
Enhanced luminescence and stability of TFMDSA nanoparticles via polymer-induced aggregation for bioimaging†
Xiang
Li‡
a,
Xue
Ren‡
ad,
Yuchao
Luo
*ab,
Haotian
Shi
a,
Zhigang
Xie
 c,
Bin
Xu
a and
Wenjing
Tian
*a
c,
Bin
Xu
a and
Wenjing
Tian
*a
aState Key Laboratory of Supramolecular Structure and Materials, Department of Chemistry, Jilin University, Qianjin Street No. 2699, Changchun, 130012, P. R. China. E-mail: luoyuchao@jlu.edu.cn; wjtian@jlu.edu.cn
bSchool of Mechanical and Aerospace Engineering, Jilin University, Changchun 130022, China
cState Key Laboratory of Polymer Physics and Chemistry, Changchun Institute of Applied Chemistry, Chinese Academy of Sciences, 5625 Renmin Street, Changchun, Jilin 130022, P. R. China
dDepartment of Gynecologic Oncology, Gynecology and Obstetrics Centre, The First Hospital of Jilin University, Changchun 130021, P. R. China
First published on 7th November 2024
Abstract
In recent years, fluorescence imaging has occupied a very important position in the life science and biomedical fields. However, achieving nanomaterials for bioimaging with both high fluorescence quantum efficiency and high stability remains a significant challenge. Herein, we synthesized mPEG5K–PCL10K@TFMDSA and mPEG5K–PLLA10K@TFMDSA nanoparticles using polymer-induced aggregation. This method significantly enhanced the luminescence efficiency of TFMDSA nanoparticles in solution, attributed to improved intermolecular interactions and restricted molecular vibrations. The resulting nanoparticles exhibited exceptional optical stability over a period of seven days and demonstrated low cytotoxicity towards HeLa cells, making them highly suitable for bioimaging applications. Cellular uptake studies indicated that these nanoparticles were more efficiently internalized by HeLa cells compared to their amorphous counterparts, likely due to their unique square morphology. Our findings highlight the potential of polymer-induced aggregation in enhancing the optical properties and stability of TFMDSA nanomaterials, suggesting their promise as biofluorescent probes for cancer diagnosis and other biomedical applications.
Introduction
As a pivotal tool in biological research, bioimaging plays an indispensable role by significantly simplifying the diagnostic process and providing an effective means for visualizing and monitoring the morphological details of cells and tissues.1–6 Over the past few decades, various imaging techniques, such as magnetic resonance imaging (MRI),7–9 computed tomography (CT),10,11 positron emission tomography (PET),12 single-photon emission computed tomography (SPECT),1,13 and fluorescence imaging,8,14–17 have significantly expanded their applications in biology. Among these, fluorescence imaging stands out due to its ability to enable real-time, non-invasive monitoring of biomolecules in their native environments with high spatial and temporal resolution.18–20 Fluorescent organic nanoparticles based on small molecules have garnered extensive attention from the research community owing to their high quantum yield, sensitivity, ease of synthesis and purification, biodegradability, and chemical customizability.21–24 These properties endow fluorescent organic nanoparticles with significant potential in bioimaging.The development of organic fluorescent molecules with aggregation-induced emission (AIE) properties has addressed the limitation of conventional organic molecules, which typically suffer from aggregation-induced quenching (ACQ).25–27 Molecules with AIE properties emit bright fluorescence in their aggregated state due to restricted intramolecular vibrational motion, which suppresses non-radiative processes, resulting in nanoparticles with high fluorescence quantum yields.28–33 These AIE nanoparticles are usually prepared via ultrasonic nanoprecipitation and exhibit excellent luminescent properties and biocompatibility. Li et al. developed TPETPAF AIEgens with a solid-state quantum efficiency of 52.5% and encapsulated TPETPAF in a DSPE–PEG polymer matrix to produce AIE nanoparticles with a fluorescence quantum efficiency of 24% in the aqueous phase.34 Additionally, Zong et al. synthesized a diimine fluorophore with AIE properties and used the Pluronic F-127 method to prepare AIE nanoparticles for bioimaging applications.35 However, most AIE nanoparticles tend to form nano-aggregates in aqueous solutions, suffering from loose molecular packing and disordered molecular structures, which result in suboptimal fluorescence properties.36
In this study, we present a method to enhance the solution-state fluorescence quantum efficiency of AIE nanoparticles through polymer-induced enhancement. We synthesized mPEG5K–PCL10K and mPEG5K–PLLA10K nanoparticles with unique square morphology for bioimaging applications using TFMDSA as the AIE luminescent material via a “one-pot” sonication process. Notably, the fluorescence quantum efficiencies of the mPEG5K–PCL10K and mPEG5K–PLLA10K nanoparticles were significantly improved compared to conventional aggregated nanoparticles, increasing from 6.67% to 40.13% and 10.70%, respectively. Furthermore, we predicted the equilibrium morphology of TFMDSA to confirm the nanoparticle assembly mechanism. Cellular imaging experiments demonstrated that mPEG5K–PCL10K and mPEG5K–PLLA10K TFMDSA nanoparticles exhibit superior imaging effects. Our findings provide an effective strategy to enhance the bioimaging performance of organic fluorescent nanoparticles, highlighting their substantial potential for future biomedical applications.
Results and discussion
Preparation and characterization of TFMDSA nanoparticles
The TFMDSA molecule was synthesized according to our previously reported procedure, as shown in Fig. S1 (ESI†).37 The molecular structure of TFMDSA was confirmed through 1H NMR, 13C NMR (Fig. S2 and S3, ESI†) and mass spectrometry analyses (Fig. S4, ESI†). As illustrated in Scheme 1, amorphous TFMDSA nanoparticles (TFMDSA NPs) were synthesized using a conventional reprecipitation method. To further enhance their fluorescence quantum yield, we employed a “one-pot” sonication emulsification technique with amphiphilic polymers mPEG5K–PCL10K and mPEG5K–PLLA10K. This method facilitated the formation of polymer-induced aggregation TFMDSA nanoparticles in solution. The hydrophilic PEG segments were positioned on the surface of the nanomaterials, enhancing their water solubility, while the hydrophobic PCL and PLLA segments interacted with the TFMDSA molecules through supramolecular interactions. This interaction promoted closer and more ordered intermolecular stacking of TFMDSA, resulting in the successful synthesis of mPEG5K–PCL10K@TFMDSA NPs and mPEG5K–PLLA10K@TFMDSA NPs with high fluorescence quantum efficiency.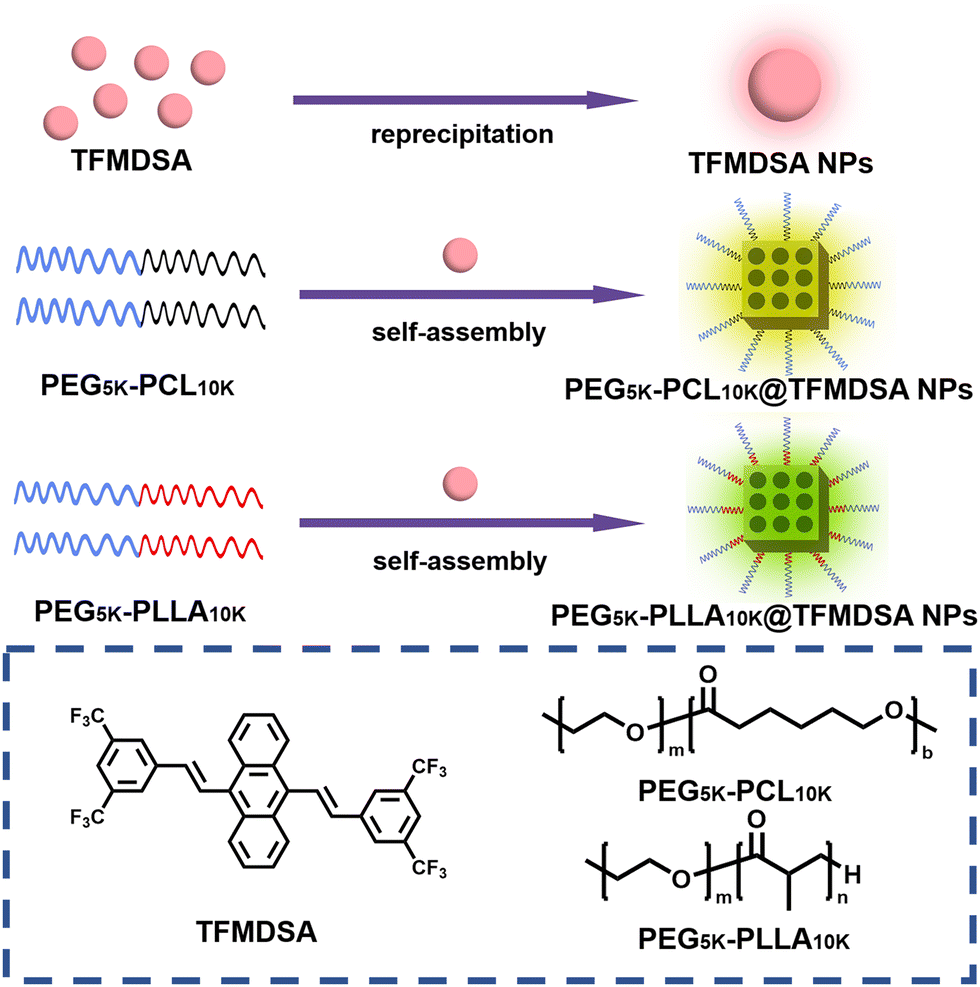 | ||
| Scheme 1 Schematic diagram of the preparation of mPEG5K–PCL10K@TFMDSA NPs, mPEG5K–PLLA10K@TFMDSA NPs, and TFMDSA NPs. | ||
The particle size distribution and microscopic morphology of the TFMDSA nanoparticles were characterized using dynamic light scattering (DLS) and transmission electron microscopy (TEM). The average particle sizes of TFMDSA NPs, mPEG5K–PCL10K@TFMDSA NPs, and mPEG5K–PLLA10K@TFMDSA NPs were determined to be 50 nm, 80 nm, and 80 nm, respectively, demonstrating good dispersion, as shown in Fig. 1a–c. TEM images of TFMDSA revealed that the TFMDSA NPs exhibited a spherical morphology with a diameter of approximately 50 nm, consistent with the DLS results. However, electron diffraction experiments did not reveal any diffraction spots, indicating that the TFMDSA NPs had a disordered and amorphous stacking pattern (Fig. 1d). Notably, the mPEG5K–PCL10K@TFMDSA NPs displayed a bulk morphology with dimensions of approximately 80 nm in length and 75 nm in width, while the mPEG5K–PLLA10K@TFMDSA NPs also exhibited a block morphology with dimensions of approximately 80 nm in length and 76 nm in width. In SAED, the well-defined diffraction spots demonstrate that the square-shaped morphology nanoparticles we prepared are crystalline for both mPEG5K–PCL10K@TFMDSA NPs and mPEG5K–PLLA10K@TFMDSA NPs (Fig. 1e and f). As shown in Fig. S5 (ESI†), under the same conditions, pure PEG–PLLA nanoparticles exhibited spherical morphology with a size of 100 nm, and the SAED pattern did not display diffraction spots. This result indicates that the unique square morphology and crystallinity of mPEG5K–PLLA10K@TFMDSA NPs are induced by PEG–PLLA promoting the aggregation of TFMDSA. On the other hand, pure PEG–PCL nanoparticles showed a rectangular morphology of around 100 nm under TEM, and the SAED pattern revealed diffraction spots, indicating that the pure PEG–PCL nanoparticles formed under these conditions exhibit crystallinity, which further promoted the formation of polymer-induced aggregation TFMDSA nanoparticles. These results validate our polymer-induced aggregation method, demonstrating its effectiveness in preparing mPEG5K–PCL10K@TFMDSA NPs and mPEG5K–PLLA10K@TFMDSA NPs with crystal structures, thereby providing a viable approach for producing nanomaterials with high fluorescence quantum efficiency.
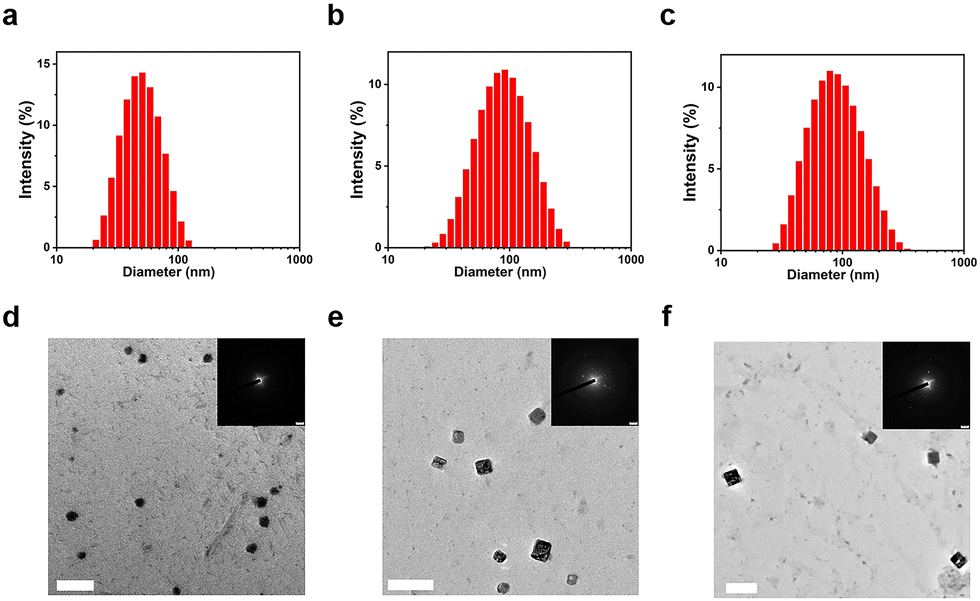 | ||
| Fig. 1 Size distribution and TEM images of the (a) and (d) TFMDSA NPs; (b) and (e) mPEG5K–PCL10K@TFMDSA; (c) and (f) mPEG5K–PLLA10K@TFMDSA. Insets: SAED image. Scale bars in all TEM images are 200 nm. | ||
Optical properties of TFMDSA nanoparticles
The optical properties of fluorescent nanomaterials are crucial for cellular imaging. The absorption peak of TFMDSA in THF solution was observed at 417 nm (Fig. S6a, ESI†). As shown in Fig. S6b (ESI†), the fluorescence intensity of the TFMDSA solution was significantly enhanced with increasing water concentration, demonstrating that TFMDSA exhibits pronounced AIE properties. This characteristic makes TFMDSA highly suitable for application as a fluorescent nanomaterial in bioimaging. We measured the absorption and emission spectra of TFMDSA NPs, mPEG5K–PCL10K@TFMDSA NPs and mPEG5K–PLLA10K@TFMDSA NPs (sample concentration: 10 μg mL−1) in water. Both mPEG5K–PLLA10K@TFMDSA NPs and mPEG5K–PCL10K@TFMDSA NPs exhibited absorption peaks at 416 nm, which were almost unchanged compared to the absorption peak of TFMDSA NPs at 419 nm (Fig. 2a). As shown in Fig. 2b, the emission maxima of mPEG5K–PLLA10K@TFMDSA NPs and mPEG5K–PCL10K@TFMDSA NPs appeared at 514 nm and 527 nm, respectively, whereas the fluorescence emission peak of TFMDSA NPs was at 517 nm. As shown in Fig. S7 (ESI†), the TFMDSA polymer-induced aggregation TFMDSA nanoparticles emitted bright yellow-green fluorescence under white light and 365 nm UV lamps, while the nanoparticles emitted weak green fluorescence. This result suggests that in amorphous TFMDSA nanoparticles, the relatively loose stacking structure leads to weak intermolecular interactions, allowing the molecules to rotate and vibrate freely. However, after the formation of polymer-induced aggregation TFMDSA nanoparticles, the dense and ordered arrangement restricts molecular vibration. Due to the strong intermolecular interactions and distorted molecular structure, the conjugation degree of the system is reduced. As a result, the fluorescence emission peaks are blue-shifted and narrowed. All three AIE nanostructures have large Stokes shifts. After the formation of polymer-induced aggregation TFMDSA nanoparticles, the self-absorption is significantly reduced, improving the signal-to-noise ratio of bioimaging and facilitating subsequent bioimaging. The excitation spectra of mPEG5K–PCL10K@TFMDSA NPs and mPEG5K–PLLA10K@TFMDSA NPs were almost identical, whereas the excitation spectra of TFMDSA exhibited a pronounced redshift. This shift was attributed to the nanocrystalline state of mPEG5K–PCL10K@TFMDSA NPs and mPEG5K–PLLA10K@TFMDSA NPs, whereas TFMDSA NPs were in a disordered form, consistent with the SAED findings (Fig. 2c). The fluorescence decay curves of TFMDSA nanoparticles were also analyzed (Fig. 2d). The fluorescence lifetime of TFMDSA NPs is 1.57 ns with a quantum yield of 6.67%, while the fluorescence lifetime of mPEG5K–PCL10K@TFMDSA NPs is 2.62 ns with a quantum yield of 40.13%. The fluorescence lifetime of mPEG5K–PLLA10K@TFMDSA NPs is 3.14 ns with a quantum yield of 10.40%. The quantum yield of polymer-induced aggregation TFMDSA nanoparticles is significantly higher than that of TFMDSA nanoparticles. All these data are collected in Table 1.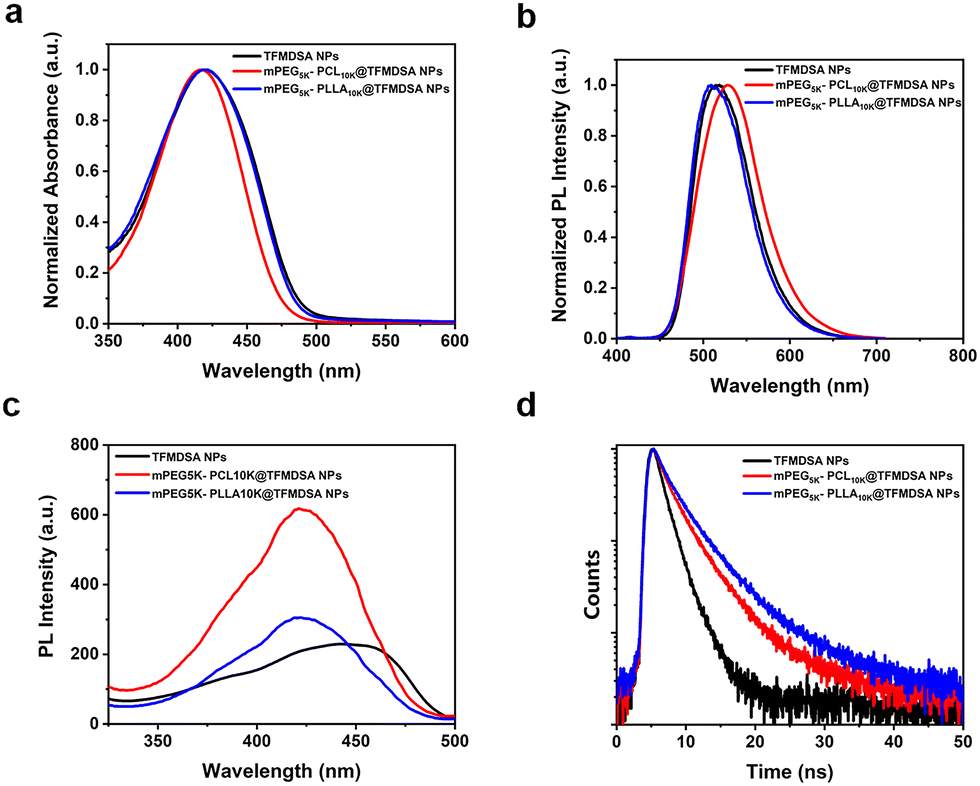 | ||
| Fig. 2 (a) Absorbance spectra of the TFMDSA NPs, mPEG5K–PLLA10K@TFMDSA NPs, and mPEG5K–PCL10K@TFMDSA NPs. (b) PL spectra of the TFMDSA NPs, mPEG5K–PLLA10K@TFMDSA NPs, and mPEG5K–PCL10K@TFMDSA NPs. (c) Excitation spectra of the TFMDSA NPs, mPEG5K–PLLA10K@TFMDSA NPs, and mPEG5K–PCL10K@TFMDSA NPs. (d) Time-resolved decay profiles of the TFMDSA NPs, mPEG5K–PLLA10K@TFMDSA NPs, and mPEG5K–PCL10K@TFMDSA NPs. | ||
| UV-vis (nm) | PL (nm) | Φ f (%) | τ FL (ns) | k r 10−9 (s−1) | k nr 10−8 (s−1) | |
|---|---|---|---|---|---|---|
| TFMDSA NPs | 419 | 517 | 6.67 | 1.57 | 4.25 | 5.9 |
| mPEG5K–PCL10K@TFMDSA NPs | 416 | 527 | 40.13 | 2.62 | 15.32 | 2.3 |
| mPEG5K–PLLA10K@TFMDSA NPs | 416 | 514 | 10.40 | 3.14 | 3.31 | 2.9 |
| TFMDSA powder | 490 | 537 | 22.32 | 2.81 | 7.01 | 2.76 |
Moreover, the radiative transition rates (kr) of polymer-induced aggregation TFMDSA nanoparticles are significantly higher than those of TFMDSA nanoparticles, and the non-radiative transition rates (knr) of TFMDSA polymer-induced aggregation TFMDSA nanoparticles are significantly lower. This is due to the dense and orderly packing structure formed between the amphiphilic polymers (mPEG5K–PCL10K and mPEG5K–PLLA10K) and TFMDSA, which enhances intermolecular interactions, restricting molecular rotation and vibration, thereby inhibiting non-radiative transition processes and increasing the radiative transition rate. The variation in the fluorescence quantum yield and lifetime between the nanoparticles and the two types of polymer-induced aggregation TFMDSA nanoparticles primarily arises from the extent of molecular motion restriction. In nanoparticles, molecular movement is somewhat constrained, but the flexible PLLA and PEG polymers offer TFMDSA molecules more freedom of movement. This increased vibrational and rotational motion promotes the dissipation of excited-state energy, resulting in a lower quantum yield and shorter fluorescence lifetime.
In contrast, mPEG5K–PCL10K@TFMDSA NPs exhibit a more tightly packed structure due to the semicrystalline nature of PCL. The glass transition temperature (Tg) of PCL (−60 °C) is significantly lower than that of PLLA (+60 °C), further restricting the motion of TFMDSA molecules. This increased restriction on molecular movement leads to stronger intermolecular interactions and less vibrational freedom, which favors radiative over non-radiative transitions. Consequently, mPEG5K–PCL10K@TFMDSA NPs display higher fluorescence quantum yield and longer fluorescence lifetime compared to nanoparticles, where the relatively loose packing structure leads to weaker intermolecular interactions and higher rates of non-radiative decay.
To further investigate the nanoparticles formed between the copolymer and TFMDSA, Fourier transform infrared spectroscopy (FT-IR) analysis was performed to elucidate the assembly mechanism of polymer-induced aggregation TFMDSA nanoparticles. As shown in Fig. 3a, the spectrum of mPEG5K–PCL10K@TFMDSA exhibits a redshift compared with that of mPEG5K–PCL10K, suggesting that the supramolecular interactions (π–π stacking and hydrophobic interactions) are relatively strong. Similarly, the infrared spectra of mPEG5K–PLLA10K and mPEG5K–PLLA10K@TFMDSA show the same redshift (Fig. 3b). These results indicate that the hydrophobic side chains of the amphiphilic polymers, mPEG5K–PCL10K and mPEG5K–PLLA10K, enhance the supramolecular interactions between chains and TFMDSA molecules, thereby inducing the formation of a stable crystalline arrangement. To further confirm the molecular stacking orientation of the polymer-induced aggregation TFMDSA nanoparticles, the equilibrium morphology of TFMDSA was predicted using the Materials Studio software package. The results (Fig. 3c and Fig. S8, ESI†) indicate that the thermodynamically stable morphology of the smallest TFMDSA cell is a rectangular structure. We propose that, following stable growth, the nanoparticles will evolve into a more stable square structure. This hypothesis is supported by the TEM morphology results of mPEG5K–PCL10K@TFMDSA NPs and mPEG5K–PLLA10K@TFMDSA NPs.
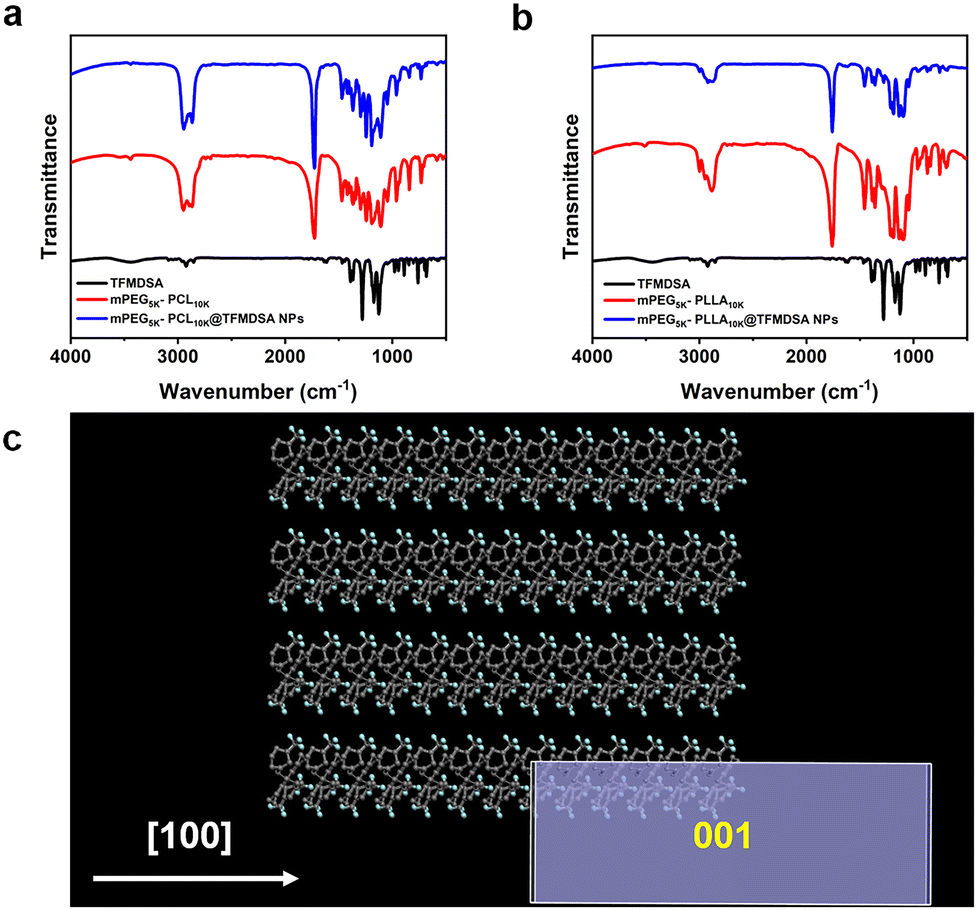 | ||
| Fig. 3 (a) FTIR spectra of the TFMDSA, mPEG5K–PCL10K, and mPEG5K–PCL10K@TFMDSA NPs. (b) FTIR spectra of the TFMDSA, mPEG5K–PLLA10K, and mPEG5K–PLLA10K@TFMDSA NPs. (c) Theoretically predicted equilibrium morphology of TFMDSA. | ||
Stability of TFMDSA nanoparticles
To thoroughly evaluate the stability of TFMDSA nanoparticles, we collected UV-visible absorption spectra and fluorescence emission spectra over a period of seven days. As shown in Fig. 4 and Fig. S9 (ESI†), the absorbance of mPEG5K–PCL10K@TFMDSA NPs and mPEG5K–PLLA10K@TFMDSA NPs slowly decreased over time, maintaining 83% and 75% of the initial intensity by the seventh day, respectively. In stark contrast, TFMDSA NPs showed a significant decrease in absorbance, which dropped to 31% of the original value by day seven. Additionally, the fluorescence intensity of mPEG5K–PLLA10K@TFMDSA NPs and mPEG5K–PCL10K@TFMDSA NPs gradually weakened, retaining 87% and 82% of their original values, respectively, on the seventh day. Conversely, the fluorescence intensity of TFMDSA NPs decreased significantly to only 43% of the original value. These results indicate that the polymer-induced aggregation TFMDSA nanoparticles exhibit superior stability compared to the amorphous nanoparticles. The enhanced stability of the nanoparticles can be attributed to their ordered molecular stacking, resulting in stronger intermolecular interactions, and the amphiphilic polymer shell, which preserves the nanoparticles' morphology by providing both hydrophilic and hydrophobic interactions. In contrast, the amorphous TFMDSA nanoparticles lack this ordered structure and protective polymer shell, leading to weaker intermolecular interactions and increased susceptibility to environmental factors, resulting in the rapid degradation of their optical properties. These findings underscore the superior stability of mPEG5K–PLLA10K@TFMDSA NPs and mPEG5K–PCL10K@TFMDSA NPs, making them more suitable for applications requiring prolonged stability in bioimaging.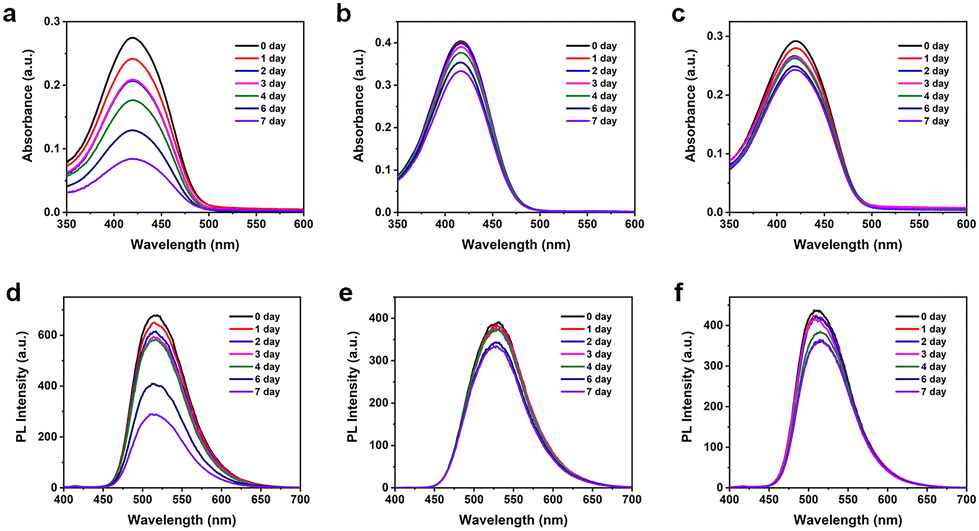 | ||
| Fig. 4 The absorbance intensity of the (a) TFMDSA NPs, (b) mPEG5K–PCL10K@TFMDSA NPs and (c) mPEG5K–PLLA10K@TFMDSA NPs over seven days, the fluorescence intensity of the (d) TFMDSA NPs, (e) mPEG5K–PCL10K@TFMDSA NPs and (f) mPEG5K–PLLA10K@TFMDSA NPs over seven days. | ||
Cellular uptake and cytotoxicity of TFMDSA nanoparticles
Biocompatibility is a crucial prerequisite for the use of fluorescent nanoparticles as bioimaging agents. We first investigated the biocompatibility of TFMDSA NPs, mPEG5K–PLLA10K@TFMDSA NPs, and mPEG5K–PCL10K@TFMDSA NPs on HeLa cells using the MTT (3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide) assay. As shown in Fig. S10 (ESI†), TFMDSA NPs, mPEG5K–PCL10K@TFMDSA NPs, and mPEG5K–PLLA10K@TFMDSA NPs exhibited low cytotoxicity to HeLa cells after 24 hours of incubation at various concentrations. These results indicate that TFMDSA NPs, mPEG5K–PCL10K@TFMDSA NPs, and mPEG5K–PLLA10K@TFMDSA NPs possess good biocompatibility, making them suitable for cellular imaging applications.Cellular uptake is essential for nanomaterials to fulfill their function, especially in live cell imaging. We studied the cellular uptake of TFMDSA nanoparticles in HeLa cells using confocal laser scanning microscopy (CLSM). TFMDSA NPs, mPEG5K–PCL10K@TFMDSA NPs, and mPEG5K–PLLA10K@TFMDSA NPs were incubated with HeLa cells stained with 4,6-diamidino-2-phenylindole (DAPI) to visualize the cell nuclei at 37 °C. As shown in Fig. S11–S13 (ESI†), all three nanomaterials demonstrated significant fluorescence in the cytoplasm, indicating that TFMDSA NPs, mPEG5K–PCL10K@TFMDSA NPs, and mPEG5K–PLLA10K@TFMDSA NPs could cross the cell membrane and enter the cytoplasm. These results demonstrate that TFMDSA nanoparticles were effectively internalized by cancer cells.
We investigated the effect of different nanoparticle morphologies on the cellular uptake efficiency in HeLa cells cultured with TFMDSA NPs, mPEG5K–PCL10K@TFMDSA NPs, and mPEG5K–PLLA10K@TFMDSA NPs. As shown in Fig. 5, the intensity of intracellular fluorescence increased progressively with the extension of the incubation time from 1 to 6 hours, suggesting sustained cellular uptake of these AIE NPs in HeLa cells. Additionally, the CLSM data of the three nanomaterials co-cultured with HeLa cells for 6 h was compared. Both mPEG5K–PCL10K@TFMDSA NPs and mPEG5K–PLLA10K@TFMDSA NPs exhibited strong green fluorescence, whereas TFMDSA NPs showed weaker fluorescence. This indicates that mPEG5K–PCL10K@TFMDSA NPs and mPEG5K–PLLA10K@TFMDSA NPs were more readily taken up by cells compared to TFMDSA NPs. We propose that this phenomenon is due to the unique square morphology of the polymer-induced aggregation TFMDSA nanoparticles, which provides multiple contact points with the cellular membrane, resulting in stronger adhesion and higher uptake relative to spherical nanoparticles. These experiments confirm that our mPEG5K–PCL10K@TFMDSA NPs and mPEG5K–PLLA10K@TFMDSA NPs, prepared using polymer induction, possess excellent cellular imaging capabilities. Therefore, these nanoparticles are expected to be used as biofluorescent probes for applications in cancer diagnosis and other fields.
 | ||
| Fig. 5 CLSM images of HeLa cells incubated with the DPP NRs for 6 h at 37 °C. Cells are viewed in the blue channel for DAPI, and the green channel for TFMDSADSA. Scale bars represent 20 mm in all images. | ||
Conclusions
In this study, we successfully synthesized mPEG5K–PCL10K@TFMDSA NPs and mPEG5K–PLLA10K@TFMDSA NPs via polymer-induced aggregation, achieving high luminescence efficiency and stability. The incorporation of amphiphilic polymers enhanced intermolecular interactions and constrained molecular vibrations, thereby significantly augmenting the luminescence efficiency of TFMDSA from 6.67% to 40.13% and 10.40%, respectively. These nanoparticles exhibited excellent photostability over seven days and demonstrated low cytotoxicity to HeLa cells, suggesting their potential for bioimaging applications. Cellular uptake studies revealed that these nanoparticles were more efficiently internalized by HeLa cells compared to amorphous nanoparticles due to their unique cuboidal morphology. Our findings indicate that polymer-induced aggregation method improves the optical properties and stability of TFMDSA nanomaterials, positioning them as promising biofluorescent probes for cancer diagnostics and other biomedical applications.Author contributions
X. L., Y. L., B. X., and W. T. conceived and designed the project. X. L. and X. R. synthesized the TFMDSA nanoparticles and performed the experiments on the optical properties. H. S. performed experiments on Fourier transform infrared spectroscopy analysis. X. L., H. S., and Y. L. performed experiments on cellular uptake and cytotoxicity of TFMDSA nanoparticles. Y. L., B. X. and W. T. co-supervised the molecular design and live cell imaging applications. X. L., Y. L. and W. T. wrote the manuscript with comments and suggestions from all co-authors.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (22275065 and 52073116), the Science and Technology Project of Jilin Provincial Department of Education (JJKH20241257KJ) and the Natural Science Foundation of Jilin Province (20240101003JJ).References
- S. Kunjachan, J. Ehling, G. Storm, F. Kiessling and T. Lammers, Chem. Rev., 2015, 115, 10907–10937 CrossRef CAS.
- W. W.-W. Hsiao, Y. Y. Hui, P.-C. Tsai and H.-C. Chang, Acc. Chem. Res., 2016, 49, 400–407 CrossRef CAS.
- K. Shou, C. Qu, Y. Sun, H. Chen, S. Chen, L. Zhang, H. Xu, X. Hong, A. Yu and Z. Cheng, Adv. Funct. Mater., 2017, 27, 1700995 CrossRef.
- Z. Zheng, D. Li, Z. Liu, H.-Q. Peng, H. H. Y. Sung, R. T. K. Kwok, I. D. Williams, J. W. Y. Lam, J. Qian and B. Z. Tang, Adv. Mater., 2019, 31, 1904799 CrossRef CAS PubMed.
- W. Wang, Y. Kong, J. Jiang, Q. Xie, Y. Huang, G. Li, D. Wu, H. Zheng, M. Gao, S. Xu, Y. Pan, W. Li, R. Ma, M. X. Wu, X. Li, H. Zuilhof, X. Cai and R. Li, Angew. Chem., Int. Ed., 2020, 59, 22431–22435 CrossRef CAS PubMed.
- R. Xu, D. Dang, Z. Wang, Y. Zhou, Y. Xu, Y. Zhao, X. Wang, Z. Yang and L. Meng, Chem. Sci., 2022, 13, 1270–1280 RSC.
- X. Liang, Y. Li, X. Li, L. Jing, Z. Deng, X. Yue, C. Li and Z. Dai, Adv. Funct. Mater., 2015, 25, 1451–1462 CrossRef CAS.
- H. Gao, Y. Bi, X. Wang, M. Wang, M. Zhou, H. Lu, J. Gao, J. Chen and Y. Hu, ACS Biomater. Sci. Eng., 2017, 3, 3628–3634 CrossRef CAS.
- S. K. Mishra and S. Kannan, ACS Biomater. Sci. Eng., 2017, 3, 3607–3619 CrossRef CAS PubMed.
- N. Lee, S. H. Choi and T. Hyeon, Adv. Mater., 2013, 25, 2641–2660 CrossRef CAS PubMed.
- L. Cheng, J. Liu, X. Gu, H. Gong, X. Shi, T. Liu, C. Wang, X. Wang, G. Liu, H. Xing, W. Bu, B. Sun and Z. Liu, Adv. Mater., 2014, 26, 1886–1893 CrossRef CAS PubMed.
- X. Ning, W. Seo, S. Lee, K. Takemiya, M. Rafi, X. Feng, D. Weiss, X. Wang, L. Williams, V. M. Camp, M. Eugene, W. R. Taylor, M. Goodman and N. Murthy, Angew. Chem., Int. Ed., 2014, 53, 14096–14101 CrossRef CAS PubMed.
- J. Zhang, C. Yang, R. Zhang, R. Chen, Z. Zhang, W. Zhang, S.-H. Peng, X. Chen, G. Liu, C.-S. Hsu and C.-S. Lee, Adv. Funct. Mater., 2017, 27, 1605094 CrossRef PubMed.
- K. Li, Z. Zhu, P. Cai, R. Liu, N. Tomczak, D. Ding, J. Liu, W. Qin, Z. Zhao, Y. Hu, X. Chen, B. Z. Tang and B. Liu, Chem. Mater., 2013, 25, 4181–4187 CrossRef CAS.
- X. Ji, F. Peng, Y. Zhong, Y. Su, X. Jiang, C. Song, L. Yang, B. Chu, S.-T. Lee and Y. He, Adv. Mater., 2015, 27, 1029–1034 CrossRef CAS PubMed.
- R. Vankayala, C.-L. Kuo, K. Nuthalapati, C.-S. Chiang and K. C. Hwang, Adv. Funct. Mater., 2015, 25, 5934–5945 CrossRef CAS.
- C. Guo, J. Hu, L. Kao, D. Pan, K. Luo, N. Li and Z. Gu, ACS Biomater. Sci. Eng., 2016, 2, 860–870 CrossRef CAS.
- L. Yan, H. Wang, A. Zhang, C. Zhao, Y. Chen and X. Li, J. Mater. Chem. B, 2016, 4, 5560–5566 RSC.
- A. Nicol, W. Qin, R. T. K. Kwok, J. M. Burkhartsmeyer, Z. Zhu, H. Su, W. Luo, J. W. Y. Lam, J. Qian, K. S. Wong and B. Z. Tang, Chem. Sci., 2017, 8, 4634–4643 RSC.
- Y. Cui, R. Zhang, L. Yang and S. Lv, Chin. Chem. Lett., 2019, 30, 1078–1082 CrossRef CAS.
- D. Huang, S. Lin, Q. Wang, Y. Zhang, C. Li, R. Ji, M. Wang, G. Chen and Q. Wang, Adv. Funct. Mater., 2019, 29, 1806546 CrossRef.
- J. Chen, S. Feng, M. Chen, P. Li, Y. Yang, J. Zhang, X. Xu, Y. Li and S. Chen, Small, 2020, 16, 2002054 CrossRef CAS.
- C. Li, G. Chen, Y. Zhang, F. Wu and Q. Wang, J. Am. Chem. Soc., 2020, 142, 14789–14804 CrossRef CAS PubMed.
- X. Wang, Y. Song, G. Pan, W. Han, B. Wang, L. Cui, H. Ma, Z. An, Z. Xie, B. Xu and W. Tian, Chem. Sci., 2020, 11, 10921–10927 RSC.
- J. Luo, Z. Xie, J. W. Y. Lam, L. Cheng, H. Chen, C. Qiu, H. S. Kwok, X. Zhan, Y. Liu, D. Zhu and B. Z. Tang, Chem. Commun., 2001, 1740–1741 RSC.
- B. Xu, J. He, Y. Mu, Q. Zhu, S. Wu, Y. Wang, Y. Zhang, C. Jin, C. Lo, Z. Chi, A. Lien, S. Liu and J. Xu, Chem. Sci., 2015, 6, 3236–3241 RSC.
- J. Hwang, P. Nagaraju, M. J. Cho and D. H. Choi, Aggregate, 2023, 4, e199 CrossRef CAS.
- C. Yan, J. Dai, Y. Yao, W. Fu, H. Tian, W.-H. Zhu and Z. Guo, Nat. Protoc., 2023, 18, 1316–1336 CrossRef CAS PubMed.
- Z. Zhang, Z. Deng, L. Zhu, J. Zeng, X. M. Cai, Z. Qiu, Z. Zhao and B. Z. Tang, Regenerative Biomater., 2023, 10, rbad044 CrossRef CAS.
- S. Huang, S. Bai, T. Luo, F. Zheng, D. Fan, Y. Zhou, J. Dong, F. Chen and W. Zeng, Adv. Funct. Mater., 2024, 2409292 CrossRef CAS.
- J.-L. Wang, Y. Chen, J.-X. Song, B.-W. Guo, F.-W. Xia, Y. Wan, W.-X. Wu, C. Zhang, S. Feng and M.-Y. Wu, Adv. Funct. Mater., 2024, 34, 2312162 CrossRef CAS.
- S.-Y. Yang, Y. Chen, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Chem. Soc. Rev., 2024, 53, 5366–5393 RSC.
- Y. Zuo, R. T. K. Kwok, J. Sun, J. W. Y. Lam and B. Z. Tang, Chem. Phys. Rev., 2024, 5, 011309 CrossRef CAS.
- K. Li, W. Qin, D. Ding, N. Tomczak, J. Geng, R. Liu, J. Liu, X. Zhang, H. Liu, B. Liu and B. Z. Tang, Sci. Rep., 2013, 3, 1150 CrossRef.
- L. Zong, H. Zhang, Y. Li, Y. Gong, D. Li, J. Wang, Z. Wang, Y. Xie, M. Han, Q. Peng, X. Li, J. Dong, J. Qian, Q. Li and Z. Li, ACS Nano, 2018, 12, 9532–9540 CrossRef CAS.
- C. Yang, Q. T. Trinh, X. Wang, Y. Tang, K. Wang, S. Huang, X. Chen, S. H. Mushrif and M. Wang, Chem. Commun., 2015, 51, 3375–3378 RSC.
- Y. Dong, B. Xu, J. Zhang, H. Lu, S. Wen, F. Chen, J. He, B. Li, L. Ye and W. Tian, CrystEngComm, 2012, 14, 6593–6598 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4tb01825g |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |