3–5% of the globally extracted natural gas, and emits >400 Mt of CO2 annually13 – more than a quarter of the total chemical industry emissions.4,14,15 Although modern HB plants convert part of the CO2 (150 Mt per year) into urea ammonium nitrate (UAN), which is also used as fertilizer, this still cannot offset the carbon emission.16 Other strategies to reduce CO2 emissions include full or partial process electrification, such as decarbonizing the hydrogen source (produced electrolytically from water) and utilizing renewable energy to power the HB process.16 However, it's worth noting that the energy cost of NH3 produced in such electrified HB processes is almost 1.5 times higher than that of traditional fossil fuel-based processes.16–18 Nevertheless, despite the rise of renewable energy, current HB plants, due to their high inertia, cannot accommodate the intermittency of fluctuating energy sources, such as wind and solar.19Because of the highly energy-intensive conditions required for the HB process operation and its heavy reliance on CH4, HB becomes economically feasible only on large industrial scales in regions with established natural gas supplies.20 This results in massive centralized production, subsequent costly distribution of the produced NH3, as shown in Fig. 1a, and a large dependency on price volatility in the energy market. Furthermore, the distribution of NH4NO3 produced via the combined HB-Ostwald process is often done in the most economically feasible form, i.e., as a dry solid salt, whose explosive properties resulted in multiple lethal accidents during transportation and storage in the last decade alone.21 The recent geopolitical events also showed that an unstable supply of CH4 (to be used for fertilizers production in a specific region) and the ready-made fertilizers delivery (into a country) create an unsustainable network with unpredictable performance.22–24 Nonetheless, it has to be acknowledged that due to the extremely extensive optimization of HB for over a century of its existence, the energy cost (EC) of the HB-produced NH3 is a meager 0.48 MJ mol−1 (0.70 MJ mol−1 for electrified HB), which corresponds to a theoretical minimum obtainable with the employed pathway of NF.16,17,25
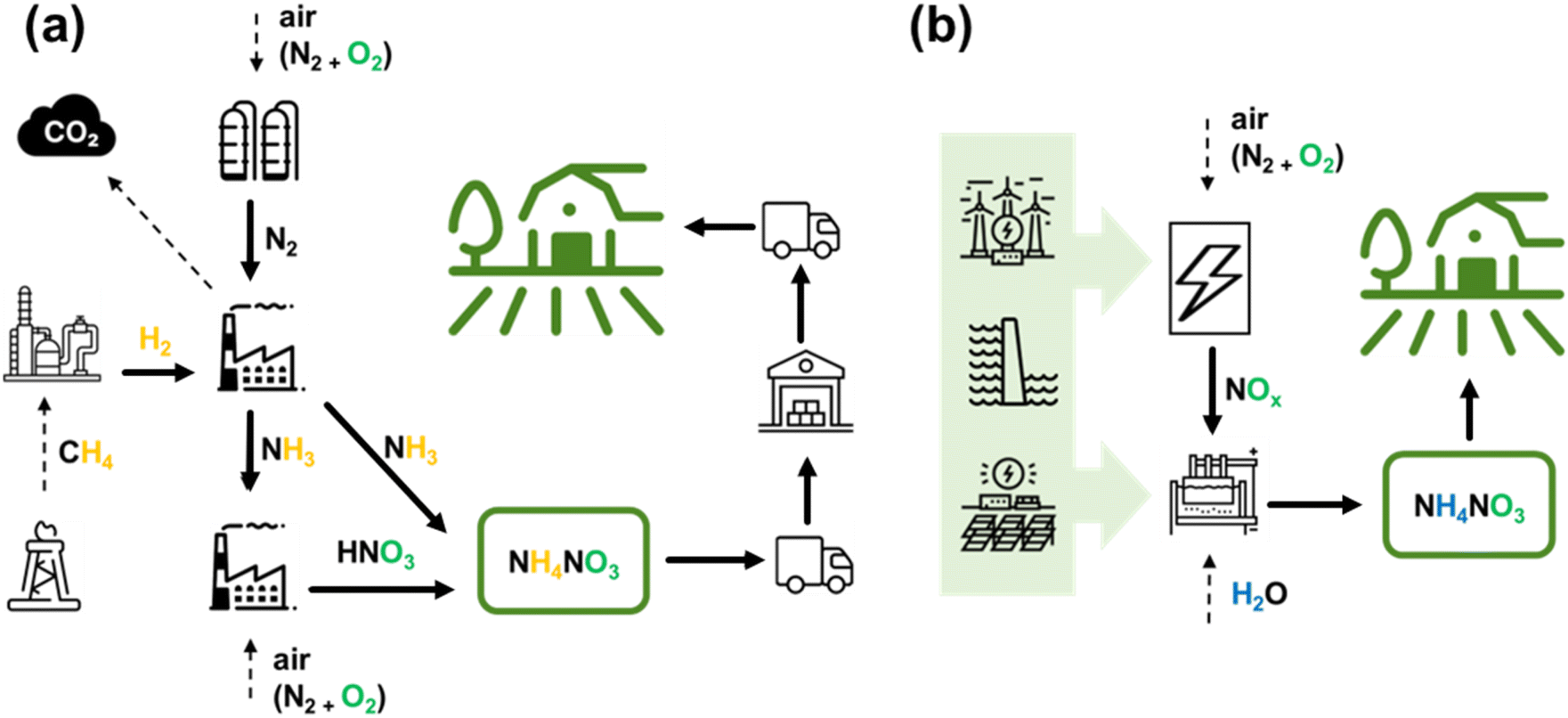 | ||
| Fig. 1 Schematic representation of the fertilizer production and delivery chain for HB-based (a) and plasma-based (b) NH4NO3. The plasma-based route used as an example here involves electrocatalytic reduction as one step to directly obtain an aqueous solution of NH4NO3. | ||
Finally, one more important point motivating the change in the paradigm of the current NF industry is that, at present, more than 50% of the applied N in the form of fertilizer is lost to the atmosphere and water streams,19,26 damaging aquatic organisms and causing air pollution.27,28
2.4 Nitrogen fixation via electrified plasma technology
To this extent, we can summarize that a future alternative should be independent of rare resources and has to be carbon-neutral to address the current ecological crisis. In line with this goal, significant efforts are directed toward developing alternatives to industrial-scale NF due to its associated challenges, which are expected to worsen with increasing demand. These alternatives include complementary NF processes, such as enzyme-based, plasma-based, and (electro-)catalytic methods using both heterogeneous and homogeneous catalysts.5,29–32 Among these technologies, the plasma-based NF approach stands out due to the following advantages, which are also schematically depicted in Fig. 1b:• Plasma or electrical discharge allows the direct conversion of electrical energy into gas-phase chemical products.
• Plasma is a plug-and-play technology, making it fully compatible with renewable energy sources that fluctuate over time.17,33,34
• Theoretical estimations suggest plasma NF can be less energy-intensive than the HB process: the minimum theoretical energy cost of plasma-based NF is estimated at only 0.2 MJ mol−1 (discussed in detail in the following sections).35
• Plasma enables on-site fertilizer production, potentially reducing nutrient loss and ammonia pollution by up to 25%.36
Therefore, plasma technology can provide a sustainable and eco-friendly small-scale process that is compatible with renewable energy sources, completely fossil fuel-free, and carbon-free. It fulfils requirements for producing fertilizers directly on-site where they will be used and in amounts they are needed. It is important to note that this approach does not seek to completely copy the performance metrics of HB (e.g., to reach its EC values). Instead, a completely decentralized alternative, which eliminates all need for distribution, is very sought-after.
Nowadays, two of the most used N-containing fertilizers are urea (CO(NH2)2) and ammonium nitrate (NH4NO3) because of their high nitrogen content (46 and 34% by weight, respectively), cost-effectiveness, and high solubility in water. The major differences between them, from application point of view, are related to their handling, characteristics, and timing of nitrogen release. NH4NO3 requires more attention for storage and transport due to its hygroscopic properties, i.e., the ability to absorb atmospheric moisture. Nitrogen release to plants undergoes hydrolysis, and in the case of CO(NH2)2, it converts to ammonium (NH4+) and bicarbonate ions, while NH4NO3 provides nitrogen in two forms of NH4+ and NO3−. This affects the timing of their application due to the different uptake mechanisms of NO3− and NH4+ ions by plants. CO(NH2)2 is applied during rain or irrigation to assist the movement of nitrogen into the soil. NH4+ is retained in the soil, but nitrogen losses can occur due to NH3 gas volatilization. However, NH4NO3 can be applied at any time. NO3− is readily available to plants, and NH4+ is retained in the soil and slowly consumed.
The most effort has been made in the plasma community towards either nitrates or ammonia production, whereas urea (and UAN) is much less addressed. One of the reasons is that the biological cycle of urea results in CO2 emission, which makes it environmentally a lot less attractive and compromises the global challenge of reducing CO2 emissions from the agricultural sector. This brings us to the concept of plasma-based NF into NOx and NH3, and ultimately into a fertilizer – NH4NO3.
The plasma-based NF approach has yielded numerous publications and reviews with various degrees of detalization.29,37–43 However, an easily accessible, up-to-date overview of plasma-based NF processes towards fertilizers, which could serve as a tutorial, has not been available so far. Therefore, this work aims to fill this knowledge gap, especially for those who are new in the field, underlying what is already well-understood and established, as well as guiding towards new, promising research directions.
3 Fundamentals of plasma and plasma-based nitrogen fixation
This section provides an overview of plasma's key metrics and explores the unique characteristics that make plasma a powerful driver of non-conventional chemistry. The section begins with the definition of plasma and the fundamental principles behind plasma generation. The discussion then moves to the classification of different plasma types, emphasizing their distinct properties. Key elementary processes that underpin plasma chemistry are introduced, followed by an analysis of its advantages and limitations in industrial and scientific applications. This foundation sets the stage for understanding plasma's role in NF.3.1 Basics of electrical discharges known as plasma
Irving Langmuir first introduced the term plasma in his article published in the Proceedings of the National Academy of Sciences in 1928.44 Plasma is a quasi-neutral, ionized gas consisting of electrons, ions, photons, and neutrals in the ground and excited states. Plasma is referred to as the fourth state of matter due to its abundant presence in nature (>99% of the visible universe is plasma). In laboratory conditions, plasma can be generated in any gas by different means of providing energy to ionize the gas, including (i) heating the gas, (ii) applying a strong electrical field, (iii) focusing laser or microwave (MW) radiation, and some others. Depending on the energy of the electrons and heavy particles, plasmas can be divided into two large classes:• Plasmas at local thermodynamic (or thermal) equilibrium (LTE);
• Plasmas at non-local thermodynamic equilibrium (non-LTE).45
LTE, or thermal plasmas, are characterized by an established equilibrium between every collision balanced by its inverse process, i.e., excitation/de-excitation, ionization/recombination. In this case, the temperature of heavy particles or gas temperature (Tg) is equivalent to the temperature of the electrons (Te). Here and later in the paper, the Maxwellian distribution of particle energy is expected to hold for both electrons and heavy species, so the macroscopic parameter, temperature, can be used to describe the energy of the species. In the more general case, the electron energy distribution function (EEDF) has to be used instead of Te, and deviation from the Maxwellian distribution can be considerable, especially in low pressure plasmas.46
In turn, de-excitation processes in non-LTE, or non-thermal, plasmas are characterized by different energy distributions between energetic electrons and heavy particles, and typically, Te is much higher than Tg and the ion temperature (Tions). Both thermal and non-thermal plasmas can be generated at low and atmospheric pressure, whereas high pressure operation often results in fast thermalization of the discharge due to a high number of collisions between particles. Plasma operation at low pressure provides a convenient way to control the energy of electrons, as the latter can only participate in a few collisions with other species and can gather substantial energy. Such plasmas can be very uniform and reach complete ionization, i.e., fusion plasmas in tokamaks, but they are not adapted for NF on an industrial scale due to the requirement of vacuum equipment. Thus, in this overview, the primary focus lies on atmospheric (or high) pressure plasmas as the only feasible way to implement plasmas for NF.
3.2 Elementary processes in plasmas
At high pressure (∼1 atm), electrons are the first to receive energy from the electric field and distribute it among the other plasma components. The energy transfer from an externally supplied electrical field to electrons and consequently to other species (ions, metastables, radicals, etc.) provides a possibility to create a mixture rich in reactive species and so initiate unique chemistry with the use of electrical energy. As such, plasmas of high pressure are often considered a sort of plasma-chemical reactor where unique chemical reactions take place. The main elementary processes initiated in plasma, important for gas conversion chemistry, are schematically presented in Table 1.| Process | Scheme | |
|---|---|---|
| P1 | Momentum transfer | AB + e−(p1) → AB + e−(p2) |
| P2 | Electron dissociation | AB + e− → A + B + e− |
| P2.1 | Heavy particle dissociation | AB* + AB → AB + A + B |
| P3 | Excitation | AB + e− → AB* + e− |
| P3.1 | Electronic excitation | AB(g,Ei) + e− → AB(Ej) + e− |
| P3.2 | Vibrational excitation | AB(g,vi) + e− → AB(vj) + e− |
| P3.3 | Vibrational–vibrational energy exchange |
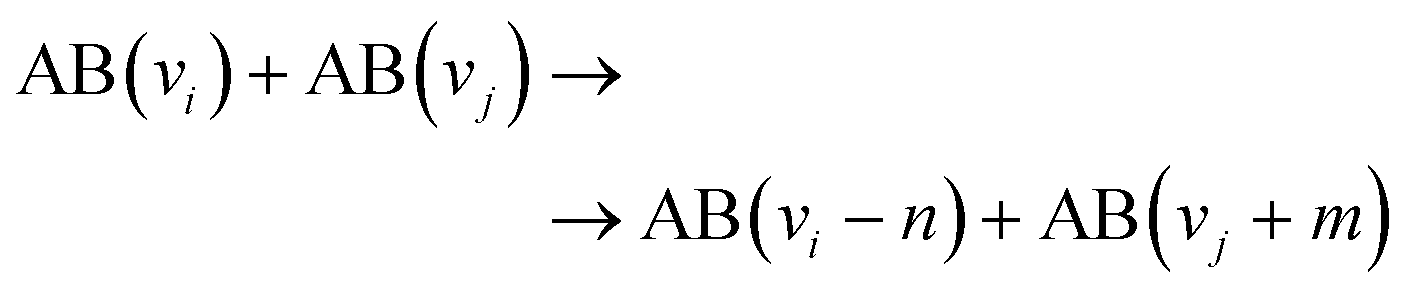
|
| P3.4 | Vibrational-translational energy exchange | AB(vi) + M → AB(vi − n) + M |
| P4 | Electron ionization | A + e− → A+ + 2e− |
| P4.1 | Ion induced ionization | AB + C+ → AB+ + C |
| P4.2 | Dissociative ionization | AB + e− → A+ + B + 2e− |
| P5 | Dissociative attachment | AB + e− → A− + B |
| P6 | Detachment | A + e− → A + 2e− |
| P7 | Photon emission | AB(Ej) → AB(Ei) + hv |
The main electron-initiated processes are P1–P6. These processes are generally divided into two groups, representing elastic and inelastic collisions. The first group, described as P1, changes the kinetic energy of the neutral species. At high pressure, due to the high frequency of collisions, the P1 process is attributed to gas heating. The second group, P2–7, is a set of processes determining plasma's unique properties, creating a manifold of highly reactive species and so capable of initiating plasma chemical reactions. These inelastic collisions between electrons and heavy particles can dissociate molecules (P2), modify the electronic structure of the neutral species, i.e., excite (P3, electronically (E) and/or vibrationally (v)) or ionize (P4) them, and can lead to electron attachment/detachment (P5–P6). Electron collisions with heavy particles, which lead to the formation of excited species, have a particular interest in gas conversion (P3.1–P3.4). This is because these formed species can also have enough energy to overcome a reaction's activation energy barrier (Ea) and participate in gas conversion, particularly in NF. Electrons can transfer their energy to other species, resulting in the formation of ions (P4, e.g., N2+), electronically excited (P3.1, e.g., N2(A, B, C)), and vibrationally excited species (P3.2, e.g., N2(vx)). These energetic particles will also transfer their energy in processes such as P2.1 and P4.1, significantly contributing to gas conversion. However, the same energetic species can also exchange (more often lose) their energy in vibrational–vibrational (V–V, P3.3) and vibrational–translational (V–T, P3.4) energy exchange reactions, through which vibrational energy is converted to heat. Finally, electronically excited species can lose their energy by radiation (P7, spontaneous emission process). The main N2 active species generated in plasma conditions and important for NF are listed in Table 2, together with possible mechanisms of their formation.
| Typical activated N species and their electronic configurations47 | |||||
|---|---|---|---|---|---|
| N2 | N | N2+ | |||
| Electronic state | Potential energy [eV] | Electronic state | Potential energy [eV] | Electronic state | Potential energy [eV] |
| a 1 Td = 10–21 V m2. | |||||
| N2(X1Σg+, v) | 0 | N(S4) | 0 | N2+(X2Σg+, v) | >15.45 |
| N2(X1Σg+, v = 1) | <0.3 | N(D2) | 2.39 | N2+(A2Πu, v) | >16.5 |
| N2(A3Σu+, v) | >6.17 | N(P2) | 3.57 | N2+(B2Σu+, v) | >18.35 |
| N2(B3Πg, v) | >7.35 | N+2(C2Σu+, v) | >23.2 | ||
| N2(C3Πu, v) | >11.03 | ||||
| Nitrogen activation mechanisms in plasmas47,62 | |||
|---|---|---|---|
| Reaction | *– electronic configuration | Minimum activation energy [eV] | |
| Electron impact dissociation and excitation (dominant at E/n > 100 Td ) | |||
| e + N2 → N(*) + N(*) + e | (R2.1) | S4 + S4 | 9.75 |
| D2 + S4 | 12.15 | ||
| P2 + S4 | 13.3 | ||
| D2 + D2 | 14.6 | ||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
|||
| Vibrational excitation, aka. vibrational ladder climbing (dominant at E/n < 100 Td) | |||
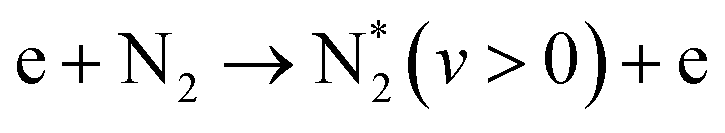
|
(R2.2) | (X1Σg+, v) | <0.29 |
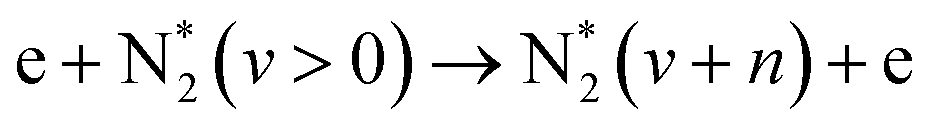
|
(R2.3) | ||
| e + N2(X1Σg+, v) → N + N(*) + e | (R2.4) | <9.75 | |
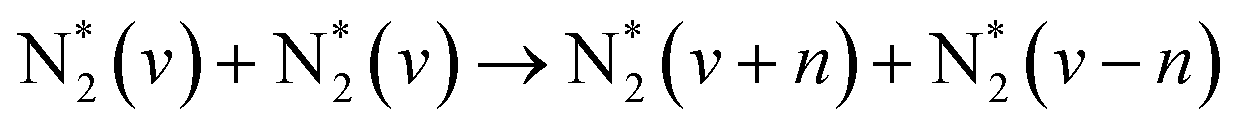
|
(R2.5) | ||
|
|||
| Electronic excitation (dominant at E/n > 100 Td) | |||
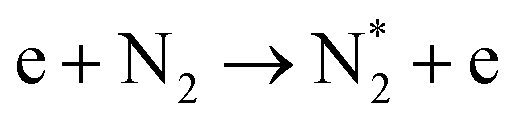
|
(R2.6) | A3Σu+ | 6.17 |
| B3Πg | 7.35 | ||
| C3Πu | 11.03 | ||
|
|||
| Ionization (dominant at E/n ≫ 100 Td) | |||
| e + N2 → N2+* + 2e | (R2.7) | (X2Σg+, v) | 15.58 |
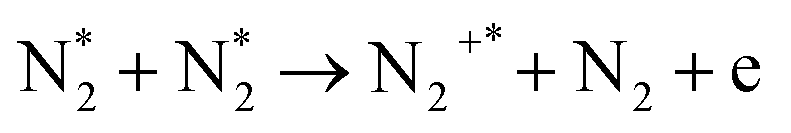
|
(R2.8) | (B2Σu+, v) | 18.75 |
|
|||
| Photon excitation (dominant at E/n ≫ 100 Td) | |||
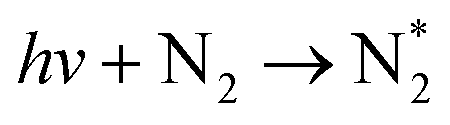
|
(R2.9) | ≈200 nm | Also possible with multi-photon excitation |
| hv + N → N* | (R2.10) | ≈103 nm | |
In contrast to “conventional chemistry,” the reaction rates of electron-involved processes in plasma chemistry are determined by a reaction threshold energy (in [eV]), cross-section (in [m2]), and the electron energy distribution function, or Te, if the Maxwell energy distribution approximation is fulfilled. The threshold energy depends on the nature of the heavy particles and determines the minimum energy of the electron to initiate the reaction. The cross-section defines the probability of the process and depends on the electron energy. Finally, the electron energy is determined by the reduced electric field (E/n), which is the ratio of the electric field E over gas number density n. Consequently, plasma chemistry can be precisely controlled by influencing the electron properties, which is exactly the aim of researchers working in the field of plasma chemistry: to control the plasma-initiated reactions through the tuning of plasma properties.
3.3 Advantages and limitations of plasma-based nitrogen fixation from a physical perspective
It is important to emphasize that in the context of NF, thermal plasmas, where the P1 process is dominant, cannot overcome the thermodynamic limit and so cannot compete with HB or other catalytical processes in terms of energy costs. A thermal arc was historically one of the first methods applied for industrial NF in 1903 (Norway). Birkeland and Eyde developed a method (BE) for the artificial synthesis of HNO3 from atmospheric air using electrical arcs.48,49 Soon after their invention, the method was replaced by HB due to economic reasons mainly related to the high energy costs of the BE process. Indeed, the theoretical energy consumption minimum in thermal plasmas is 0.86 MJ mol−1, which is almost twice the theoretical minimum of 0.48 MJ mol−1 for HB.29 Moreover, it can only be achieved at high pressure and a fast cooling rate of the gas (ca. 107 K s−1), preventing the reverse formation of initial molecular components.29,35 Despite numerous efforts to optimize the arc reactor, the best performance obtained in thermal plasmas so far is 1.8–4.1 MJ mol−1 (at different N2:O2 ratios and reactor configurations), which again makes it difficult to compete with HB on a large scale but still can be of interest for small scale production and use of abundant resources, i.e., air as feedstock.50
On the other hand, the advantage of non-thermal plasmas is related to the pathway of N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N bond dissociation and the possibility of overcoming the energy efficiency of thermal processes because of unique chemistry realized under such conditions, namely at low Tg and high Te. The N2 dissociation limit is 9.7 eV, requiring very energetic electrons to be present in the discharge. This can only be achieved at very high E/n strength, which makes the process energy-demanding and inefficient from an industrial point of view. An alternative to this is the step-wise N2 vibrational excitation. This mechanism is initiated by low-energy electrons that can vibrationally excite N2 (process P3.2 in Table 1). More importantly, these N2(vi) states can also exchange energy (process P3.3 in Table 1), forming N2(vj) with a high vibrational number (j > i). This process is called vibrational ladder climbing. The energy difference between low vibrational levels in N2(Δvx = 1) is < 0.29 eV and decreases with every level according to the anharmonic oscillator theory.51 As a result of ladder climbing, the vibrationally excited states of N2 generated because of P3.2 and P3.3 will induce an increase in the vibrational population of N2 higher states that can efficiently drive NF, decreasing the N2 dissociation limit. Very importantly, the N2 vibrational states do not need to be in equilibrium with the rest of the gas, and so such a process can be driven by electron impact at low Tg. Therefore, NF driven by the vibrational ladder-climbing mechanism is superior to direct electron dissociation or thermal dissociation because:
N bond dissociation and the possibility of overcoming the energy efficiency of thermal processes because of unique chemistry realized under such conditions, namely at low Tg and high Te. The N2 dissociation limit is 9.7 eV, requiring very energetic electrons to be present in the discharge. This can only be achieved at very high E/n strength, which makes the process energy-demanding and inefficient from an industrial point of view. An alternative to this is the step-wise N2 vibrational excitation. This mechanism is initiated by low-energy electrons that can vibrationally excite N2 (process P3.2 in Table 1). More importantly, these N2(vi) states can also exchange energy (process P3.3 in Table 1), forming N2(vj) with a high vibrational number (j > i). This process is called vibrational ladder climbing. The energy difference between low vibrational levels in N2(Δvx = 1) is < 0.29 eV and decreases with every level according to the anharmonic oscillator theory.51 As a result of ladder climbing, the vibrationally excited states of N2 generated because of P3.2 and P3.3 will induce an increase in the vibrational population of N2 higher states that can efficiently drive NF, decreasing the N2 dissociation limit. Very importantly, the N2 vibrational states do not need to be in equilibrium with the rest of the gas, and so such a process can be driven by electron impact at low Tg. Therefore, NF driven by the vibrational ladder-climbing mechanism is superior to direct electron dissociation or thermal dissociation because:
• It requires low E/n strength, corresponding to a mean electron energy of about 2 eV, resulting in reasonable energy consumption;
• Vibrational excitation decreases the N2 dissociation energy in electron impact reactions because the net reaction can take a path as follows: N2(vx > 0) + e(Ee < 9.7 eV) → N + N(*) + e, where * stands for the energy surplus, which leads to the formation of an excited N atom.
• Finally, an important reaction for N2 oxidation: N2 + O → NO + N, having Ea of 3.06 eV, can be initiated by vibrationally excited N2(vx > 13). As such, the mechanism can be more energy efficient compared to direct dissociation.35,52–56
Theoretical estimations based on the dissociation mechanism of N2 by vibrational ladder climbing suggest that NF, namely oxidation, carried out via non-thermal plasmas can be 2.5 times more efficient than the currently used HB process (0.2 and 0.48 MJ mol−1, respectively).29,35 This is the most important aspect of non-thermal plasma-induced NF, as the aforementioned mechanism provides a way to overcome the thermodynamic limit of thermal chemical processes, and so is able to compete with HB and other catalytic pathways. Unfortunately, vibrational ladder climbing is often coupled with vibrational energy relaxation in V–T processes (P3.4, Table 1), leading to gas heating and increasing the costs of NN bond dissociation. Moreover, V–T relaxation accelerates with temperature and vibrational state numbers. This is why the most pronounced vibrational excitation is observed in low pressure plasmas, making it difficult to achieve at elevated pressure.35,57–59 However, a number of methods were proposed in the literature to reach high vibrational excitation of N2 in plasmas operating at high pressure.54,58,60,61
In summary, depending on the mean electron energy or Te, plasma-induced NF processes can be dominated either by (i) elastic collisions, with typical examples of thermal plasmas; (ii) vibrational excitation, or (iii) electron impact dissociation. As the contribution of the listed processes is defined by the energy of the electrons, it can be tuned depending on a type of electrical discharge, more specifically depending on the value of E/n required to sustain the discharge and heat the electrons. In that regard, it is very convenient to classify electrical discharges based on their E/n value, as shown in Table 3. The electronically excited species (e.g., N2(A, B, C)) are typically produced under conditions when the E/n strength is >100 Td (in an N2 atmosphere). This mechanism of N2 excitation is dominant in dielectric barrier discharges (DBDs), corona discharges (where also photon-related processes take place), and short-pulse plasmas. At the same time, the favorable vibrational excitation, i.e., N2(vx) formation, can take place in plasmas operating at E/n < 50 Td. These are plasmas operating in glow discharge mode (mostly direct current (DC)), radiofrequency (RF), and MW plasmas. It is generally assumed that both electronic excitation and vibrational excitation can be initiated in pulsed plasmas (pulse duration from 10 μs to 100 ms).62,63 At E/n below 5 Td, the contribution of elastic collisions starts to be dominant, and plasmas tend to transfer to thermal equilibrium. An overview of different plasma types used for NF is provided in Table 3.
| Plasma type | Reduced electric field, E/n [Td] | Mean electron energy [eV] | Electron density, ne [m−3] | Gas temperature, Tg [K] | Dominant N2 excitation pathway |
|---|---|---|---|---|---|
| a Values strongly depending on the experimental arrangement; AC stands for alternating current; APPJ stands for atmospheric pressure plasma jet | |||||
| Corona discharge (DC or pulsed) | ≫100 | >10 | 1015–1019 | <400 | Photoexcitation |
| DBD | >100 | ≈10 | 1018–1021 | <700 | Electronic excitation |
| Glow discharge | <50 | 1–2 | 1019–1021 | 300–1000 | Electronic and vibrational excitation |
| Arc discharge (DC) | ≪10 | 1–2 | 1021–1026 | T e = Tg = 8000–14000 | Thermal excitation |
| Gliding arc (GA) | >100 at breakdown | 1–2 | 1020–1022 | 2000–3500 | Vibrational and thermal excitations |
| ≪100 when sustained | |||||
| Spark discharge (AC, DC pulses) | ≫100 at breakdown | <10 at breakdown | 1021–1024 | >1000 | Electronic, vibrational, and thermal excitations |
| ≪100 when sustained | 1–2 when sustained | ||||
| MW plasmas | 1017–1022 | 2400–10000 | Dissociative excitation, vibrational, thermal excitations | ||
| RF discharges | 1021–1026 | T e = Tg = 600–3000 K | |||
| Pulsed RF | <400 | ||||
| RF APPJ | 1017–1018 | <600 | |||
In this section, the basic aspects of plasma physics in the frame of the NF approach were introduced, to help readers understand how different types of plasma can be used for the initiation of desirable chemistry. This knowledge is essential for any plasma researcher embarking on the design of plasma-based NF experiments. Armed with this knowledge, researchers can address key questions such as: What type of plasma should I use? What activated species does my plasma produce? What gas temperature and degree of non-equilibrium can I achieve? To provide a guideline in answering such key questions, the following sections will discuss the current state-of-the-art in two primary directions: N2 oxidation and N2 reduction, with a focus on mechanisms of reactions initiated by plasma.
4 Plasma nitrogen fixation: chemistry
This section explores the chemical pathways involved in nitrogen fixation, specifically focusing on the nitrogen oxidation and reduction processes in various gas atmospheres. Before delving into these processes, it is essential to establish the foundational concepts, including the common logic, terminology, and denotations used throughout the discussion.In general, plasma-based NF starts with feedstock activation through a reaction between energetic electrons and heavy particles or between several heavy particles with redundant energy. These processes depend on the gas mixture composition and can be generally introduced as follows:
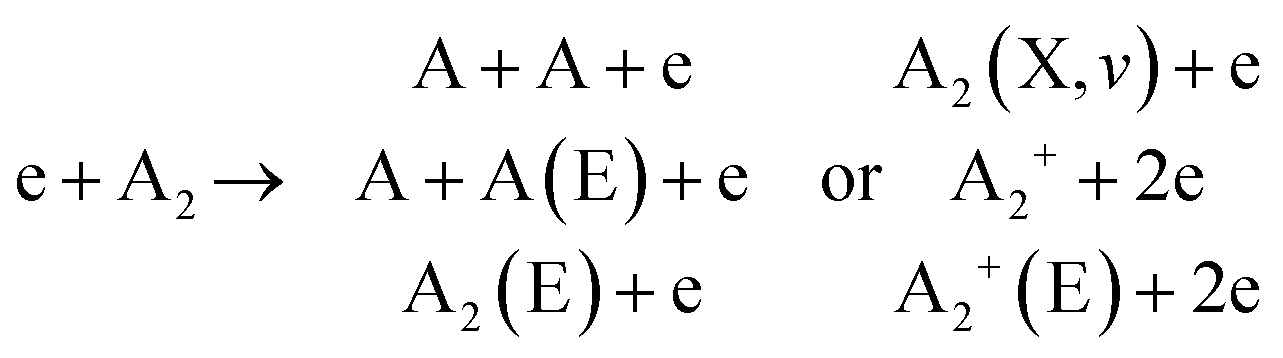
Further, for contingency, all electronically excited states (E) and vibrationally excited states (X, v) will be denoted with an asterisk *, while an asterisk enclosed in brackets, (*), will mean a species that can be in ground as well as in excited state,
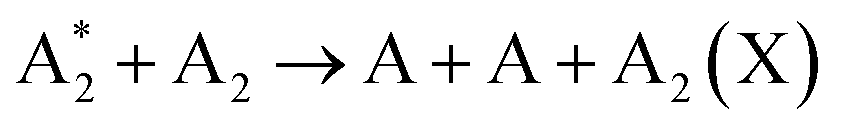
There is no general agreement on the importance of each activation channel facilitating NOx and especially NH3 synthesis. Direct electron impact dissociation,64 ionization,64,65 electronic excitation,64,66,67 and vibrational excitation68,69 have all been proposed as important mechanisms taking place in different plasmas.70 However, it is well accepted that the larger the contribution of vibrational excitation in N2 dissociation, the more energy-efficient the NF. In turn, the gas-activated species initiate the formation of initial products, for example as:
| A + B → AB |
As a final step, further oxidation or hydrogenation takes place for N2 oxidation and N2 reduction processes, respectively.
The overall rate-determining and most energy-consuming step of NF in the plasma environment is electron impact activation of the N2 molecule, i.e., (R2.1)–(R2.10) reactions in Table 2. Such an initial step is common for all plasma-based NF processes, oxidation or reduction, but the plasma type determines selectivity towards the generation of specific intermediates and so the final reactive species. It must also be emphasized that the mechanisms discussed in Sections 3.1 and 3.2 serve only the purpose of introducing divergence in NF chemistry. From this perspective and the purpose of the paper, they are given in a very generalized way (although supported by literature and reaction rate analysis), while real chemistry is much more complicated and may involve thousands of reactions from radical chemistry, photochemistry, etc., but discussing these would lie outside the scope of our discussion.
4.1 Plasma nitrogen fixation: oxidation pathways
The following section introduces readers to the key chemical reaction pathways leading to (H)NOx synthesis. It is organized by the type of feedstock used, allowing readers to focus on the processes most relevant to their interests.The understanding of N2 oxidation mechanisms has been built upon the combustion processes. N2 oxidation chemistry at thermal conditions (in combustion) has been well-known since the 1940s and is described below via reactions (R3.1)–(R3.3), known as the extended Zeldovich mechanism.71
| N2 + O ⇆ NO + N | (R3.1) |
| N + O2 ⇆ NO + O | (R3.2) |
| N + OH ⇆ NO + H | (R3.3) |
Reaction (R3.1) is the rate-determining step due to the need to break the strong triple bond of the N2 molecule. As such, in combustion, the final concentration of NO is notably influenced by the composition of the fuel mixture and the temperature used. In O2-rich gas mixtures, the NO concentration reaches equilibrium through the reversible reaction (R3.2). This entails a forward reaction at low NO concentrations and a reverse reaction at higher NO concentrations. However, under specific conditions, particularly when the oxygen content is low in the gas mixture, the NO concentration is governed by reaction (R3.3), which involves OH radicals. The latter can be generated in significant quantities within the combustion mixture through the oxidation of hydrocarbons (the fuel).71–73
Building upon initial attempts in NOx synthesis at plasma conditions via the thermal Birkeland–Eyde process and emerging insights into NO formation chemistry from combustion studies, a broad range of plasma sources have been explored for the dissociation and oxidation of the NN bond.35 Several literature reports delve into the underlying oxidation pathways.17,62,74–79 The chemistry of N2 oxidation differs significantly and exhibits greater diversity in plasma compared to combustion, owing to the non-equilibrium conditions that can be realized in plasmas.
:O2 gas mixtures, NOx formation can be described following the aforementioned steps: activation – initial product formation – oxidation, as shown in Table 4 and schematically illustrated in Fig. 2a.
| Nitrogen oxidation in N2 + O2 (air) mixtures | |
|---|---|
Feedstock (N
2
, O
2
) activation (N, N*,
 , O, O*,
, O, O*,
 )
)
|
|
| (R2.1)–(R2.10) (N2 activation, as described in Table 2) | |
| e + O2 → O + O* + e | (R3.4) |
| e + O → O* + e | (R3.5) |

|
(R3.6) |
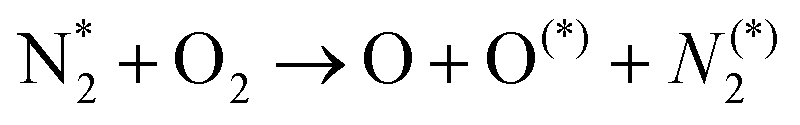
|
(R3.7) |
| Initial oxidation of N | |
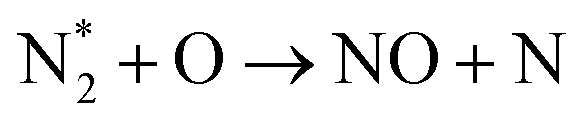
|
(R3.8) |
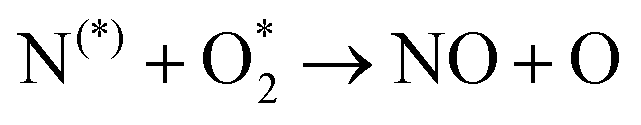
|
(R3.9) |
| Further N oxidation | |
| NO + O + M → NO2 + M | (R3.10) |
| NO + O3 → NO2 + O2 | (R3.11) |
| Decomposition reaction of NOx | |
| NOx + O + M → NOx−1 + O2 + M | (R3.12) |
| Nitrogen oxidation in N2 + H2O and N2 + O2 (air) + H2O mixtures | |
|---|---|
Feedstock (N
2
, O
2
, H
2
O) activation (N, N*,
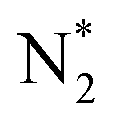 , O, O*,
, O, O*,
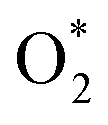 ,OH, H) in the gas phase
,OH, H) in the gas phase
|
|
| (R2.1)–(R2.10) (N2 activation, as described in Table 2) | |
| (R3.4)–(R3.7) (O2 activation) | |
| e + H2O → OH + H + e | (R3.13) |
| O* + H2O → OH + OH | (R3.14) |
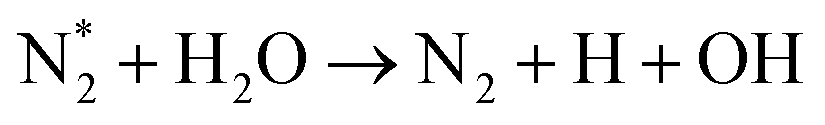
|
(R3.15) |
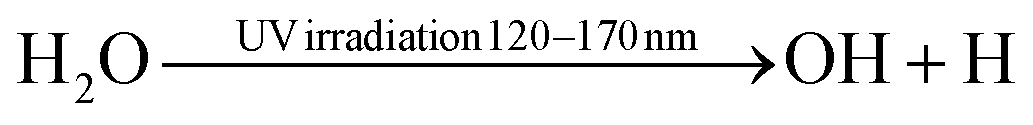
|
(R3.16) |
| e + OH → H + O + e | (R3.17) |
| Initial oxidation of N: (R3.8) and (R3.9) | |
| N* + OH → NO + H | (R3.18) |
| Further N oxidation: (R3.10) and (R3.11) | |
| NO + H + M → HNO + M | (R3.19) |
| NO2 + HNO + M → HNO2 + NO + M | (R3.20) |
| NO + OH + M → HNO2 + M | (R3.21) |
| H2O + NO2 → OH + HNO2 | (R3.22) |
| H2O + NO2 + NO → HNO2 + HNO2 | (R3.23) |
|
|
Feedstock (N
2
, O
2
, H
2
O) activation (N, N*,
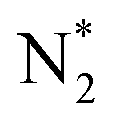 , O, O*,
, O, O*,
 ,OH, H) in the plasma/liquid interface
,OH, H) in the plasma/liquid interface
|
|
| (R2.1)–(R2.10) (N2 activation described in Table 2) | |
| (R3.4)–(R3.7) (O2 activation) | |
| (R3.13)–(R3.17) (H2O activation) | |
| OH + OH → H2O2 | (R3.24) |
| H2O2 + O → HO2 + OH | (R3.25) |
| OH + O3 → HO2 + O2 | (R3.26) |
| OH + H2O2 → HO2 + H2O | (R3.27) |
| Initial oxidation of N in the plasma/liquid interface: (R3.8), (R3.9) and (R3.18) | |
| N + HO2 → NO + OH | (R3.28) |
| Further N oxidation in the plasma/liquid interface: (R3.10), (R3.11), (R3.19)–(R3.23) | |
| NO2 + O + M → NO3 + M | (R3.29) |
| HO2 + NO2 → HNO2 + O2 | (R3.30) |
| OH + NO2 + M → HNO3 + M | (R3.31) |
| H2O2 + NO2 + NO2 → HNO3 + HNO3 | (R3.32) |
| HO2 + NO + M → HNO3 + M | (R3.33) |
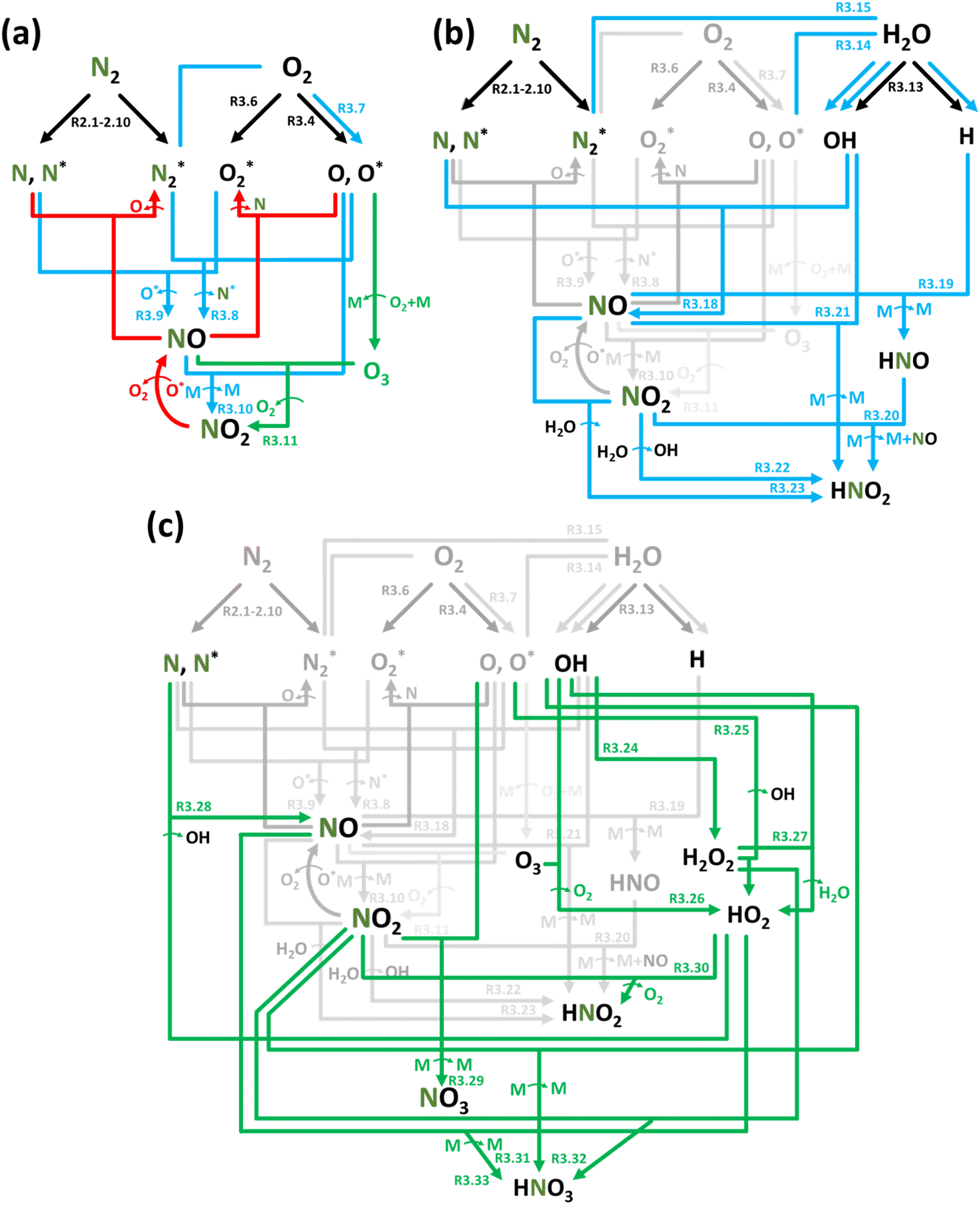 | ||
| Fig. 2 Reaction diagrams of the N2 oxidation processes in (a) N2:O2 (air), (b) N2 + H2O and N2 + O2 (air) + H2O feedstocks, where (c) highlights the chemistry in the plasma/liquid interface. Black, blue, red, and green arrows indicate electron impact reactions, forward reactions, reverse reactions at high temperature, and reactions pronounced at low temperature, respectively. | ||
The plasma type strongly affects the occurring chemistry, namely during feedstock activation ((R2.1)–(R2.10) for N2 and (R3.4)–(R3.7) for O2, Tables 2 and 4, respectively) and precursor formation steps (R3.8) and (R3.9). Plasmas with high electric field, i.e., E/n > 100 Td (such as a DBD), generate electronically excited species, while plasmas with E/n < 100 Td can initiate strong vibrational excitation (see Table 2 for details). In general, the generation of NO, a primary precursor for the formation of further, higher oxides, happens via the so-called non-thermal Zeldovich mechanism. This term is often met in the plasma community, and it refers to reactions (R3.8) and (R3.9) driven by highly reactive species formed in plasma.
It is also generally accepted that the most energy-efficient pathway for NF is promoted by vibrational excitation of ground state to N2(X, v) (see Section 2). Vibrational excitation decreases the activation energy barrier for nitrogen dissociation, facilitating reaction (R2.1) through (R2.5), as shown in Table 2. More importantly, for N2 oxidation, the Zeldovich mechanism can be efficiently exploited by overpopulating the N2 vibrational levels, and reaction (R3.1), shown earlier, can be driven by N2(X, v > 13). However, often, vibrational excitation is suppressed by undesirable V–T energy transfer processes, which lead to a rise in gas temperature, inherently initiating NO formation following the thermal Zeldovich mechanism (R3.1) and (R3.2). This means that in warm plasmas (with E/n < 100 Td, such as GA), NO generation can occur through a combination of both non-thermal and thermal Zeldovich mechanisms. In this case, NO formation via thermal mechanism can be considered a waste of energy because it is less efficient than its non-thermal counterparts. In addition, the reverse processes (R3.8) and (R3.9) become more pronounced at high temperatures, restricting NO formation (red arrows pathway in Fig. 2a). Therefore, the specific contribution of non-thermal and thermal Zeldovich mechanism in plasmas with E/n < 100 Td remains uncertain and subject to ongoing scientific debate.
The ultimate oxidation of NO to NO2 is shown in reactions (R3.10) and (R3.11). Ozone (O3) production primarily occurs through the reaction O2 + O + M → O3 + M, which consumes reactive oxygen atoms (M stands for the third body). O3 is crucial for the formation of NO2; nevertheless, in plasmas with elevated gas temperatures, the significance of (R3.11) diminishes as O3 becomes unstable at high temperatures (green arrows pathway in Fig. 2a). It is worth noting that reaction N + O3 → NO + O2 cannot contribute at the initial oxidation state stage due to its low reaction rate constant.80
The effect of the N2:O2 ratio on the production rate (PR) and energy cost was investigated in various plasma systems, including MW, DBD, nanosecond pulse plasma, and GA.50,59,62,81 The best performance is consistently achieved with a gas mixture ratio of 1:1 (rather than 4:1 as in air). However, the enhancement in the yield of oxidation products is not significant, considering the energy expenses associated with the production of either pure N2 or O2.
The most industrial-relevant question is achieving the theoretical minimum EC of NF. It can be realized by utilizing the non-thermal Zeldovich mechanism, which is a key objective in the plasma community. Alongside direct NOx formation, two common challenges are:
• Minimizing its decomposition via reverse reactions;
• Maximizing the fraction of gas treated by the plasma.48,50,74,82
These issues are often addressed by using pulsed spark plasmas,83,84 incorporating specialized output gas nozzles (utilizing the adiabatic and Joule–Thomson effects to cool the gas through expansion),85–87 modifying the gas flow dynamics,85,87 and introducing increased pressure to favor NO oxidation into NO2 rather than NOx decomposition (i.e., favoring (R3.10) over (R3.12)).49 It is, however, important to note that the produced NOx species are not fertilizers but rather fertilizer building blocks.
When liquid H2O is introduced into the system, NF products accumulate in the liquid phase. These products may include NH3 in the form of NH4+, as well as oxidation products of nitrogen, such as nitrite (NO2−) and nitrate (NO3−) ions. Therefore, the plasma-based synthesis of reactive oxygen species (ROS) and reactive nitrogen species (RNS) in water is often referred to as plasma-treated water (PTW) or plasma-activated water (PAW).90
There is no general agreement on the reaction locus for NOx formation in the presence of H2O. Opinions are divided among the gas phase, plasma–liquid interface, and liquid phase. It is well known that an increase in NF species production rate can be achieved by increasing the plasma–liquid surface area.91 Considering this, designs with an increasing interface surface have been intensively studied: aerosol droplets,92 water film, and vapour.91,93 The source of the reactive species responsible for plasma–liquid chemistry is still under debate. In the case of plasma jets interacting with liquid water, the contradictory findings by various research groups indicate that there may or may not be a direct interaction of plasma with molecules of liquid water. The formation of NHx (which will be discussed in Section 3.2) and NOx species by N2-containing plasma over water was suggested to occur (1) in the gas phase from the evaporated water92,93 or (2) through direct interaction of plasma species with the top layer molecules of liquid H2O.94,95 Thus, the nature of plasma interaction with liquid likely depends on the specific plasma–liquid system, including the properties of the gaseous discharge and the geometry of the plasma–liquid interface.
Overall, conclusions about the reaction locus strongly depend on the reactor arrangement and the way water is introduced. Given that NF chemistry in the presence of H2O is still under debate, it will be discussed considering distinguishable cases: (i) water in the gas phase, (ii) plasma–liquid interface, and (iii) in the bulk.
Fig. 2b depicts the NF oxidation chemistry in the presence of water, underlying the changes in the chemistry compared to the dry N2:O2 case in color. For both N2:O2 + H2O and N2 + H2O systems, the feedstock activation stage is the same, and H2O homolytically dissociates into H and OH ((R3.13), see Table 4) under the electron impact and reactions with electronically excited states of  (and O*, in the case of N2:O2 + H2O, (R3.15) and (R3.14), respectively).96,97 Photodissociation of H2O (R3.16) may also take place in plasmas, especially when using high E/n values, such as in the case of corona or DBD.98 Furthermore, the full dissociation of H2O into H and O may also be considered following reaction (R3.17). However, the probability of this latter process is likely small because of two main reasons. Firstly, it involves a two-step process: two electron impact reactions (R3.13) and (R3.17) or heavy-particle (R3.14) or (R3.15) and electron impact (R3.17). Secondly, the reaction rate will be determined by the concentration of reactants, especially OH and electrons, which may be in limited supply. The concentration of the latter strongly depends on the plasma type (ne(DBD) ≪ 1021 m−3, and ne(spark) < 1024 m−3, at atmospheric pressure conditions).
(and O*, in the case of N2:O2 + H2O, (R3.15) and (R3.14), respectively).96,97 Photodissociation of H2O (R3.16) may also take place in plasmas, especially when using high E/n values, such as in the case of corona or DBD.98 Furthermore, the full dissociation of H2O into H and O may also be considered following reaction (R3.17). However, the probability of this latter process is likely small because of two main reasons. Firstly, it involves a two-step process: two electron impact reactions (R3.13) and (R3.17) or heavy-particle (R3.14) or (R3.15) and electron impact (R3.17). Secondly, the reaction rate will be determined by the concentration of reactants, especially OH and electrons, which may be in limited supply. The concentration of the latter strongly depends on the plasma type (ne(DBD) ≪ 1021 m−3, and ne(spark) < 1024 m−3, at atmospheric pressure conditions).
In the gas phase, NO formation should dominantly follow the reactions (R3.8), (R3.9) and (R3.18). However, in the N2 + H2O gas medium, the precursor formation process (R3.9) is significantly suppressed due to the lack of O2 in the gas mixture but can be replaced through (R3.18), involving OH radicals, as highlighted in Fig. 2b. The effect of the latter on NO formation in the gas phase has been shown through experiments in many studies.62,81,92,97,99–102
In addition to (R3.10) and (R3.11), further N oxidation processes can be enhanced by the reactions (R3.19)–(R3.23). In the N2 + H2O gas mixture, due to a shortage of O/O2, the selectivity of stable products is strongly shifted towards HNO2 formation because further N oxidation is mostly governed by OH radicals (R3.21).103 Several important points should be noted here. Due to its high reactivity, among different products of the NF process, HNO cannot be isolated as a product. It reacts rapidly with other molecules or undergoes decomposition into simpler compounds. Therefore, HNO is primarily encountered as an intermediate in chemical reactions. Nitric acid (HNO3) formation in the gas phase can occur only under very specific conditions, such as low pressure and temperature. At atmospheric pressure in the gas phase, HNO3 can be formed as OH + NO2 + M → HNO3 + M with k(298 K) = 4.75 × 10−11 cm3 s−1 (reduced to the second-order reaction),80 which decreases with temperature, slowing it down. Meanwhile, one of the major HNO3 removal processes, namely OH + HNO3 → H2O + NO2, has k(298 K) = 2 × 10−13 cm3 s−180 and exponentially increases with temperature. Therefore, HNO3 is unstable and prone to decompose, ending up as NO2. The same effect can take place with NO3, whose removal processes, namely NO3 + O → O2 + NO2 and NO + NO3 → NO2 + NO2, are faster than the formation ones.
The NF chemistry at the plasma/liquid interface remains unclear and has yet to be completely revealed. The lack of systematic studies is the primary reason for this uncertainty. Most research focuses on the behavior of a few radicals, neglecting the full range of possibilities. Additionally, studies of chemical pathways are often lacking due to the absence of thorough computational chemical modeling. The lack of necessary reaction rate constants (or their high uncertainty), as well as the difficulty in matching three different phases where water exists in different states (gas phase – vapor, plasma/liquid interface – mist, liquid phase – water bulk), are major obstacles preventing the development of a chemical kinetics model for plasma–liquid systems. While some works address the chemistry in such systems, focusing on gas chemistry, plasma/liquid interface chemistry, and liquid chemistry separately, much more research is needed for further advancements.104,105
Fig. 2c shows N2 oxidation chemistry in the plasma/liquid interface. Due to the high concentration of gaseous H2O at the plasma/liquid interface region, its activation following reactions (R3.13)–(R3.17) becomes more significant than in the gas phase, increasing OH concentration. Moreover, this activation can significantly enhance the production of reactive species such as hydroxyl peroxide (H2O2) and hydroperoxyl radical (HO2) via reactions (R3.24)–(R3.27). The latter opens an additional pathway for precursor formation, namely NO (R3.28). Finally, the abundance of OH, H2O2, and HO2 can further boost HNO2 formation (R3.30) and, more importantly, facilitate the formation of HNO3 through reactions (R3.31)–(R3.33) at the plasma/liquid interface. Besides these often addressed reactions, the possibility of other reactions, such as NO + OH−, is unknown, requiring further investigation.98
The plasma/liquid interface primarily serves as the site for solvation reactions, following Henry's law. The solubility of (H)NOx components, arranged from weakest to strongest, follows NO → NO2 → NO3 → HNO2 → HNO3. Considering the high Henry's constants of HNO3 and NO3, the plasma/liquid interface can indeed be regarded as the crucial site for their formation (R3.29), (R3.31)–(R3.33), where they are initially generated and then solvated.
The products of N2 oxidation in liquids are NO2− and NO3− ions. Liquid-phase chemistry in this context has been extensively researched, with numerous reviews available on this topic.98,106–108 However, uncertainty persists regarding the reaction pathways that lead to the formation of these products. The liquid phase becomes increasingly dominated by NO2− in plasmas with high concentrations of HNO2 and NO in the gas phase, achievable in N2 + H2O and dry N2:O2 feedstocks, respectively. Meanwhile, the selectivity shift toward NO3− is pronounced in plasmas with large plasma/liquid interface area, due to the elevated solubility of HNO3 and NO3 and at increased concentrations of NO2. The latter, high NO2 concentrations, is most pronounced in dry N2:O2 plasmas, reaching 50–70% selectivity, and can be further enhanced up to 100% by introducing an additional oxidation source, such as ozone, or performing the synthesis at elevated pressure.50,109
In line with the objective of this work, focusing on plasma-based NF for fertilizer production, it is imperative to maintain a low concentration of NO2− in the output solution due to its detrimental effects on plants. Elevated NO2− levels can disrupt various cellular processes and inhibit essential enzymes, leading to oxidative stress, chlorosis (yellowing of leaves), and even plant death. From a chemical perspective, the most efficient method to convert NO2− present in the output into NO3− is through reaction with H2O2. The latter can be effectively generated in plasmas with electronic excitation at low gas temperatures. In warm plasmas, such as GA (E/n < 100 Td), the high temperature inhibits H2O2 generation. In such cases, NO2− can be converted into NO3− by bubbling O2 gas through the liquid.
To summarize, plasma-based nitrogen oxidation is typically carried out in a dry N2:O2 (air) atmosphere, resulting in NOx production, or N2:O2 (air) + H2O or N2 + H2O atmospheres, leading to the formation of (H)NOx. It is important to note that in the latter cases, product formation occurs in both gas and liquid phases, which must be considered during analysis. Using N2 + H2O feedstock represents mainly a scientific interest in determining the chemical reaction pathways due to the high cost of pure N2. Similar to the Oswald process (R1.2)–(R1.5), the ultimate aim of plasma-based nitrogen oxidation for fertilizer production is to generate HNO3. This is achievable only if HNO3 is preserved in the liquid phase as NO3−, making the selectivity of its generation crucial. The most efficient performance in terms of EC is attainable in plasmas with significant vibrational excitation of N2, as shown in Table 6. However, while the presence of the liquid phase is essential for the accumulation of NO3−, the plasma–liquid interface can adversely affect the NF process from a physical standpoint. Firstly, the presence of water vapor can diminish vibrational excitation, thereby hindering the activation of N2 molecules.78,110 Secondly, the activation of water (R3.13)–(R3.17) is energy-intensive, diverting electron energy away from NF, specifically N2 activation. Finally, the presence of a plasma–liquid interface involves the transfer of H2O from the liquid to the gas phase (evaporation), where electrons can react with it, further increasing the energy demands of the process. From a chemical kinetics standpoint, N2 oxidation in the presence of water can provide extra pathways for NOx generation and shift process selectivity towards HNOx formation. At the same time, the chemistry becomes diverse and more complicated, as visually seen in Fig. 2. The formation of numerous components in the system can lead to electron energy losses during their formation and subsequent excitation. Thus, N2 oxidation in the presence of a plasma–liquid interface can significantly alter process selectivity, while simultaneously leading to a notable reduction in process energy efficiency (by some estimates up to 20%).101
4.2 Plasma nitrogen fixation: reduction pathways
This section provides an overview of nitrogen reduction, focusing on the synthesis of NH3 (or NH4+ in the liquid medium). The process has been less explored due to several fundamental and practical challenges, which will be discussed in detail within each sub-section, focusing on nitrogen reduction in different feedstocks.NH3 synthesis in conditions of electrical discharge has been conducted utilizing different N- and H-containing feedstocks introduced as gases (N2, N2 + O2 (air), H2, H2O, CH4, alcohols) and liquids (H2O, alcohols). The presence of different phases does not bring consensus on the reaction mechanisms for NH3 formation. Opinions are divided between the liquid phase,111–114 gas phase,115,116 catalyst surface,116 or the gas/liquid111,114 interface, based on the focus of the research.
Generalizing, plasma-based NH3 formation chemistry follows similar basic steps as NOx generation: feedstock activation, formation of initial products, and further hydrogenation. In the context of N2 reduction, the use of a catalyst can significantly promote precursor formations via heterogeneous processes on the surface of catalysts through Eley–Rideal or Langmuir–Hinshelwood mechanisms.117 The latter is of particular interest because, in this case, the catalyst surface acts as a “trap” for activated species, followed by their reaction to form products and their later desorption from the surface.118 It is often hypothesized that a synergistic combination of a non-equilibrium plasma and catalysis affords higher reaction productivity than conventional thermal catalysis.118,119 However, the presence of a catalyst in the plasma zone can significantly impact the plasma properties, making plasma–catalytic processes interconnected, which is often not considered despite its crucial importance, as it strongly influences the plasma power density and, consequently, the chemistry and process efficiency.120,121
NH3 (or NH4+ in liquid) synthesis can be performed using different feedstocks. In the subsequent sections, the main reaction pathways leading to N2 reduction in various plasma forming gases are summarized.
| Nitrogen reduction in N2 + H2 mixtures | |
|---|---|
Feedstock (N
2
, H
2
) activation (N,
 ,N
2
+
, H, H*,
,N
2
+
, H, H*,
 )
)
|
|
| (R2.1)–(R2.10) (N2 activation, as described in Table 2) | |
| e + H2 → H + H* + e | (R3.34) |
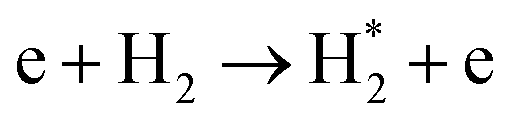
|
(R3.35) |
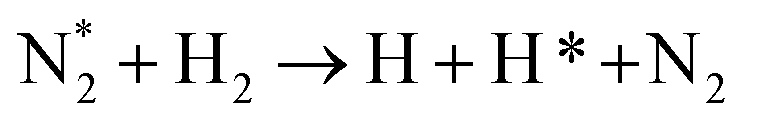
|
(R3.36) |
| Initial reduction of N | |
| N + H → NH | (R3.37) |
| N* + H2 → NH + H | (R3.38) |
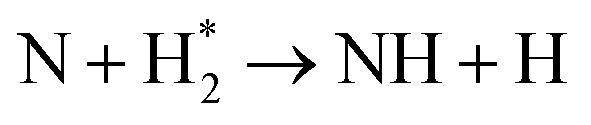
|
(R3.39) |
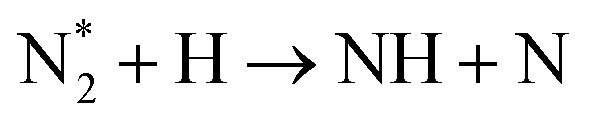
|
(R3.40) |
| Further hydrogenation | |
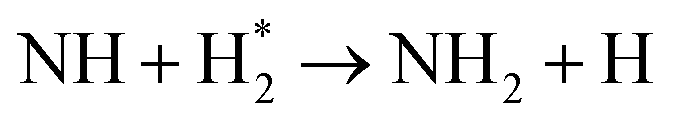
|
(R3.41) |
| NH + H → NH2 | (R3.42) |
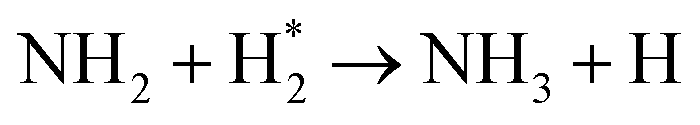
|
(R3.43) |
| NH2 + H → NH3 | (R3.44) |
| Nitrogen reduction in N2 + H2O mixtures | |
|---|---|
Feedstock (N
2
, H
2
O) activation (N, N*,
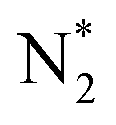 ,N
2
+
, H, H*,
,N
2
+
, H, H*,
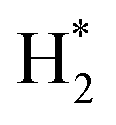 ,OH, H
2
O*)
,OH, H
2
O*)
|
|
| (R2.1)–(R2.10) (N2 activation, as described in Table 2) | |
| (R3.13)–(R3.17) (H2O activation, as described in Table 4) | |
| Initial reduction of N: (R3.37), (R3.40) | |
| N + OH → NH + O | (R3.45) |
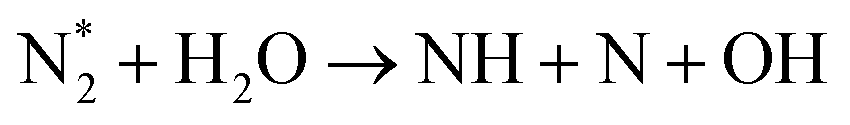
|
(R3.46) |
| Further hydrogenation: (R3.42), (R3.44) | |
| NH + OH → NH2 + O | (R3.47) |
| NH2 + OH → NH3 + O | (R3.48) |
| Decomposition reaction of NHx in the presence of water | |
| OH + NH → H + HNO | (R3.49) |
| OH + NH2 → H2O + NH | (R3.50) |
| OH + NH3 → H2O + NH2 | (R3.51) |
| Nitrogen reduction in N2 + CH4 mixtures | |
|---|---|
Feedstock (N
2
, CH
4
) activation (N, N*,
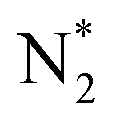 ,N
2
+
, H, H*,
,N
2
+
, H, H*,
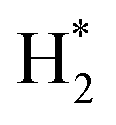 , CH
3
, CH
2
, CH, CH*)
, CH
3
, CH
2
, CH, CH*)
|
|
| (R2.1)–(R2.10) (N2 activation, as described in Table 2) | |
| e + CH4 → products (CH3,CH2,CH, H,H2,C) + e | (R3.52) |
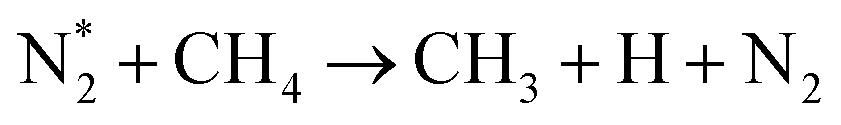
|
(R3.53) |
| Initial reduction of N: (R3.37) and (R3.40) | |
| N* + CHx → CHx−1 + NH | (R3.54) |
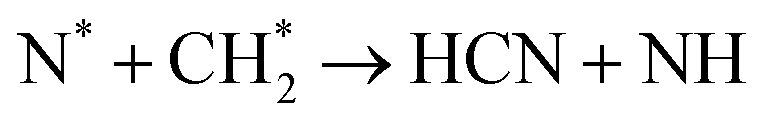
|
(R3.55) |
| N* + CH → C + NH | (R3.56) |
| Further hydrogenation: (R3.42) and (R3.44) | |
| CH4 + NH → CH3 + NH2 | (R3.57) |
| CH4 + NH2 → CH3 + NH3 | (R3.58) |
| HCN formation (dangerous but value-added chemical (NOT FOR NF)) | |
| CH3 + N → HCN + H2 | (R3.59) |
| CH2 + N → HCN + H | (R3.60) |
| Nitrogen reduction in N2 + EtOH mixtures | |
|---|---|
Feedstock (N
2
, C
2
H
5
OH) activation (N, N*,
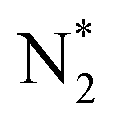 ,N
2
+
, H, H*,H
2
+
, CH
3
, CH
2
, CH, CH*)
,N
2
+
, H, H*,H
2
+
, CH
3
, CH
2
, CH, CH*)
|
|
| (R2.1)–(R2.10) (N2 activation, as described in Table 2) | |
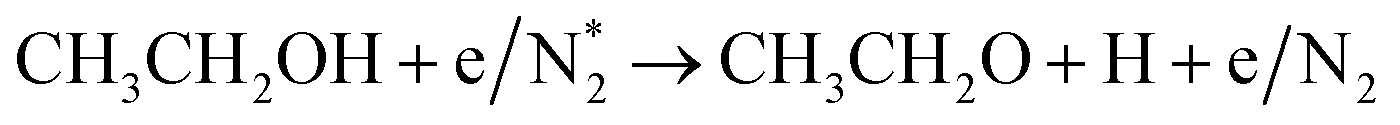
|
(R3.61) |
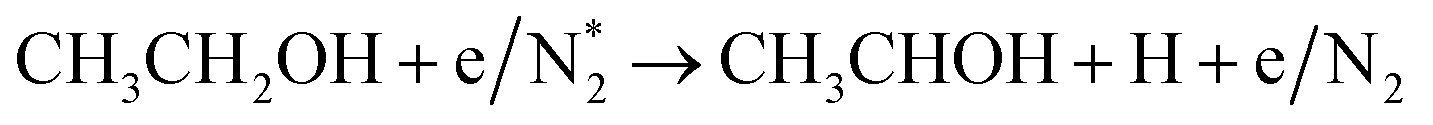
|
(R3.62) |
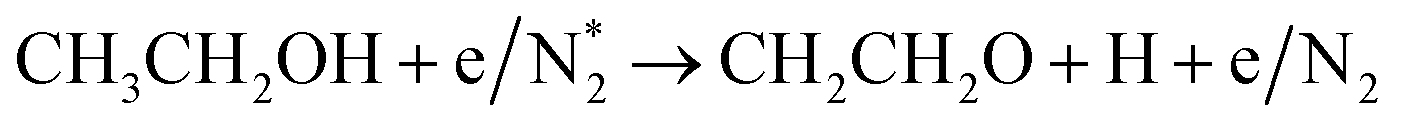
|
(R3.63) |
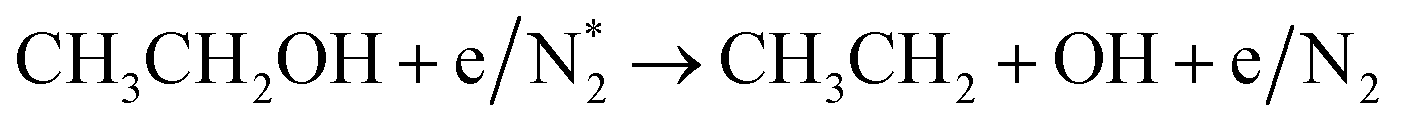
|
(R3.64) |
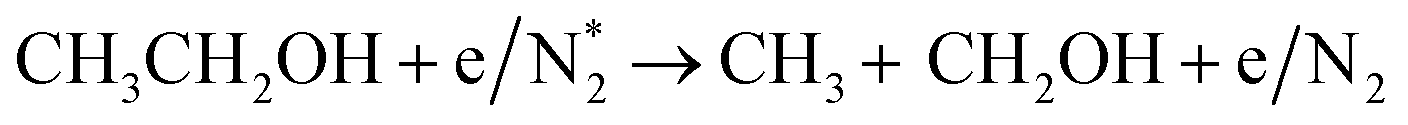
|
(R3.65) |
| Initial reduction of N: (R3.37), (R3.40), (R3.3.45) and (R3.54)–(R3.56) | |
| CH3CH2 + N* → NH + CH2CH2 | (R3.66) |
| CH3CH2OH + N* → NH + CH3CHOH | (R3.67) |
| Further hydrogenation: (R3.42), (R3.44), (R3.47), (R3.48), (R3.57) and (R3.58) | |
| CH2O + NH2 → HCO + NH3 | (R3.68) |
| Reverse processes in an N2 + CxHyOz system | |
| C2H5 + NH2 → C2H6 + NH | (R3.69) |
| CH3O + NH3 → CH3OH + NH2 | (R3.70) |
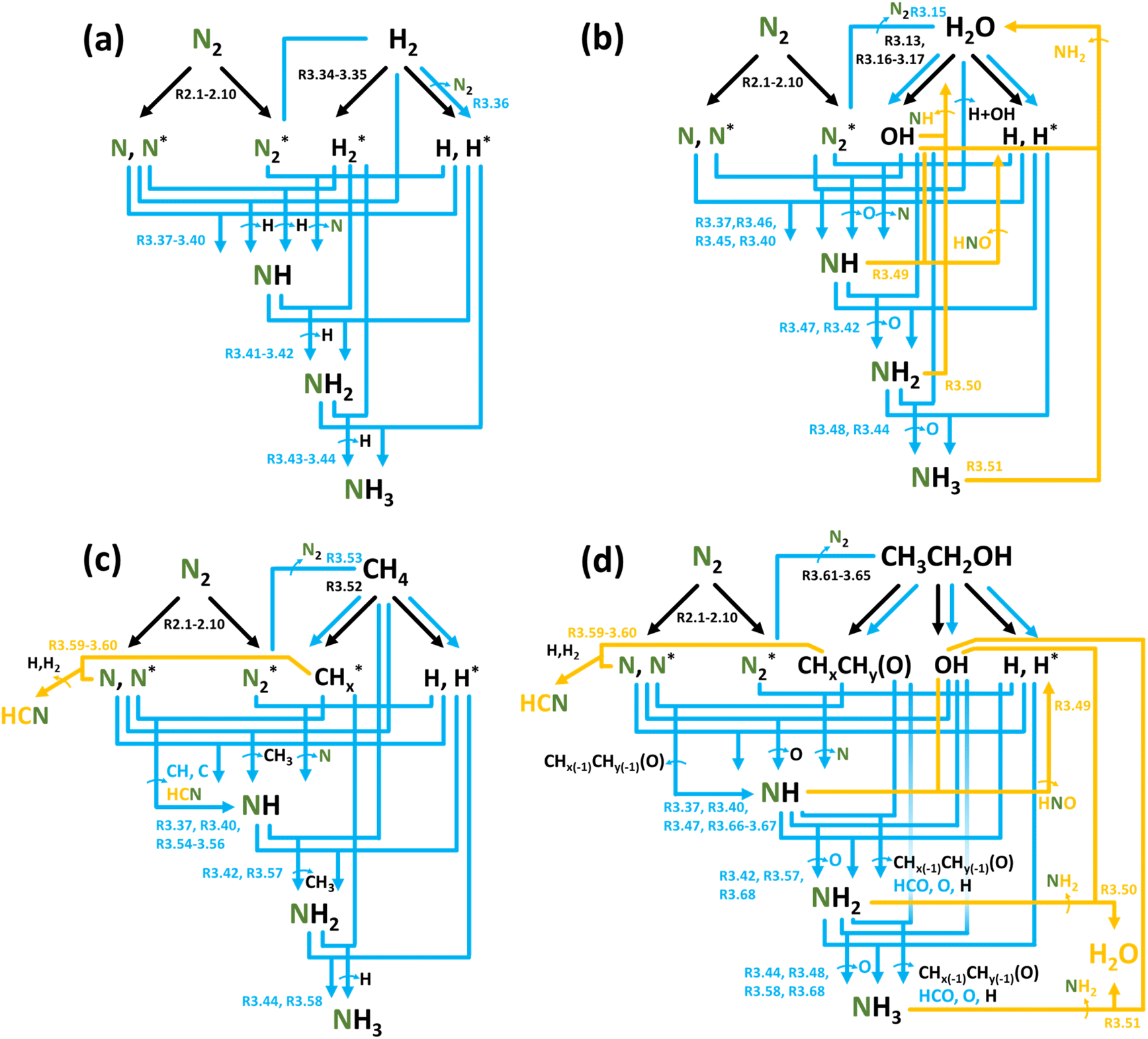 | ||
| Fig. 3 Reaction diagrams of the N2 reduction processes in (a) N2:H2, (b) N2 + H2O, (c) N2 + CH4, (d) and N2 + EtOH feedstocks. Black, blue, and yellow arrows indicate electron impact reactions, forward reactions, and reverse reactions, respectively. | ||
N2 reduction processes can be significantly reinforced using a combination of non-thermal plasmas and a catalyst. In this case, conceptually, the chemistry remains the same but is shifted onto the catalyst surface, improving reaction activity and selectivity. Hence, the aforementioned reactions, specifically activation, and further hydrogenation stages, dominantly take place in accordance with Eley–Rideal and Langmuir–Hinshelwood mechanisms (with the main mechanism depending on the nature of the active metal site126,127).
Plasma-based NH3 synthesis in an N2 + H2 gas mixture is of fundamental research interest because it contradicts the concept of plasma-based NF. The utilized gases, N2 and H2, are relatively expensive feedstocks, i.e., more expensive than simple air needed for NOx generation, as described in Section 3.1. Therefore, further utilization seems to be hindered by this.128 Nonetheless, such studies help to understand the chemistry of NH3 formation and assess the importance of the heterogeneous process on the catalyst surface. The overall performance of such systems in terms of EC [MJ mol−1] and PR [mg h−1] is concatenated in Table 6. Evidently, the use of a catalyst appears beneficial for process performance, allowing a decrease in EC and a significant gain in PR (note that PR for catalytic systems is reported in mg per h per gram of catalyst).
| Type of NF | Plasma type | Energy cost [MJ mol−1] | Production rate [mg h−1] | Employment details | ||
|---|---|---|---|---|---|---|
| NH3 | NOx | NH3 | NOx | |||
| a Production rates reported in mg per hour per g of a catalyst; “In liquid” or “in gas” highlights where the products were observed. Abbreviations “cat.” and “el.” stand for catalyst and electrolyte, respectively | ||||||
| N 2 + O 2 (air) | ||||||
| Oxidation | Pulsed DC spark83 | — | 0.4 | — | 300 | In air |
| DC glow129 | — | 2.8 | — | 1900 | In air | |
| Low-current coaxial plasmatron130 | — | 3.4 | — | 3000 | In air | |
| Thermal arc131 | — | 10.5 | — | 128000 |
In air | |
| Rotating GA50 | — | 2.3 | — | 32000 |
In air. 3 barg | |
| Rotating GA50 | — | 1.8 | — | 68900 |
N2:O2 1:1.3 barg |
|
|
||||||
| N 2 + H 2 | ||||||
| Reduction | DBD132 | 102 | 154 | No cat | ||
| DBD + cat133 | 437 | — | 8a | — | Ni/Al2O3 cat | |
| DBD + cat134 | 74 | — | 25.5a | — | Co–Ni/Al2O3 cat | |
| DBD + cat135 | 32 | — | 38a | — | Ru/Al2O3 cat | |
| DBD + cat136 | 58 | — | 75a | — | Ni–Mg/SBA-15-IWI cat | |
| DBD + cat121 | 80 | — | 40a | — | CoCe or CoLA cat | |
|
||||||
| N 2 + H 2 O | ||||||
| Reduction and oxidation | DBD jet + H2O droplets92 | 7854 | 3010 | 0.2 | 3.1 | In liquid |
| GA + above H2O69 | 2601 | 1579 | 0.6 | 6.1 | In liquid | |
| Spark jet + above water93 | 52 | 187 | 0.2 | In liquid | ||
| Spark jet + H2O vapour103 | 771 | 73 | 0.2 | 2.3 | In gas phase | |
|
||||||
| N 2 + C x H y O z | ||||||
| Reduction and oxidation | Packed bed-DBD in N2+CH465 |
2.7 | — | 5210 | — | Borosilicate glass beds |
| N2 jet above H2O + EtOH113 | 352 | 9534 | 52 | 5.3 | In liquid 20 vol% aqueous EtOH solution | |
|
||||||
| N 2 + O 2 (air) + H 2 O | ||||||
| Oxidation and reduction | Plasma electrolytic system137 | 14.1 | 8.6 | 3 | — | 0.1 M PBS with 500 ppm NO2− |
| Hybrid plasma-electrocatalytic system138 | 15.5 | 13.7 | 23.2 | 0.3 | In liquid, Cu nanowires cat | |
| Hybrid plasma- electrocatalytic system139 | — | 117 | 3 | 69 | In liquid, LaFeO3 cat., 0.1 M KOH el | |
| Hybrid plasma- electrocatalytic system140 | 40.5 | 40.2 | 39.6 | 5.9 | In liquid, Co3O4 nanoparticles cat., 0.11 M NOx− + 1 M NaOH el | |
| Hybrid plasma- electrocatalytic system141 | 3.1 | 2.4 | 3 | 1 | In liquid Co Sas/N–C cat. 0.1 M KOH el | |
As mentioned above, for systems with H2O, the reaction locus is a subject of debate and likely depends on a specific plasma–liquid configuration. Still, in the case of systems comprised of plasma interacting with a liquid interface, extracting H from the liquid surface is considered the rate-limiting step for NH3 formation in plasma–liquid systems.91 To overcome this limitation, an additional activation mechanism via ultraviolet (UV) radiation of the water surface is a way used to increase the available H+ at the plasma/liquid interface through H2Oaq → Haq+ + OHaq− (120–170 nm, (R3.16) in Table 4), resulting in a more efficient hydrogenation. It can increase NH4+ formation (up to 4 fold) compared to conditions without UV.91,95,113
Unfortunately, although N2 + H2O plasma systems are characterized by a rich environment with a variety of reactive species, namely additional OH and H2O2, these species also have negative effects on NH3 or NH4+ generation from a physical and chemical point of view. First of all, the presence of water in the plasma zone initiates the quenching of vibrationally excited N2(X, v) and electronically excited N2(E).78,99,110 This decreases the N2 activation efficiency. Secondly, at non-thermal conditions, OH is “wasted” on H2O2 generation instead of contributing to NH3 production, while at thermal conditions, OH radicals can trigger reverse reactions ((R3.49)–(R3.51), Table 5), which negatively affects NH and NH2 generation.96 Furthermore, even assuming the complete two-step dissociation of H2O into H + H + O ((R3.13)–(R3.15) and (R3.17)) from which both H react to form NHx, the required O–H bond dissociation energy in H2O is 4.81 eV (464 kJ mol−1), whereas in H2 it is 4.52 eV (436 kJ mol−1). This means that to produce the same amount of NH3, more energy is inherently required when using H2O instead of H2. This is clearly seen when looking at the performance of N2 + H2O plasma systems with respect to their N2 + H2 counterparts presented in Table 6, where the EC is two orders of magnitude higher, and the PR is two orders of magnitude lower for N2 + H2O systems.
Interestingly, N2 + H2O reduction with H2O vapor in the presence of a catalyst in a DBD plasma can reduce the contribution of the oxidation pathway and increase the contribution of the reduction pathway.143 However, the practical importance of these systems for N2 reduction is low due to the high EC and low PR.128
NH3 formation in hydrocarbon-containing gas mixtures or plasma/liquid systems is poorly investigated but has recently received considerable scientific attention. The major problem lies in the low process selectivity towards NH3 due to the complexity of plasma-initiated reactions with the involvement of numerous highly reactive species forming various side products of low importance for NF.146
Evidently, the reaction set expands in the presence of CH4 as shown in Fig. 3c and Table 5, and the plasma-induced chemistry of CH4 activation, (R3.52) and (R3.53), can be strongly affected by a variety of parameters: pressure, reactor geometry, and the type of plasma. In plasmas of high E/n value, like a DBD, CH4 activation mainly occurs via electron impact reactions and interaction with electronically excited heavy particles. In “warm” plasmas of medium-range E/n, such as a GA, thermal dissociation of CH4 is expected to be predominant 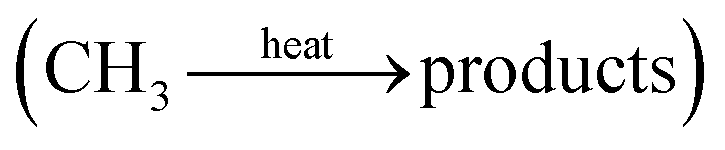 . However, under high-temperature conditions, NH3 formation is highly unlikely, and thus, it is expected to occur only in the lower-temperature plasma compartments, e.g., in the afterglow zone.
. However, under high-temperature conditions, NH3 formation is highly unlikely, and thus, it is expected to occur only in the lower-temperature plasma compartments, e.g., in the afterglow zone.
Another significant feature of N2 + CH4 plasmas is the formation of hydrogen cyanide (HCN), e.g. (R3.55), (R3.59) and (R3.60).147,148 HCN is extremely toxic and poses a significant risk of rapid harm. Its concentration in N2 + CH4 plasmas can be comparable to or even higher than the concentration of NH3.149 Consequently, stringent safety measures are essential when dealing with such an NF method. Nonetheless, HCN is also a value-added chemical, and its synthesis is a topic of many studies.150
Alcohols, including EtOH can also be potentially utilized for NH3 production (see Fig. 3d and Table 5 for details), being introduced into the plasma zone through various means such as vapor, steam, or liquid interface. In plasma, it undergoes activation via electron impact and N2 excited states (R3.61)–(R3.65) and consequent product recombination, yielding a range of reactive species, including CH3, H, OH, H2, CH3CHOH, etc., ultimately resulting in the production of H2, CO, CO2, NH3, C, and other products.151–153 The selectivity towards particular species is the subject of numerous studies and is significantly influenced by experimental conditions (pressure, gas mixture composition), plasma type (non-thermal, warm, thermal), and the presence of different phases (gas, liquid, solid (catalyst)). Some of the possible routes of NH3 formation specific to ethanol are described below. Given the low evaporation temperature of EtOH (351 K at atmospheric pressure), the gas phase is always enriched by gaseous EtOH. Considering this, thermal activation mechanisms must also be considered 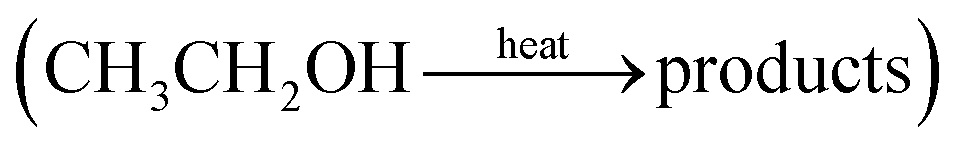 .154 Overall, exposing plasma above liquid EtOH-water mixtures can enhance the production rate of NH4+. This is generally attributed to the increased lability of the H atom and OH radical at the central CH2 group (R3.61)–(R3.64).113
.154 Overall, exposing plasma above liquid EtOH-water mixtures can enhance the production rate of NH4+. This is generally attributed to the increased lability of the H atom and OH radical at the central CH2 group (R3.61)–(R3.64).113
To summarize, plasma-based NH3 synthesis in N2 + CxHyOz feedstock systems is feasible, but the process selectivity strongly depends on the type of plasma. Under non-thermal and strongly non-equilibrium conditions, where activation primarily occurs through non-elastic collisions (as in DBDs), NH3 can be generated in significant quantities in the gas phase for the N2 + CH4 system and promoted in the liquid phase (comparing N2 + H2O and N2 + H2O + EtOH), as illustrated in Table 6. However, under elevated temperature conditions (such as GA nd MW), the selectivity shifts towards the generation of H2, olefins, and C (soot). This shift is likely due to reverse processes with highly temperature-dependent reaction rate constants, e.g., (R3.69) and (R3.70), and NH3 thermal decomposition.
To conclude this section, NH3 formation chemistry across various feedstocks, including N2 + H2, N2 + H2O, N2 + CxHyOz, and N2 + O2(air) + H2O is also examined. The proposed reaction schemes offer simplified representations, enabling an understanding of fundamental mechanisms. However, plasma-induced chemistry poses prediction challenges, involving hundreds to thousands of chemical reactions. Nonetheless, analysis suggests that process selectivity is influenced by feedstock activation; the more energy demanding the activation, the higher the EC and the lower the PR. Furthermore, the presence of numerous active species complicates the chemistry, presenting both advantages and disadvantages. This includes the potential for enhanced initial reduction pathways (to form NH3), as well as the risk of reverse processes, especially pronounced at elevated temperatures.
5 From plasma nitrogen fixation to NH4NO3 fertilizer
In general, NF processes, including plasma-based approaches, do not directly produce ready-to-use fertilizers but instead generate precursors, or “building blocks”, such as NH3 (see Section 3.2) or (H)NOx (see Section 3.1). These core materials serve as essential components in the subsequent synthesis of nitrogen-rich fertilizers that can effectively enhance soil nitrogen content for plant growth. However, due to the wide variety of plant nutrient requirements, this discussion focuses exclusively on nitrogen-based fertilizers. Fertilizers designed to supply other essential elements, such as phosphorus (P) or potassium (K), fall outside the scope of this review.This subsection aims to provide a broad perspective on the development of plasma-based fertilizer technologies, focusing on general scientific and technological principles rather than delving into economic feasibility or industrial-scale implementation. By narrowing the scope, this part serves as a foundational introduction to the challenges and opportunities in plasma-based fertilizer production, catering specifically to readers who are new to the field. It offers a conceptual framework to understand how plasma technologies can contribute to advancing sustainable agriculture while leaving more detailed discussions on economics and industrialization to specialized studies. For newcomers, this serves as an entry point into the intricate problematics of plasma-based fertilizer synthesis, setting the stage for more in-depth exploration in future studies.
5.1 Targeted fertilizer
The wide variety of synthetic fertilizers employed nowadays ranges from ammonium phosphates or sulphates (where N is present in the cationic form) to nitrates (with N in the anionic form, or both in cationic and anionic form, i.e., NH4NO3) and even organic molecules such as urea (Fig. 4). As already discussed in Section 1, urea, although possessing the highest N content, compromises the decarbonization strategy due to the unavoidable CO2 emissions when being used.156 Of all other fertilizers, ammonium nitrate has the highest N content and is carbon-free. Thus, it is chosen as the target compound in this work to illustrate a plasma-based approach in fertilizer production.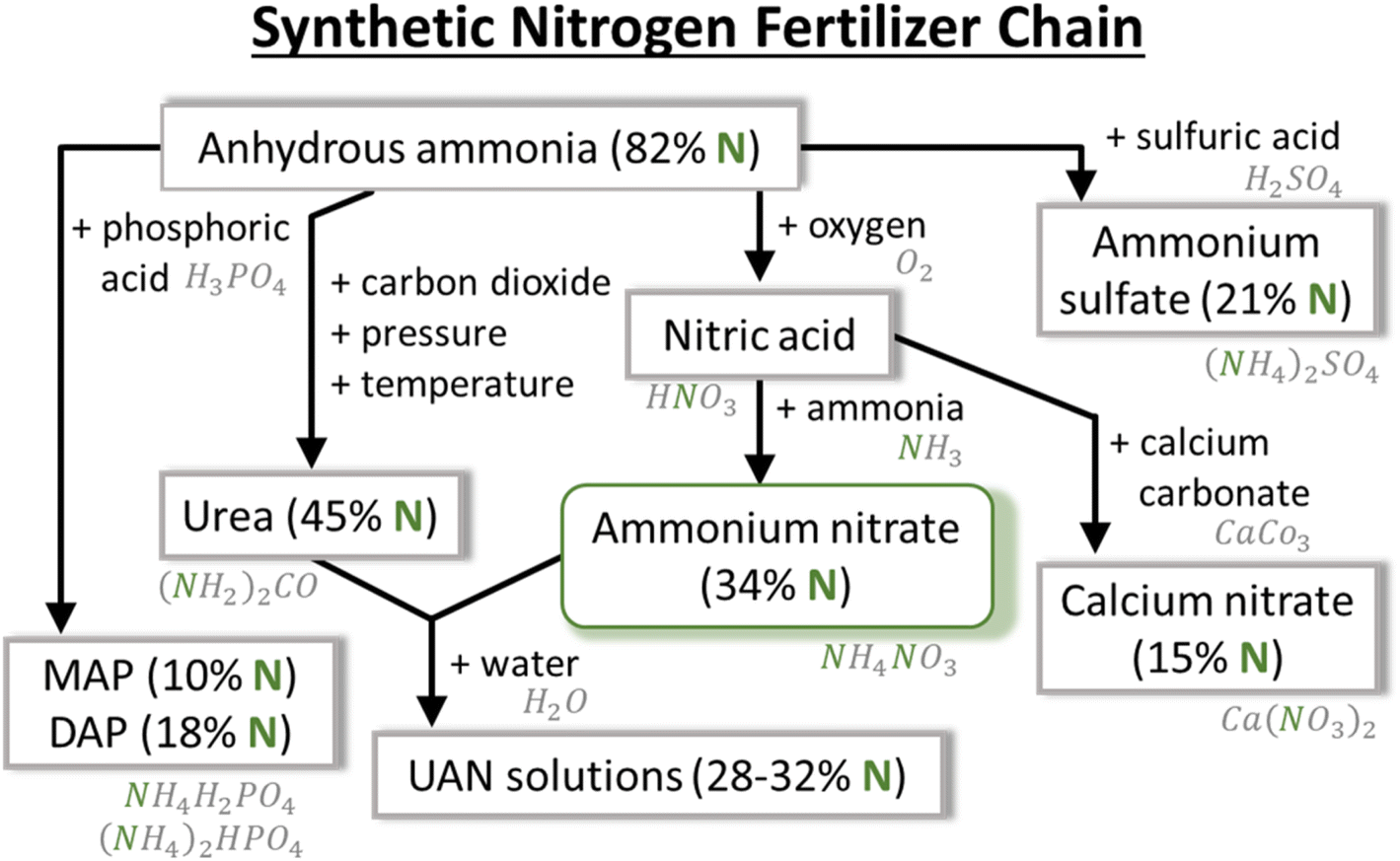 | ||
| Fig. 4 Commonly used fertilizers, their production pathways and conditions, and nitrogen content. | ||
In Sections 3.1 and 3.2, the plasma-driven chemical processes involved in N2 oxidation and reduction in various reactive systems, with and without O2, H2O, etc., were detailed. However, neither of the two processes (oxidation and reduction) individually leads to the desired fertilizer—NH4NO3.
5.2 Possible pathways of NH4NO3 synthesis involving electrified plasma technology
Here, we will focus on the ways of obtaining NH4NO3, which involve plasma NF to substitute, or at least reduce the contribution of the industry-dominating HB. So far, the majority of the researchers have focused on improving NOx and NH3 synthesis separately to achieve the best process performance in terms of lowest EC and highest PR. Often, these two parameters are counter-effective: the lowest EC is accompanied by low PR and vice versa, with a proper balance yet to be found. This bottleneck must be solved to achieve a wide application of this technology.Let us first consider several pathways of NH4NO3 production involving plasma NF. Considering the demands of the agricultural sector and the capacity of plasma technology to facilitate the synthesis of both HNO3 and NH3, the in-place production of NH4NO3 can be accomplished if “green” energy supplies are available directly on the farm field. For the NH4NO3 production, the process can be subdivided into two steps, namely (i) HNO3 and (ii) NH3 generation. Techno-economic analysis of the approach has been carried out recently, showing importance of energy costs of NF in the overall value chain of fertilizers synthesis.157
Naturally, the most direct alternative to HB is the direct synthesis of NH3 and its subsequent partial oxidation to HNO3, followed by combining NH3 and HNO3 into NH4NO3 as the ultimate output product (Fig. 5a). As discussed in Section 3, the route using C-containing H sources is not feasible in the long term within the new decarbonization policies. Therefore, the reduction agents are limited to H2 and H2O. Furthermore, the choice of plasma type is also restricted to highly non-equilibrium, non-thermal plasmas – e.g., DBD. However, DBD plasmas, even in combination with catalysts, typically have a low throughput and high EC. The EC usually belongs in the range of 10–100 MJ mol−1 (Table 6), while the estimated requirement for plasma NF to be competitive with HB is 1–1.5 MJ mol−1.17,18,128 Moreover, since NH3 cannot be used as a fertilizer as-is, it would need to be further oxidized into HNO3. Although distillation of N2 from the air is not strictly needed because NH3 can also be produced by plasma from the air with, e.g., H2O as the H source, the EC and PR values remain below the required levels. Thus, such a route is not the most feasible.
 | ||
| Fig. 5 Pathways of NH4NO3 production via plasma-based NF: (a) plasma production of NH3 coupled with its subsequent (partial) oxidation to HNO3; (b) plasma production of (H)NOx coupled with plasma-based production of NH3; (c) plasma production of (H)NOx coupled with its subsequent (partial) reduction to NH3. | ||
An alternative route involves the production of NH3via the aforementioned plasma N2 reduction and (separately) production of NOx/HNOx, which can later be combined with NH3 to produce NH4NO3 (Fig. 5b). Here, the production of NOx can be done from air in direct plasma N2 oxidation. This oxidative plasma NF can be performed in a variety of ways, which are described in detail in Section 3.1 and can be implemented in non-equilibrium plasmas such as GA, DC arc, and MW. However, it should be emphasized that plasma N2 oxidation of dry air in more thermal plasmas has the lowest EC and the highest PR of all reported routes of plasma-based NF (∼2.3 MJ mol−1, ∼3.2 g h−1, Table 6).50,131 Moreover, the data presented in Table 6 for N2 + O2 systems is for systems with air (N2:O2 ratio 4:1), whereas with oxygen-enriched mixtures, the values of EC and PR can be further improved,42,50 although at the EC of oxygen production. The plasma-formed NOx can be further dissolved in H2O, which, in the presence of unreacted O2 from the air, results ultimately in an HNO3 solution. Here, two other points must be also addressed. Although technically, HNOx can be formed directly in the liquid phase when creating direct contact between liquid H2O and plasma, part of the energy is inevitably lost to the evaporation of H2O and the quenching of excited N2, which has a negative impact on the EC.101,158 Thus, although this route has a higher electrification potential due to the strong feasibility of using plasma for N2 oxidation than the one described in Fig. 5a,direct plasma-based N2 reduction is still a limiting factor.
A plasma-based pathway to NH4NO3, which does not involve plasma N2 reduction, is shown in Fig. 5c. Here, the only substrate is air, without the need for distillation of N2. Air is used to produce NOx using warm (or thermal) plasmas, which have significantly higher throughput and lower EC than non-thermal (e.g., DBD) plasmas. After this, the NOx needs to be reduced to NH3. One of the ways proposed in the literature is thermocatalytic over-reduction of NOx over lean NOx trap catalysts commonly used in the automotive industry in the presence of H2.24,159 Such a process does not require a separation step because the catalyst acts as a NOx adsorber itself. However, it does require producing H2 first, either from CH4 (again, compromising decarbonization) or from the electrolysis of H2O, and a heat source to activate the thermocatalytic reaction.24 Although a very low EC was reported for such a combined plasma oxidation/thermocatalytic reduction process, this EC is accompanied by a low PR (2.1 MJ mol−1, 17.3 mg h−1; Table 6), and the activity range may be limited to low concentrations of the initially plasma-produced NOx.
Here, it is acknowledged that if the goal is fertilizers without producing NH4NO3 specifically, the plasma N2 oxidation step is nearly sufficient to produce nitrates, which already possess fertilizer properties. However, to turn them from acidic solutions into applicable fertilizers, neutralization is still required, e.g., with caustic soda or other chemicals. This, together with the increased N content of NH4NO3, makes it a more desirable product.
5.3 From plasma oxidation to electrochemical reduction
Electrochemical reduction of the plasma-produced NOx is another alternative way to NH3.38,118,160 The electrochemical reduction is normally carried out in a two-compartment H-type cell, where an ion-exchange membrane separates both compartments.161 The cell is typically equipped with a three-electrode configuration: a working electrode, where the reduction occurs; a counter electrode, which provides the current needed for the reaction; and a reference electrode, which monitors the electrode potential. A catalyst, often in the form of nanoparticles or single atoms, is immobilized on the surface of the working electrode or dispersed within the electrolyte solution. It serves to lower the activation energy barrier for the reduction reactions, thereby increasing the reaction rate and improving selectivity. A typical performance of hybrid plasma-electrocatalytic systems is presented in Table 6. The EC in such systems is the sum of N2 oxidation and N2 reduction steps, performed in series via plasma and electrochemical approaches, respectively. The electrocatalytic NOx into NH3 process illustrates the best state-of-the-art EC values, ranging between 3–15 MJ per mol NH3, and reasonable PRs for laboratory scale systems.Electrochemical reduction reactions are well-known and do not specifically involve plasma chemistry. Therefore, these are not addressed here. However, it should be noted that although HNO3 in an aqueous solution can be reduced to NH3, the reduction of HNO2 occurs faster.137 The ratio of HNO2/HNO3 in the water after passing the plasma exhaust gasses through it depends on the initial ratio of NO/NO2 produced during plasma N2 oxidation and can be controlled by varying plasma parameters. Thus, several concepts can be proposed. In one, the plasma-produced NOx mixture is dominated by NO2, which, accompanied by continuous recirculation through a water solution with O2 from the air, forms NO3− in the solution. Without any separation steps, this NO3− is further partially reduced electrochemically to NH4+, in the same solution. Such one-pot synthesis is the simplest on-site fertilizer production concept, utilizing only renewable energy (plasma and electrochemical cells can be powered by, e.g., solar panels) and ubiquitous materials such as air and water. On the other hand, if the NOx−containing plasma output stream contains lower fractions of NO2, the liquid phase can be shifted to mostly NO2−, which requires less energy to be reduced to NH4+. To avoid dividing the NOx stream to produce HNO3 in one of them and HNO2-to-NH3 in another, one can envision that NO2− is first partially reduced to NH4+, and then the remaining NO2− in the solution is oxidized to NO3− by recirculating air through it. Of course, at this stage, the comparison between the two concepts is purely speculative: the exact values of EC and PR of the whole process need to be assessed in dedicated future works.
6 Conclusion and Outlook
This tutorial discusses the fundamentals of plasma-based nitrogen fixation for fertilizers as a sustainable alternative to conventional methods. Through this work, the reader can acquire knowledge about elementary processes in plasmas (Section 2), the role of vibrational excitation (Section 2), the chemistry of nitrogen oxidation (Section 3.1) and nitrogen reduction (Section 3.2) in various feedstocks, and how these processes can be integrated to produce the final product – a fertilizer (Section 4). Finally, we provide key conclusions, perspectives, and challenges.6.1 Plasma technology is a sustainable alternative
The nitrogen fertilizer industry aims to meet the demands of modern agriculture while being independent of fossil fuels due to related economic and ecological problems. Given these premises, plasma technology presents an attractive alternative for converting N2 into nitrogen-based fertilizers in a manner that aligns with current sustainability goals. However, implementing the plasma-based approach necessitates a reconsideration of the existing soil fertilization paradigm, specifically a pivotal shift from large-scale centralized production to on-site direct synthesis. This shift is highly challenging, but the potential collective benefits could be immense. In this approach, valuable nitrogen species (NO3− and/or NH4+) do not require separation or recycling, thereby reducing associated energy costs. Instead, they can be synthesized directly from inexpensive feedstock (air) using plasma oxidation or reduction processes, as described in previous sections, and applied shortly thereafter.6.2 Which type of plasma should be used?
So far, warm plasmas show the best EC and NOx yield. This is often attributed to a significant vibration excitation of N2. However, the contribution of vibrational states to NO production in such plasmas is still unclear and, based on modeling, often considered to be comparable with thermal N2 dissociation. Thus, an experimental study investigating the N2 vibrational excitation degree in atmospheric pressure air plasmas is of high interest and can provide insights into non-thermal plasma chemistry. The enhancement of vibrational excitation can be achieved by tailoring the applied voltage waveform, e.g., combining ns-pulses with DC bias and ns-pulses with RF or MW, leading to a unique unrivaled chemistry.6.3 Challenges in plasma-based reduction
In contrast to HB's industrial scale N2 processing to NH3, the plasma-initiated reduction route is less developed and studied. The underlying chemistry of reduction is based on the reaction of very short-lived intermediates, such as OH, NH, and N radicals, particles difficult to detect. The future direction of research should tackle the complex chemical kinetics behind N2 reduction to reveal the full potential of this route in NF.6.4 Complexities in liquid phase processes
Although water is recognized as a green source of hydrogen, the challenges of enhancing plasma nitrogen fixation become even greater when liquids are present in the process environment. A combined oxidation and reduction process can take place both in the gas and liquid phases, complicating pathway elucidation. Insight into mechanisms is lacking, and there is no consensus on the role of H2O. In addition, the transport of plasma species into the liquid is poorly understood.6.5 Plasma–catalytic processes
As a possibility in both reduction and oxidation routes in NF, the plasma-catalytic approach is an important step toward industrial applications. It is well known that catalysts cannot directly facilitate N2 dissociation under milder conditions due to the inherent stability of the nitrogen triple bond. However, plasma-based nitrogen oxidation can still be aided by promoting O2 dissociation via heterogeneous catalysts. Atomic O-enriched medium can positively contribute to NOx formation by boosting the Zeldovich mechanism that can take place on the surface of a catalyst. Despite foreseen benefits in plasma–catalytic processes, the best-performing catalysts have to be yet defined and tested in industry-relevant conditions.6.6 Perspective and challenges
Based on the current state-of-the-art, it has been indicated that the most attractive route to the on-site, small-scale, decentralized production of NH4NO3 for fertilizer applications is initial plasma oxidation directly from air, followed by accumulating the products in aqueous solutions (in the form of HNOx), and their subsequent electrochemical or catalytical reduction. Moreover, the oxidation and reduction steps can be time-differentiated to accommodate the difference in the production rate in each step. Another important feature of such a combined plasma oxidation/electrocatalytic reduction approach is that the resulting product, NH4NO3, is already in solution and ready to be applied on farming sites. In contrast, the centralized HB production implies the distribution of NH4NO3, which is done in solid form as the most concentrated method to reduce transport costs.162 However, this, in turn, means the necessity of storage facilities, which, in combination with the explosivity of NH4NO3, sometimes leads to hugely destructive accidents.163As a final remark, several other approaches to fertilizer production via plasma-based NF will be briefly mentioned. Many natural fertilizers (e.g., cattle and pig waste) can be effectively recycled. The manure contains N (2–8.1 g kg−1)164 with an NH4+ content of about 70%, which is released over time due to the high pH in the animal waste. This volatilization is estimated in the range of tens of kg per hectare of fertilized soil.165 Industrial-level attempts, including recent research in N2 applied, are made to reduce such NH3 losses by producing NOxvia plasma N2 oxidation from the air and further applying this NOx to create NH4NO3 in the waste slurry.166,167
Overall, plasma technology enables a large variety of approaches, directions, and possibilities in NF for fertilizers, making them compatible with the electrification and decarbonization policies. The NH4NO3 synthesis routes discussed in this work (see Section 4 for details) have a solid foundation and should be the focus of future investigations. Beyond fundamental scientific information, a detailed techno-economic analysis of the proposed approaches can reveal the potential capital costs of the product.17,18,168–171
Data availability
No experimental data were used in the preparation of this manuscript.Author contributions
M. Gromov: conceptualization, data curation, investigation, formal analysis, visualization, writing – original draft, writing – review & editing. Y. Gorbanev: conceptualization, data curation, visualization, writing – original draft, writing – review & editing. E. Vervloessem: investigation, formal analysis. R. Morent: funding acquisition, writing – review & editing. R. Snyders: funding acquisition, writing – review & editing. N. De Geyter: funding acquisition, writing – review & editing. A. Bogaerts: funding acquisition, writing – review & editing. A. Nikiforov: conceptualization, data curation, visualization, writing – original draft, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The graphical icons used in the creation of Fig. 1 are from free subscription to Icons8. com. This research was supported by the Excellence of Science FWO-FNRS project NITROPLASM (EOS ID 30505023), and the Fund for Scientific Research (FWO) Flanders Bioeconomy project (grant G0G2322N) funded by the European Union-NextGenerationEU.References
- V. Smil, Sci. Am., 1997, 277, 76–81 CrossRef CAS.
- A. H. Sheudzhen, V. T. Kurkaev and N. S. Kotlyarov, Agrochemistry: Textbook, Afisha, Moscow, 2nd edn, 2006, vol. 2 (In Russian) Search PubMed.
- D. E. Canfield, A. N. Glazer and P. G. Falkowski, Science, 2010, 330(1979), 192–196 CrossRef CAS PubMed.
- C. P. Chanway, R. Anand and H. Yang, in In Advances in Biology and Ecology of Nitrogen Fixation, InTechOpen, 2014 Search PubMed.
- B. Bergman, G. Sandh, S. Lin, J. Larsson and E. J. Carpenter, FEMS Microbiol. Rev., 2013, 37, 286–302 CrossRef CAS PubMed.
- M. M. M. Kuypers, H. K. Marchant and B. Kartal, Nat. Rev. Microbiol., 2018, 16, 263–276 CrossRef CAS PubMed.
- V. Smil, Ambio, 2002, 31, 126–131 CrossRef PubMed.
- J. N. Galloway and E. B. Cowling, Ambio, 2002, 31, 64–71 CrossRef PubMed.
- J. W. Erisman, M. A. Sutton, J. Galloway, Z. Klimont and W. Winiwarter, Nat. Geosci., 2008, 1, 636–639 CrossRef CAS.
- J. Humphreys, R. Lan and S. Tao, Adv. Energy Sustainability Res., 2021, 2, 2000043 CrossRef CAS.
- M. Kamphus, Nitrogen + Syngas, 2014, 328, 48–53 CAS.
- V. Hessel, G. Cravotto, P. Fitzpatrick, B. S. Patil, J. Lang and W. Bonrath, Chem. Eng. Process., 2013, 71, 19–30 CrossRef CAS.
- R. M. Nayak-Luke and R. Bañares-Alcántara, Energy Environ. Sci., 2020, 13, 2957–2966 RSC.
- B. S. Patil, Q. Wang, V. Hessel and J. Lang, Catal. Today, 2015, 256, 49–66 CrossRef CAS.
- J. Baltrusaitis, ACS Sustain. Chem. Eng., 2017, 5, 9527 CrossRef CAS.
- C. Smith, A. K. Hill and L. Torrente-Murciano, Energy Environ. Sci., 2020, 13, 331–344 RSC.
- K. H. R. Rouwenhorst, F. Jardali, A. Bogaerts and L. Lefferts, Energy Environ. Sci., 2021, 14, 2520–2534 RSC.
- K. H. R. Rouwenhorst, F. Jardali, A. Bogaerts and L. Lefferts, Energy Environ. Sci., 2023, 16, 6170–6173 RSC.
- L. R. Winter and J. G. Chen, Joule, 2021, 5, 300–315 CrossRef CAS.
- G. Hochman, A. S. Goldman, F. A. Felder, J. M. Mayer, A. J. M. Miller, P. L. Holland, L. A. Goldman, P. Manocha, Z. Song and S. Aleti, ACS Sustain. Chem. Eng., 2020, 8, 8938–8948 CrossRef CAS.
- W. Pittman, Z. Han, B. Harding, C. Rosas, J. Jiang, A. Pineda and M. S. Mannan, J. Hazard. Mater., 2014, 280, 472–477 CrossRef CAS PubMed.
- R. Ingels, D. Graves, S. Anderson and R. Koller, in Proceedings-International Fertiliser Society, International Fertiliser Society, 2015, pp. 1–28 Search PubMed.
- B. S. Patil, F. J. J. Peeters, G. J. van Rooij, J. A. Medrano, F. Gallucci, J. Lang, Q. Wang and V. Hessel, AIChE J., 2018, 64, 526–537 CrossRef CAS.
- L. Hollevoet, E. Vervloessem, Y. Gorbanev, A. Nikiforov, N. De Geyter, A. Bogaerts and J. A. Martens, ChemSusChem, 2022, 15, e202102526 CrossRef CAS PubMed.
- K. H. R. Rouwenhorst, P. M. Krzywda, N. E. Benes, G. Mul and L. Lefferts, Ullmann’s Encyclopedia of Industrial Chemistry, 2020, pp. 1–20 Search PubMed.
- B. L. Bodirsky, A. Popp, H. Lotze-Campen, J. P. Dietrich, S. Rolinski, I. Weindl, C. Schmitz, C. Müller, M. Bonsch and F. Humpenöder, Nat. Commun., 2014, 5, 3858 CrossRef CAS PubMed.
- P. M. Vitousek, J. D. Aber, R. W. Howarth, G. E. Likens, P. A. Matson, D. W. Schindler, W. H. Schlesinger and D. G. Tilman, Ecol. Appl., 1997, 7, 737–750 Search PubMed.
- Z. Li, Z. Zeng, D. Tian, J. Wang, Z. Fu, F. Zhang, R. Zhang, W. Chen, Y. Luo and S. Niu, Global Change Biol., 2020, 26, 4147–4157 CrossRef PubMed.
- N. Cherkasov, A. O. Ibhadon and P. Fitzpatrick, Chem. Eng. Process., 2015, 90, 24–33 CrossRef CAS.
- A. Nuntagij, C. de Lassus, D. Sayag and L. André, Biol. Wastes, 1989, 29, 43–61 CrossRef CAS.
- S. Gouda, R. G. Kerry, G. Das, S. Paramithiotis, H.-S. Shin and J. K. Patra, Microbiol. Res., 2018, 206, 131–140 CrossRef PubMed.
- A. O. Gezerman, KUI, med. kratica Kem. Ind., 2022, 71, 57–66 CAS.
- K. H. R. Rouwenhorst, S. Mani and L. Lefferts, ACS Sustain. Chem. Eng., 2022, 10, 1994–2000 CrossRef CAS.
- K. H. R. Rouwenhorst, Y. Engelmann, K. Van’t Veer, R. S. Postman, A. Bogaerts and L. Lefferts, Green Chem., 2020, 22, 6258–6287 RSC.
- V. D. Rusanov, A. A. Fridman and G. V. Sholin, Sov. Phys.-Usp., 1981, 24, 447–474 CrossRef.
- S. Huang, W. Lv, S. Bloszies, Q. Shi, X. Pan and Y. Zeng, Field Crops Res., 2016, 192, 118–125 CrossRef.
- H. Chen, D. Yuan, A. Wu, X. Lin and X. Li, Waste Dispos. Sustain. Energy, 2021, 201–207 CrossRef CAS PubMed.
- J. Zhang, X. Li, J. Zheng, M. Du, X. Wu, J. Song, C. Cheng, T. Li and W. Yang, Energy Convers. Manage., 2023, 293, 117482 CrossRef CAS.
- S. Li, J. Medrano Jimenez, V. Hessel and F. Gallucci, Processes, 2018, 6, 248 CrossRef CAS.
- P. Peng, P. Chen, C. Schiappacasse, N. Zhou, E. Anderson, D. Chen, J. Liu, Y. Cheng, R. Hatzenbeller, M. Addy, Y. Zhang, Y. Liu and R. Ruan, J. Cleaner Prod., 2018, 177, 597–609 CrossRef CAS.
- D. Zhou, R. Zhou, R. Zhou, B. Liu, T. Zhang, Y. Xian, P. J. Cullen, X. Lu and K. Ken) Ostrikov, Chem. Eng. J., 2021, 421, 129544 CrossRef CAS.
- L. R. Winter and J. G. Chen, Joule, 2021, 5, 300–315 CrossRef CAS.
- M. Eisa, D. Ragauskaite, S. Adhikari, F. Bella and J. Baltrusaitis, ACS Sustainable Chem. Eng., 2022, 10, 8997–9001 CrossRef CAS.
- I. Langmuir, Proc. Natl. Acad. Sci. U. S. A., 1928, 14, 627–637 CrossRef CAS PubMed.
- C. Tendero, C. Tixier, P. Tristant, J. Desmaison and P. Leprince, Spectrochim. Acta, Part B, 2006, 61, 2–30 CrossRef.
- Y. P. Raizer and J. E. Allen, Gas Discharge Physics, Springer, Berlin, 1997, vol. 2 Search PubMed.
- V. N. Ochkin, Spectroscopy of Low Temperature Plasma, John Wiley & Sons, Weinheim, 2009 Search PubMed.
- S. Eyde, Ind. Eng. Chem., 1912, 4, 771–774 CrossRef CAS.
- J. Kenelem, Bachelor Thesis, Oregon State University, 1911 Search PubMed.
- I. Tsonev, C. O'Modhrain, A. Bogaerts and Y. Gorbanev, ACS Sustain. Chem. Eng., 2023, 11, 1888–1897 CrossRef CAS.
- C. N. Banwell and E. M. McCash, Fundamentals of Molecular Spectroscopy, Indian Edition, 2017 Search PubMed.
- D. Burnette, A. Montello, I. V. Adamovich and W. R. Lempert, Plasma Sources Sci. Technol., 2014, 23, 45007 CrossRef CAS.
- M. Uddi, N. Jiang, I. V. Adamovich and W. R. Lempert, J. Phys. D Appl. Phys., 2009, 42, 075205 CrossRef.
- C. Richards, E. Jans, I. Gulko, K. Orr and I. V. Adamovich, Plasma Sources Sci. Technol., 2022, 31, 034001 CrossRef CAS.
- E. R. Jans, K. Frederickson, T. A. Miller and I. V Adamovich, J. Mol. Spectrosc., 2019, 365, 111205 CrossRef CAS.
- A. Montello, Z. Yin, D. Burnette, I. V. Adamovich and W. R. Lempert, J. Phys. D Appl. Phys., 2013, 46, 464002 CrossRef.
- A. Bogaerts and E. C. Neyts, ACS Energy Lett., 2018, 3, 1013–1027 CrossRef CAS.
- T. Kim, S. Song, J. Kim and R. Iwasaki, Jpn. J. Appl. Phys., 2010, 49, 126201 CrossRef.
- O. S. Bahnamiri, C. Verheyen, R. Snyders, A. Bogaerts and N. Britun, Plasma Sources Sci. Technol., 2021, 30, 065007 CrossRef CAS.
- P. Barboun, P. Mehta, F. A. Herrera, D. B. Go, W. F. Schneider and J. C. Hicks, ACS Sustain. Chem. Eng., 2019, 7, 8621–8630 CrossRef CAS.
- E. Vervloessem, M. Aghaei, F. Jardali, N. Hafezkhiabani and A. Bogaerts, ACS Sustain. Chem. Eng., 2020, 8, 9711–9720 CrossRef CAS.
- J. Liu, L. Nie, D. Liu and X. Lu, Plasma Processes Polym., 2024, 21, 2300153 CrossRef CAS.
- C. D. Pintassilgo, O. Guaitella and A. Rousseau, Plasma Sources Sci. Technol., 2009, 18, 025005 CrossRef.
- A. Gómez-Ramírez, A. M. Montoro-Damas, J. Cotrino, R. M. Lambert and A. R. González-Elipe, Plasma Processes Polym., 2017, 14, e1600081 CrossRef.
- M. Bai, Z. Zhang, M. Bai, X. Bai and H. Gao, Plasma Chem. Plasma Process., 2008, 28, 405–414 CrossRef CAS.
- J. Hong, S. Prawer and A. B. Murphy, IEEE Trans. Plasma Sci., 2014, 42, 2338–2339 Search PubMed.
- J. Hong, S. Pancheshnyi, E. Tam, J. J. Lowke, S. Prawer and A. B. Murphy, J. Phys. D Appl. Phys., 2017, 50, 154005 CrossRef.
- K. H. R. Rouwenhorst, H.-H. Kim and L. Lefferts, ACS Sustain. Chem. Eng., 2019, 7, 17515–17522 CrossRef CAS.
- B. Indumathy, J. Ananthanarasimhan, L. Rao, S. Yugeswaran and P. V. Ananthapadmanabhan, J. Phys. D Appl. Phys., 2022, 55, 395501 CrossRef CAS.
- I. Adamovich, S. Agarwal, E. Ahedo, L. L. Alves, S. Baalrud, N. Babaeva, A. Bourdon, P. J. Bruggeman, C. Canal, I. Adamovich, S. Agarwal, E. Ahedo, L. L. Alves, S. Baalrud and T. Plasma, J. Phys. D: Appl. Phys., 2022, 55, 373001 CrossRef CAS.
- Y. B. Zeldvich, J. Acta Phytochimica, 1946, 21, 577 Search PubMed.
- G. A. Lavoie, J. B. Heywood and J. C. Keck, Combust. Sci. Technol., 1970, 1, 313–326 CrossRef CAS.
- D. Thompson, T. D. Brown and J. M. Beér, Combust. Flame, 1972, 19, 69–79 CrossRef CAS.
- F. Jardali, S. Van Alphen, J. Creel, H. Ahmadi Eshtehardi, M. Axelsson, R. Ingels, R. Snyders and A. Bogaerts, Green Chem., 2021, 23, 1748–1757 RSC.
- N. Rehbein and V. Cooray, J. Electrost., 2001, 51, 333–339 CrossRef.
- M. S. Bak and M. A. Cappelli, IEEE Trans. Plasma Sci., 2015, 43, 995–1001 CAS.
- Y. S. Mok and S. W. Ham, Chem. Eng. Sci., 1998, 53, 1667–1678 CrossRef CAS.
- M. Capitelli, C. M. Ferreira, B. F. Gordiets and A. I. Osipov, Plasma Kinetics in Atmospheric Gases, Springer Series on Atomic, Optical, and Plasma Physics, 2013, vol. 31, https://link.springer.com/book/10.1007/978-3-662-04158-1 Search PubMed.
- S. Teodoru, Y. Kusano and A. Bogaerts, Plasma Processes Polym., 2012, 9, 652–689 CrossRef CAS.
- J. A. Manion, R. E. Huie, R. D. Levin, D. R. Burgess Jr, V. L. Orkin, W. Tsang, W. S. McGivern, J. W. Hudgens, V. D. Knyazev, D. B. Atkinson, E. Chai, A. M. Tereza, C. Y. Lin, T. C. Allison, W. G. Mallard, F. Westley, J. T. Herron, R. F. Hampson, D. H. Frizzell, NIST chemical kinetics database, National Institute of Standards and Technology, Gaithersburg, 2008, available online at https://kinetics.nist.gov/kinetics/, accessed: 01 October 2024 Search PubMed.
- N. C. Roy, C. Pattyn, A. Remy, N. Maira and F. Reniers, Plasma Processes Polym., 2021, 18, e2000087 CrossRef.
- W. Wang, B. Patil, S. Heijkers, V. Hessel and A. Bogaerts, ChemSusChem, 2017, 10, 2110 CrossRef CAS.
- N. Britun, V. Gamaleev and M. Hori, Plasma Sources Sci. Technol., 2021, 30, 08LT02 CrossRef CAS.
- E. Vervloessem, Y. Gorbanev, A. Nikiforov, N. De Geyter and A. Bogaerts, Green Chem., 2022, 24, 916–929 RSC.
- S. Van Alphen, H. A. Eshtehardi, C. O'Modhrain, J. Bogaerts, H. Van Poyer, J. Creel, M.-P. Delplancke, R. Snyders and A. Bogaerts, Chem. Eng. J., 2022, 443, 136529 CrossRef CAS.
- S. J. Andrews, H. Ogden and J. Marshall, Some Experiments on an Effusion Cooled Turbine Nozzle Blade, 1956 Search PubMed.
- I. Tsonev, H. A. Eshtehardi, M.-P. Delplancke and A. Bogaerts, Sustainable Energy Fuels, 2024, 8, 2191 RSC.
- Z. Liu, Y. Tian, G. Niu, X. Wang and Y. Duan, ChemSusChem, 2021, 14, 1507–1511 CrossRef CAS PubMed.
- J. Hong, T. Zhang, R. Zhou, L. Dou, S. Zhang, R. Zhou, B. Ashford, T. Shao, A. B. Murphy, K. K. Ostrikov and P. J. Cullen, Green Chem., 2022, 24, 7458–7468 Search PubMed.
- F. Tampieri, Y. Gorbanev and E. Sardella, Plasma Processes Polym., 2023, 20, e2300077 CrossRef CAS.
- T. Haruyama, T. Namise, N. Shimoshimizu, S. Uemura, Y. Takatsuji, M. Hino, R. Yamasaki, T. Kamachi and M. Kohno, Green Chem., 2016, 18, 4536–4541 RSC.
- J. R. Toth, N. H. Abuyazid, D. J. Lacks, J. N. Renner and R. M. Sankaran, ACS Sustain. Chem. Eng., 2020, 8, 14845–14854 CrossRef CAS.
- Y. Gorbanev, E. Vervloessem, A. Nikiforov and A. Bogaerts, ACS Sustainable Chem. Eng., 2020, 8, 2996–3004 Search PubMed.
- S. Yoshida, N. Murakami, Y. Takatsuji and T. Haruyama, Green Chem., 2022, 25, 579–588 RSC.
- T. Sakakura, S. Uemura, M. Hino, S. Kiyomatsu, Y. Takatsuji, R. Yamasaki, M. Morimoto and T. Haruyama, Green Chem., 2018, 20, 627–633 RSC.
- A. Komuro, R. Ono and T. Oda, J. Phys. D Appl. Phys., 2013, 46, 175206 Search PubMed.
- M. Gromov, K. Leonova, N. Britun, N. De Geyter, R. Morent, R. Snyders and A. Nikiforov, React. Chem. Eng., 2022, 7, 1047–1052 RSC.
- S. Ž. Corina Bradu, K. Kutasi, M. Magureanu and N. Puac, J. Phys. D Appl. Phys., 2020, 53, 223001 Search PubMed.
- M. Gromov, K. Leonova, N. Britun, N. De Geyter, R. Morent, R. Snyders and A. Nikiforov, React. Chem. Eng., 2022, 7, 1047–1052 Search PubMed.
- A. Komuro, R. Ono and T. Oda, J. Phys. D Appl. Phys., 2013, 46, 175206 Search PubMed.
- M. Gromov, N. Kamarinopoulou, N. De Geyter, R. Morent, R. Snyders, D. Vlachos, P. Dimitrakellis and A. Nikiforov, Green Chem., 2022, 24, 9677 RSC.
- F. Fresnet, G. Baravian, L. Magne, S. Pasquiers, C. Postel, V. Puech and A. Rousseau, Plasma Sources Sci. Technol., 2002, 11, 152–160 CrossRef CAS.
- E. Vervloessem, M. Gromov, N. De Geyter, A. Bogaerts, Y. Gorbanev and A. Nikiforov, ACS Sustain. Chem. Eng., 2023, 11, 4289–4298 CrossRef CAS.
- T. Murakami, in APS Annual Gaseous Electronics Meeting Abstracts, 2023, pp. EW4-003 Search PubMed.
- O. Sakai, S. Kawaguchi and T. Murakami, Jpn. J. Appl. Phys., 2022, 61, 070101 CrossRef CAS.
- A. Fridman, Y. Yang and Y. I. Cho, Plasma Discharge in Liquid: Water Treatment and Applications, CRC press, 2012 Search PubMed.
- P. Bruggeman and C. Leys, J. Phys. D Appl. Phys., 2009, 42, 053001 CrossRef.
- X. Zhao and Y. Tian, Cell Rep. Phys. Sci., 2023, 4, 101618 CrossRef CAS.
- D. K. Dinh, I. Muzammil, W. S. Kang, D. Kim and D. H. Lee, Plasma Sources Sci. Technol., 2021, 30, 055020 CrossRef CAS.
- I. Sremački, M. Gromov, C. Leys, R. Morent, R. Snyders and A. Nikiforov, Plasma Processes Polym., 2019, 17, e1900191 CrossRef.
- P. Peng, P. Chen, C. Schiappacasse, N. Zhou, E. Anderson, D. Chen, J. Liu, Y. Cheng, R. Hatzenbeller, M. Addy, Y. Zhang, Y. Liu and R. Ruan, J. Cleaner Prod., 2018, 177, 597–609 CrossRef CAS.
- T. Sakakura, N. Murakami, Y. Takatsuji and T. Haruyama, J. Phys. Chem. C, 2020, 124, 9401–9408 CrossRef CAS.
- Y. Kubota, K. Koga, M. Ohno and T. Hara, Plasma Fusion Res., 2010, 5, 042 CrossRef.
- T. Sakakura, N. Murakami, Y. Takatsuji, M. Morimoto and T. Haruyama, ChemPhysChem, 2019, 20, 1467–1474 CrossRef CAS PubMed.
- P. Peng, C. Schiappacasse, N. Zhou, M. Addy, Y. Cheng, Y. Zhang, E. Anderson, D. Chen, Y. Wang, Y. Liu, P. Chen and R. Ruan, J. Phys. D Appl. Phys., 2019, 52, 494001 CrossRef CAS.
- T. Mizushima, K. Matsumoto, J. I. Sugoh, H. Ohkita and N. Kakuta, Appl. Catal., A, 2004, 265, 53–59 CrossRef CAS.
- J. N. Armor, Catal. Today, 2011, 163, 3–9 CrossRef CAS.
- Z. Qu, R. Zhou, J. Sun, Y. Gao, Z. Li, T. Zhang, R. Zhou, D. Liu, X. Tu, P. Cullen and K. Ostrikov, ChemSusChem, 2023, 17, e202300783 CrossRef PubMed.
- S. Tanaka, H. Uyama and O. Matsumoto, Plasma Chem. Plasma Process., 1994, 14, 491–504 CrossRef CAS.
- R. De Meyer, Y. Gorbanev, R.-G. Ciocarlan, P. Cool, S. Bals and A. Bogaerts, Chem. Eng. J., 2024, 488, 150838 CrossRef CAS.
- C. Ndayirinde, Y. Gorbanev, R. G. Ciocarlan, R. De Meyer, A. Smets, E. Vlasov, S. Bals, P. Cool and A. Bogaerts, Catal. Today, 2023, 419, 114156 CrossRef CAS.
- Y. Wu, C. M. Limbach, A. A. Tropina and R. B. Miles, AIAA Scitech 2019 Forum, 2019, pp. 2066 Search PubMed.
- A. Lo, A. Cessou, P. Boubert and P. Vervisch, J. Phys. D Appl. Phys., 2014, 47, 5201 Search PubMed.
- W. Yang, Q. Zhou, Q. Sun, Z. Dong and E. Yan, AIP Adv., 2020, 10, 10 Search PubMed.
- A. Garscadden and R. Nagpal, Plasma Sources Sci. Technol., 1995, 4, 268 CrossRef CAS.
- K. H. R. Rouwenhorstm and L. Lefferts, ChemCatChem, 2023, 15, e202300078 CrossRef CAS.
- J. A. Andersen, M. C. Holm, K. van ’t Veer, J. M. Christensen, M. Østberg, A. Bogaerts and A. D. Jensen, Chem. Eng. J., 2023, 457, 141294 CrossRef CAS.
- Y. Gorbanev, I. Fedirchyk and A. Bogaerts, Curr. Opin. Green Sustainable Chem., 2024, 47, 100916 CrossRef.
- X. Pei, D. Gidon and D. B. Graves, J. Phys. D Appl. Phys., 2019, 53, 044002 CrossRef.
- Y. D. Korolev, O. B. Frants, N. V Landl and A. I. Suslov, IEEE Trans. Plasma Sci., 2012, 40, 2837–2842 CAS.
- E. D. Mccollum and F. Daniels, Ind. Eng. Chem., 1923, 15, 1173–1175 CrossRef CAS.
- M. Bai, Z. Zhang, X. Bai, M. Bai and W. Ning, IEEE Trans. Plasma Sci., 2003, 31, 1285–1291 CrossRef CAS.
- Y. Wang, M. Craven, X. Yu, J. Ding, P. Bryant, J. Huang and X. Tu, ACS Catal., 2019, 9, 10780–10793 CrossRef CAS PubMed.
- Y. Liu, C. W. Wang, X. F. Xu, B. W. Liu, G. M. Zhang, Z. W. Liu, Q. Chen and H. B. Zhang, Plasma Chem. Plasma Process., 2022, 42, 267–282 CrossRef CAS.
- S. Li, T. van Raak and F. Gallucci, J. Phys. D Appl. Phys., 2020, 53, 014008 CrossRef CAS.
- S. Li, Y. Shao, H. Chen and X. Fan, Ind. Eng. Chem. Res., 2022, 61, 3292–3302 CrossRef CAS.
- Y. Luo, H. Jiang, L.-X. Ding, S. Chen, Y. Zou, G.-F. Chen and H. Wang, ACS Sustain. Chem. Eng., 2023, 11, 11737–11744 CrossRef CAS.
- J. Sun, D. Alam, R. Daiyan, H. Masood, T. Zhang, R. Zhou, P. J. Cullen, E. C. Lovell, A. R. Jalili and R. Amal, Energy Environ. Sci., 2021, 14, 865–872 RSC.
- Y. Cui, H. Yang, C. Dai, P. Ren, C. Song and X. Ma, Ind. Eng. Chem. Res., 2022, 61, 4816–4823 CrossRef CAS.
- Z. Meng, J. X. Yao, C. N. Sun, X. Kang, R. Gao, H. R. Li, B. Bi, Y. F. Zhu, J. M. Yan and Q. Jiang, Adv. Energy Mater., 2022, 12, 2202105 CrossRef CAS.
- A. Wu, J. Yang, B. Xu, X. Y. Wu, Y. Wang, X. Lv, Y. Ma, A. Xu, J. Zheng, Q. Tan, Y. Peng, Z. Qi, H. Qi, J. Li, Y. Wang, J. Harding, X. Tu, A. Wang, J. Yan and X. Li, Appl. Catal., B, 2021, 299, 120667 CrossRef CAS.
- P. J. Bruggeman, M. J. Kushner, B. R. Locke, J. G. E. Gardeniers, W. G. Graham, D. B. Graves, R. C. H. M. Hofman-Caris, D. Maric, J. P. Reid, E. Ceriani, D. Fernandez Rivas, J. E. Foster, S. C. Garrick, Y. Gorbanev, S. Hamaguchi, F. Iza, H. Jablonowski, E. Klimova, J. Kolb, F. Krcma, P. Lukes, Z. MacHala, I. Marinov, D. Mariotti, S. Mededovic Thagard, D. Minakata, E. C. Neyts, J. Pawlat, Z. L. Petrovic, R. Pflieger, S. Reuter, D. C. Schram, S. Schröter, M. Shiraiwa, B. Tarabová, P. A. Tsai, J. R. R. Verlet, T. Von Woedtke, K. R. Wilson, K. Yasui and G. Zvereva, Plasma Sources Sci. Technol., 2016, 25, 053002 CrossRef.
- H. M. Nguyen, F. Gorky, S. Guthrie and M. L. Carreon, Catal. Today, 2023, 418, 114141 CrossRef CAS.
- S. Kumar, N. Singh and R. Prasad, Renewable Sustainable Energy Rev., 2010, 14, 1830–1844 CrossRef CAS.
- P. Lamichhane, N. Pourali, L. Scott, N. N. Tran, L. Lin, M. E. Gelonch, E. V. Rebrov and V. Hessel, Renewable Sustainable Energy Rev., 2024, 189, 114044 CrossRef CAS.
- E. Morais and A. Bogaerts, Plasma Processes Polym., 2024, 21, e2300149 CrossRef.
- A. Oumghar, J. C. Legrand, A. M. Diamy, N. Turillon and R. I. Ben-Aim, Plasma Chem. Plasma Process., 1994, 14, 229–249 CrossRef CAS.
- J.-C. Legrand, A.-M. Diamy, R. Hrach and V. Hrachova, Vacuum, 1999, 52, 27–32 CrossRef CAS.
- G. Horvath, N. J. Mason, L. Polachova, M. Zahoran, L. Moravsky and S. Matejcik, Plasma Chem. Plasma Process., 2010, 30, 565–577 CrossRef CAS.
- Y. Yi, X. Wang, A. Jafarzadeh, L. Wang, P. Liu, B. He, J. Yan, R. Zhang, H. Zhang and X. Liu, ACS Catal., 2021, 11, 1765–1773 CrossRef CAS.
- R. Zhou, R. Zhou, Y. Xian, Z. Fang, X. Lu, K. Bazaka, A. Bogaerts and K. Ken) Ostrikov, Chem. Eng. J., 2020, 382, 122745 CrossRef CAS.
- W. Wang, C. Zhu and Y. Cao, Int. J. Hydrogen Energy, 2010, 35, 1951–1956 CrossRef CAS.
- N. Budhraja, A. Pal and R. S. Mishra, Int. J. Hydrogen Energy, 2023, 48, 2467–2482 CrossRef CAS.
- J. Park, R. S. Zhu and M. C. Lin, J. Chem. Phys., 2002, 117, 3224–3231 CrossRef CAS.
- J. Feng, P. Ning, K. Li, X. Sun, C. Wang, L. Jia and M. Fan, ACS Sustain. Chem. Eng., 2023, 11, 804–814 CrossRef CAS.
- G. W. Kim, M. A. Alam, J. J. Lee, G. Y. Kim, P. J. Kim and M. I. Khan, Eur. J. Soil Biol., 2017, 83, 76–83 CrossRef CAS.
- F. Manaigo, K. Rouwenhorst, A. Bogaerts and R. Snyders, Energy Convers. Manage., 2024, 302, 118124 CrossRef CAS.
- M. Adhami Sayad Mahaleh, M. Narimisa, A. Nikiforov, M. Gromov, Y. Gorbanev, R. Bitar, R. Morent and N. De Geyter, Appl. Sci., 2023, 13, 7619 CrossRef.
- L. Hollevoet, F. Jardali, Y. Gorbanev, J. Creel, A. Bogaerts and J. A. Martens, Angew. Chem., 2020, 132, 24033–24037 CrossRef.
- L. Li, C. Tang, X. Cui, Y. Zheng, X. Wang, H. Xu, S. Zhang, T. Shao, K. Davey and S. Z. Qiao, Angew. Chem., Int. Ed., 2021, 60, 14131–14137 CrossRef CAS PubMed.
- C. Lee and Q. Yan, Curr. Opin. Electrochem., 2021, 29, 100808 CrossRef CAS.
- J. Osorio-Tejada, N. N. Tran and V. Hessel, Sci. Total Environ., 2022, 826, 154162 CrossRef CAS.
- G. Yu, Y. Wang, L. Zheng, J. Huang, J. Li, L. Gong, R. Chen, W. Li, J. Huang and Y.-S. Duh, Process Saf. Environ. Prot., 2021, 152, 201–219 CrossRef CAS.
- C. Font-Palma, C, 2019, 5, 27 CAS.
- M. Vilarrasa-Nogué, M. R. Teira-Esmatges, E. González-Llinàs, F. Domingo-Olivé and J. M. Villar, Sci. Total Environ., 2020, 724, 137918 CrossRef PubMed.
- R. Ingels, U.S. Pat., US011517848B2, 2022, https://patents.justia.com/patent/11517848 Search PubMed.
- R. Ingels and D. B. Graves, Plasma Med., 2015, 5, 2–4 Search PubMed.
- J. Osorio-Tejada, N. N. Tran and V. Hessel, Sci. Total Environ., 2022, 826, 154162 CrossRef CAS PubMed.
- J. L. Osorio-Tejada, E. Rebrov and V. Hessel, Int. J. Life Cycle Assess., 2023, 28, 1590–1603 CrossRef CAS PubMed.
- R. M. Nayak-Luke, C. Forbes, Z. Cesaro, R. Bañares-Alcántara and K. H. R. Rouwenhorst, Techno-Economic Challenges of Green Ammonia as Energy Vector, ed. Bañares-Alcántara, R. and Valera-Medina, A., 2020, pp. 191–208 Search PubMed.
- F. Manaigo, K. Rouwenhorst, A. Bogaerts and R. Snyders, Energy Convers. Manage., 2024, 302, 118124 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2025 |