
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMolecularly bridged design of an electron-delocalized dual redox-active organic electrode for high-efficiency capacitive deionization and water treatment†
Xinyue Zhanga,
Haoyuan Qiua,
Zhiyong Hana,
Lei Kec,
Mugilan Narayanasamyd,
Can Cuia,
Jun Yang *a,
Minjie Shia and
Edison Huixiang Ang*b
*a,
Minjie Shia and
Edison Huixiang Ang*b
aSchool of Materials Science and Engineering, Jiangsu University of Science and Technology, Jiangsu 212003, P. R. China. E-mail: iamjyang@just.edu.cn
bNatural Sciences and Science Education, National Institute of Education, Nanyang Technological University, Singapore 637616, Singapore. E-mail: edison.ang@nie.edu.sg
cJiangsu Hilong Technology Co. Ltd., Zhenjiang, 212003, P. R. China
dDepartment of Chemical Sciences and Bernal Institute, University of Limerick, Castletroy, V94 T9PX, Limerick, Ireland
First published on 28th July 2025
Abstract
Capacitive deionization (CDI) is emerging as a promising technology for efficient water treatment, where the electrochemical performance is strongly influenced by the activity of electrode materials. Among these, faradaic organic electrodes with pseudocapacitive behavior offer great potential but remain limited due to insufficient redox-active sites and poor electron affinity, factors that constrain their ion adsorption capacity and desalination rate. In this study, we present a novel electron-delocalized organic molecular architecture, PTAP, which features a rigid conjugated backbone and dual redox-active centers located at C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N and CO coordination sites. The molecular structure, optimized via extensive π-electron delocalization and a narrow HOMO–LUMO energy gap, significantly enhances Na+ adsorption kinetics and facilitates efficient charge carrier transport. A combination of comprehensive experimental techniques and density functional theory (DFT) calculations was employed to elucidate the Na+ electro-adsorption pathways and identify the active redox moieties. The resulting PTAP-integrated CDI device exhibited outstanding desalination performance, achieving an exceptionally high salt removal capacity of 91.50 mg g−1—far exceeding that of conventional organic-electrode CDI systems (typically < 60 mg g−1)—and a rapid removal rate of 3.05 mg g−1 min−1 at 1.2 V, while maintaining excellent cycling stability (∼95.55% retention over 100 cycles). Furthermore, a multi-module CDI system incorporating PTAP electrodes was developed, demonstrating high desalination efficiency in hypersaline environments. This system also enabled preliminary exploration of sustainable real-world applications, including dye-contaminated wastewater remediation and energy harvesting, underscoring the versatility and transformative potential of PTAP-based CDI technology.
N and CO coordination sites. The molecular structure, optimized via extensive π-electron delocalization and a narrow HOMO–LUMO energy gap, significantly enhances Na+ adsorption kinetics and facilitates efficient charge carrier transport. A combination of comprehensive experimental techniques and density functional theory (DFT) calculations was employed to elucidate the Na+ electro-adsorption pathways and identify the active redox moieties. The resulting PTAP-integrated CDI device exhibited outstanding desalination performance, achieving an exceptionally high salt removal capacity of 91.50 mg g−1—far exceeding that of conventional organic-electrode CDI systems (typically < 60 mg g−1)—and a rapid removal rate of 3.05 mg g−1 min−1 at 1.2 V, while maintaining excellent cycling stability (∼95.55% retention over 100 cycles). Furthermore, a multi-module CDI system incorporating PTAP electrodes was developed, demonstrating high desalination efficiency in hypersaline environments. This system also enabled preliminary exploration of sustainable real-world applications, including dye-contaminated wastewater remediation and energy harvesting, underscoring the versatility and transformative potential of PTAP-based CDI technology.
Introduction
The global freshwater crisis has reached critical levels, primarily driven by rapid population growth, industrial expansion, and the intensification of modern agriculture. These factors place immense pressure on limited freshwater resources, exacerbating water scarcity across many regions.1–5 Despite water covering approximately 71% of the Earth's surface, a staggering 97.5% is saline and unsuitable for direct human consumption or agricultural use without substantial treatment.6,7 This stark disparity between water abundance and availability has intensified the global pursuit of sustainable and efficient desalination technologies. Conventional desalination methods, including thermal-based processes (e.g., multi-stage flash distillation) and membrane-based techniques (e.g., reverse osmosis), have gained widespread adoption.8–10 However, these technologies are often hindered by high energy demands, operational complexity, and environmental concerns related to brine disposal and chemical usage.11–15 These limitations underscore the urgent need for next-generation desalination approaches that are energy-efficient, economically viable, environmentally friendly, and scalable.Capacitive deionization (CDI) has emerged as a promising electrochemical desalination technology, offering a sustainable alternative to traditional methods.16 Unlike energy-intensive systems, CDI operates on an electrosorption mechanism, where salt ions are electrostatically adsorbed onto electrodes under an applied electric field and subsequently released during regeneration.17–20 This process allows for precise control over water quality and achieves substantial energy savings, particularly when treating brackish or low-to-moderate salinity water sources.21–24 However, the performance of CDI is highly dependent on the characteristics of electrode materials. Carbon-based materials, such as carbon nanotubes, graphene nanosheets, and carbon nanofibers, have been widely employed due to their excellent electrical conductivity and chemical stability. Nevertheless, their reliance on electric double-layer capacitance limits their ion adsorption capacity (typically <50 mg g−1), constraining the overall efficiency of CDI systems.25,26 This shortcoming has prompted efforts to develop alternative materials with higher desalination performance.27–29
To address these limitations, researchers have increasingly turned to faradaic materials that exhibit pseudocapacitive behavior, enabling enhanced ion storage.30 faradaic electrode materials, which store ions through reversible redox reactions, offer significantly higher charge storage capabilities. Recent research has predominantly focused on inorganic faradaic materials, particularly transition metal oxides (e.g., MnO2, NiO) and sulfides (e.g., MoS2), due to their high theoretical capacities.31–34 However, these materials often undergo severe structural degradation during cycling due to repeated ion insertion/extraction, leading to irreversible phase transitions, metal dissolution, and rapid capacity fading.35 While inorganic intercalation compounds have shown promise, they often suffer from drawbacks such as metal ion leaching, irreversible phase transitions, and volumetric expansion during electrosorption.36–39 In contrast, organic materials have emerged as compelling candidates for faradaic CDI electrodes, thanks to their abundance, environmental sustainability, and molecular tunability.40,41 Their electronic structures can be precisely modulated via molecular design and functional group engineering, enabling tailored ion adsorption properties. These materials exhibit pseudocapacitive behaviour through redox-active functional groups, allowing efficient ion capture without the drawbacks of phase change or structural degradation.42 Despite these advantages, organic electrodes face persistent challenges, including limited electron mobility and insufficient redox activity, which restrict their overall desalination performance.43,44 Therefore, the development of novel organic materials with enhanced electronic properties and high ion storage capacity is essential for advancing CDI technologies.
In this study, we report the design and synthesis of a novel electron-delocalized dual-redox organic molecule (PTAP) via a one-step reaction, tailored as a high-performance CDI electrode. The PTAP molecule is engineered with multiple CN and CO functional groups that serve as active redox sites, supporting robust pseudocapacitive behavior. Its rigid conjugated backbone and extensive π-electron delocalization result in a remarkably low HOMO–LUMO energy gap (1.97 eV), promoting efficient electron transport and superior redox activity. When employed as a CDI electrode, PTAP demonstrates outstanding Na+ adsorption capabilities, achieving a high specific capacitance of 412.73 F g−1 and maintaining ∼92% capacity retention over 5000 charge–discharge cycles in NaCl electrolyte. The PTAP-based CDI device exhibits exceptional desalination performance, with a salt removal capacity of 91.50 mg g−1—over 50% higher than that of conventional organic electrode systems—and a time-averaged removal rate of 3.05 mg g−1 min−1 at 1.2 V, while retaining excellent regeneration stability (∼95.55% over 100 cycles). Moreover, the device also demonstrates effective organic dye removal and energy recovery, highlighting its versatility and potential for sustainable water purification and multifunctional environmental applications.
Results and discussion
Structural characterization
The PTAP organic molecule was synthesized via a straightforward one-step condensation reaction using perylene-3,4,9,10-tetracarboxylic dianhydride (PTCDA) and phenazine-2,3-diyldiamine (DAP) as precursors, as illustrated in Fig. 1a. Solid-state 13C NMR spectroscopy (Fig. 1b) provides detailed insights into the carbon environments within the PTAP molecule. The spectrum reveals multiple distinct resonances: the peaks at 121.6, 131.8, and 139.8 ppm were assigned to aromatic CC bonds in benzene rings, while the peaks at 123.0, 127.1, and 132.7 ppm were attributed to various CC environments within the aromatic framework. Signals at 141.7 and 143.9 ppm were ascribed to imine (CN) groups, and the pronounced peak at 193.3 ppm was assigned to carbonyl (CO) groups. Morphological characterization by scanning electron microscopy (SEM) reveals that the PTAP product adopted a stacked lamellar architecture with an average thickness of ∼200 nm (SEM images of PTCDA and DAP are shown in Fig. S1†). Elemental analysis via energy-dispersive X-ray spectroscopy (EDS) confirmed a homogeneous distribution of carbon (79 at%), nitrogen (17 at%), and oxygen (4 at%) across the structure, aligning well with the theoretical stoichiometry (Fig. 1c). Electronic structure analysis through partial density of states (PDOS) (Fig. 1d) revealed significant electron density near the Fermi level, primarily arising from the lone pair electrons of nitrogen and oxygen heteroatoms, which participate in conjugation and contribute to enhanced electrochemical activity. The X-ray diffraction (XRD) patterns (Fig. 1e) of the precursor materials PTCDA and DAP exhibited sharp Bragg reflections, indicative of well-defined crystallinity. In contrast, the synthesized PTAP material displayed a broad diffraction peak centered at 27.3°, suggesting disordered molecular packing dominated by π–π stacking interactions. This interpretation was further supported by reduced density gradient (RDG) analysis (Fig. 1f), which revealed characteristic low-gradient isosurfaces (−0.02 to 0.00 a.u.) corresponding to non-covalent π–π interactions.45,46 Additionally, Fourier-transform infrared (FT-IR) spectroscopy (Fig. S2†) confirmed the molecular structure, showing characteristic absorption bands at 1767 and 1689 cm−1, corresponding to the stretching vibrations of CO and CN bonds, respectively.47 Collectively, these comprehensive spectroscopic and structural analyses confirm the successful synthesis of the PTAP molecule.
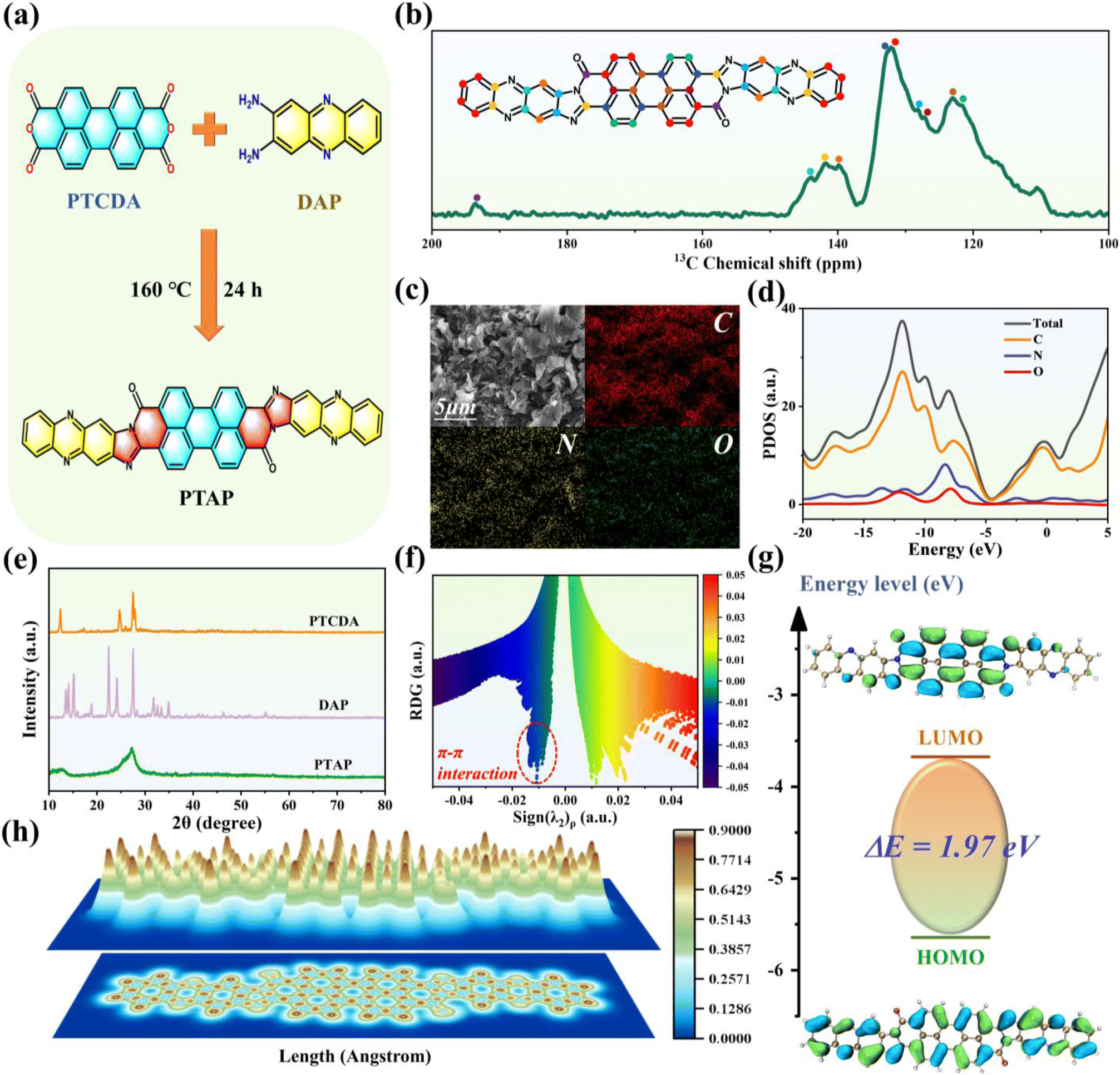 | ||
| Fig. 1 (a) Schematic illustration of the one-step condensation synthesis of the PTAP molecule. (b) Solid-state 13C NMR spectrum. (c) SEM image and corresponding EDS elemental mapping images. (d) PDOS pattern with the orbital contributions. (e) XRD pattern. (f) Plots of RDG vs. sign (λ2)ρ, and (g) HOMO–LUMO orbital distributions of the PTAP molecule. (h) LOL-π color-filled map. | ||
The optical bandgap (Eg) of the PTAP molecule was derived from UV-vis spectroscopy (Fig. S3†), representing the minimum photon energy needed to excite electrons from the valence to the conduction band. Remarkably, PTAP demonstrates an ultranarrow bandgap of ∼1.64 eV, significantly lower than those of its monomeric precursors PTCDA (2.52 eV) and DAP (2.14 eV) (Fig. S4†). This drastic reduction in Eg implies that PTAP facilitates electron excitation with lower energy input, thereby accelerating redox kinetics and enhancing charge mobility by minimizing intramolecular kinetic barriers. Further computational analysis confirms that PTAP exhibits a HOMO–LUMO gap of 1.97 eV (Fig. 1g),48 narrower than those of both its monomers and other known electroactive organic materials (Fig. S5†). The diminished HOMO–LUMO separation underscores PTAP's superior intrinsic conductivity, heightened thermodynamic stability, and an optimized electronic structure, all of which are advantageous for its performance in electroactive applications. Thermal stability analysis demonstrates that the PTAP molecule retains 83.58% of its mass at 700 °C, in stark contrast to the complete decomposition of its PTCDA and DAP precursors at the same temperature (Fig. S6†). This superior thermal stability is attributed to the robust conjugated structure of the PTAP framework. To further explore the electronic characteristics of PTAP, the localized orbital locator-π (LOL-π) method was employed to assess its electron delocalization behavior. In this analysis, a threshold LOL value of 0.5 differentiates between localized (LOL < 0.5) and delocalized (LOL > 0.5) electrons.49,50 As illustrated in Fig. 1h, the LOL-π map reveals a consistent electron density distribution along the molecular backbone, with values ranging from 0.621 to 0.862, indicative of substantial π-electron delocalization. This delocalization significantly benefits electron transport pathways and facilitates improved ion diffusion kinetics within the PTAP electrode.
Electrochemical properties
The electrochemical performance of the PTAP electrode was systematically evaluated using cyclic voltammetry (CV) and galvanostatic charge–discharge (GCD) measurements in a 0.5 M NaCl aqueous electrolyte. As shown in Fig. 2a, the CV curves exhibit well-defined redox peaks at various scan rates, indicating the presence of redox-active sites that facilitate reversible ion coordination. The broad anodic and cathodic peaks reflect the oxidation and reduction processes of the PTAP electrode. For comparison, the CV curves of the PTCDA and DAP monomers are provided in Fig. S7†. Notably, the peak potentials exhibit minimal shifts with increasing scan rates (Fig. 2d), highlighting the electrode's stable and efficient faradaic behaviour. The GCD profiles in Fig. 2b reveal high specific capacitances of 412.73, 372.19, 338.75, 319.00, and 304.27 F g−1 at current densities of 1, 2, 4, 6, and 8 A g−1, respectively. These values surpass those of previously reported Na+-capture electrode materials (Fig. S8†), underscoring the superior rate capability and capacity retention of the PTAP electrode. The PTAP electrode delivers exceptional electrochemical performance, owing to its unique molecular design and superior electronic properties. At the molecular level, PTAP features a rigid conjugated backbone integrated with a synergistic dual-redox mechanism, where CO and CN functional groups operate in tandem—outperforming conventional single-functional-group organic electrodes and significantly enhancing ion capture capacity. Furthermore, PTAP exhibits outstanding electronic characteristics, including an ultra-narrow HOMO–LUMO gap of just 1.97 eV, which ensures high electronic conductivity, rapid charge transfer efficiency, and robust thermodynamic stability. Together, these attributes make PTAP a highly efficient and stable electrode material.
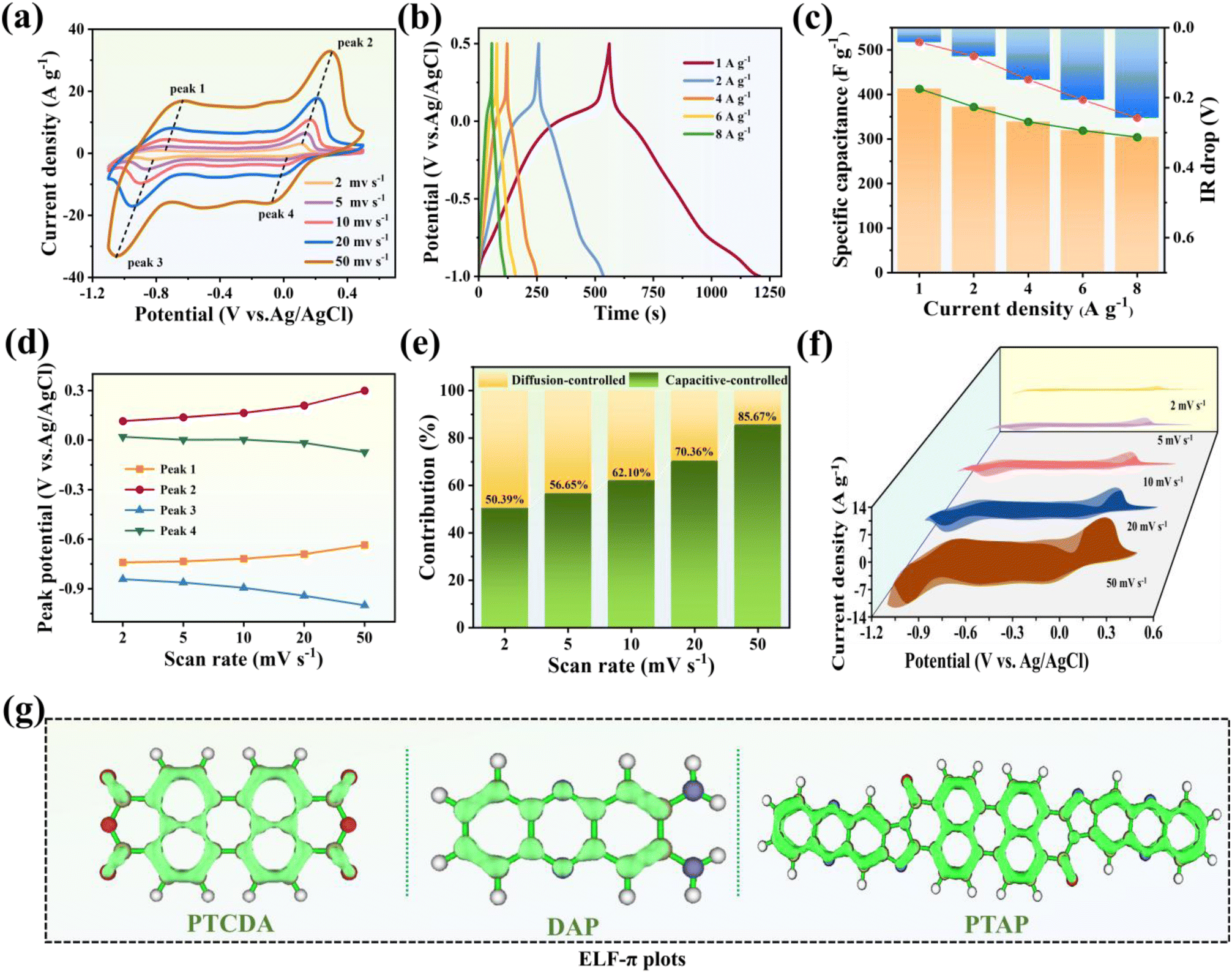 | ||
| Fig. 2 (a) Cyclic voltammetry (CV) curves of the PTAP electrode in 0.5 M NaCl solution at different scan rates. (b) Galvanostatic charge–discharge (GCD) profiles and (c) corresponding specific capacitance values and “IR drops” under various current densities. (d) Variation of anodic and cathodic peak potentials with scan rates derived from CV curves. (e) Percentage contribution of capacitive-controlled charge storage at different scan rates. (f) Separation of capacitive and diffusion-controlled contributions, with coloured regions representing capacitive effects. (g) Electron localization function for π electrons (ELF-π) of the PTAP molecule, illustrating electron delocalization characteristics. | ||
Furthermore, the IR drop remains consistently low across the range of current densities (Fig. 2c), indicating excellent charge transport and low internal resistance. To gain insight into the charge storage mechanism, a capacitive contribution analysis was conducted over a range of scan rates (Fig. 2f). The results show a progressive increase in capacitive-controlled contributions with increasing scan rate, emphasizing the electrode's strong pseudocapacitive behaviour. At a scan rate of 50 mV s−1, the surface-controlled capacitance accounts for 85.67% of the total response (Fig. 2e), demonstrating rapid electrochemical kinetics and high electrosorption capacity. Additionally, compared to the individual PTCDA and DAP monomers, the PTAP molecule exhibits significantly enhanced π-conjugation, as evidenced by continuous isosurface rings across the molecular framework (Fig. 2g). This extended conjugated network not only enhances molecular stability but also facilitates efficient electron transport throughout the electrode.
The electrochemical kinetics of the PTAP electrode were systematically investigated using electrochemical impedance spectroscopy (EIS) at various charge–discharge states, as shown in Fig. 3a (the EIS data are listed in Table S1†). The Na+ diffusion kinetics were quantitatively assessed through Warburg impedance analysis. As illustrated in Fig. 3b, the calculated diffusion coefficients ranged from 4.96 × 10−12 to 9.8 × 10−12 cm2 s−1, indicating excellent Na+ mobility within the PTAP electrode and highlighting its superior ion transport characteristics.
 | ||
| Fig. 3 (a) EIS plots of the PTAP electrode at various discharged/charged states and (b) corresponding Na+ diffusion coefficients. (c) Long-term cycling stability of the PTAP electrode over 5000 cycles. (d) The calculated HOMA values for the PTAP molecule before and after complete Na+ adsorption. (e) UV-Vis spectra and digital photographs of the electrolyte at different cycles. | ||
To assess long-term durability, a cycling stability test was conducted (Fig. 3c), revealing outstanding electrochemical stability with a capacity retention of 91.16% after 5000 cycles. In contrast, the PTCDA and DAP monomers exhibited declining performance after just 100 cycles (Fig. S9†), underscoring the enhanced structural resilience of PTAP. A deeper insight into the structural stability was obtained through the Harmonic Oscillator Model of Aromaticity (HOMA) analysis, depicted in Fig. 3d. The evolution of aromaticity in PTAP before and after Na+ insertion (PTAP and PTAP+8Na) was evaluated. The HOMA index, which ranges from 0 to 1, quantifies aromatic character, with values approaching 1 indicating strong aromaticity and structural integrity. Notably, the HOMA values remained close to 1 after full Na+ adsorption, demonstrating the preservation of the conjugated structure and confirming the excellent stability of the PTAP molecule. Further evidence of the electrode's stability was provided by UV-Vis spectroscopy (Fig. 3e). Throughout the cycling test, the electrolyte solution remained optically clear, and no characteristic absorption bands were detected in the UV-Vis spectra, indicating the absence of electrode dissolution and ensuring long-term chemical robustness. Together, these comprehensive electrochemical and spectroscopic analyses confirm the exceptional performance of the PTAP electrode, particularly its fast, stable, and efficient Na+ storage and retention capabilities.
To investigate the structural evolution of the PTAP electrode under different electrochemical conditions, ex situ X-ray photoelectron spectroscopy (XPS) was utilized, providing critical information about the fundamental electrochemical processes. Analysis of the N 1s (Fig. 4a) and O 1s (Fig. 4b) spectra in the pristine, fully discharged, and fully charged states revealed significant peak shifts, indicating reversible changes in chemical bonding during electrochemical cycling. Specifically, during discharge, the CO bond (531.8 eV) was reduced to a C–O bond (533.0 eV), while the CN bond (398.9 eV) was converted into a C–N bond (400.9 eV). These changes were fully reversed upon charging, underscoring the excellent electrochemical reversibility of the PTAP electrode.
 | ||
| Fig. 4 Ex situ XPS spectra of (a) N 1s, (b) O 1s and (c) Na 1s for the PTAP electrode in pristine, fully charged and discharged states. (d) Calculated total energies of sodium-intercalated PTAP structures, indicating stability variations with sodium incorporation, and (e) depicting key electrochemical reactions and ion transport dynamics. (f) SEM image and the corresponding EDS mapping images at the adsorption state. (g) The corresponding ELF image of a PTAP molecule. | ||
Furthermore, the ex situ Na 1s XPS spectra (Fig. 4c) showed no detectable Na signal in the pristine state. However, a distinct peak at 1071.8 eV emerged during the charging process, clearly confirming the successful capture of Na+ ions by the PTAP electrode. The redox mechanism was further elucidated through in situ Raman spectroscopy. As shown in Fig. S10,† Raman spectra collected during discharge and charge revealed dynamic changes in bonding. Prior to testing, peaks at 1254 cm−1 and 1615 cm−1 were observed, corresponding to the C–N and C–O bonds, respectively.51 As discharge progressed, new peaks appeared at 1251 and 1604 cm−1, reflecting changes in the C–N and C–O bonding environments. Simultaneously, the intensities of the original C–N and C–O peaks diminished. This suggests that during discharge, Na+ ions chelate with the C–N bonds via a coordination mechanism, while binding to the C–O bonds through an enolation process. During the subsequent charging process, the signals associated with the reduced C–N and C–O bonds weakened, while the original C–N and CO bonds re-emerged due to oxidation. These observations strongly confirm the high redox reversibility of both the imine (C–N ↔ C–N) and carbonyl (C–O ↔ CO) functional groups in PTAP during Na+ ion insertion and extraction.
The mechanism of the PTAP electrode in the desalination process was further elucidated through density functional theory (DFT) simulations (Fig. 4d). The molecular electrostatic potential (MESP) map (inset of Fig. 4d) reveals that the regions surrounding the two carbonyl groups and six imine groups in the PTAP molecule exhibit significantly higher electronegativity compared to other areas. This indicates a strong tendency for these sites to coordinate with Na+ ions, acting as redox-active centers. Further analysis identified two distinct active regions within the PTAP molecule, designated as site A and site B (Fig. 4e). The Na+ insertion process was modelled in three sequential stages: PTAP, PTAP+4Na, and PTAP+8Na, allowing evaluation of the reaction pathway and associated energy changes. The initial total energy of pristine PTAP was calculated to be −6.612 × 104 eV. During discharge, Na+ ions first interact with site A—comprising two CN and two CO bonds—to form PTAP+4Na, with a total energy of −8.377 × 104 eV. Subsequently, additional Na+ ions bind at site B, which contains four CN bonds, forming PTAP+8Na with a total energy of −10.145 × 104 eV (Fig. S11†). These computational findings are further supported by SEM and energy-dispersive X-ray spectroscopy (EDS) analyses of the fully charged PTAP electrode (Fig. 4f), which show a uniform distribution of Na across the electrode surface. Notably, the intensity of the Na 1s peak significantly decreases upon discharge, nearly reverting to its original state, providing strong evidence for the effective release of Na+ ions from the PTAP electrode.52–55 Additionally, electron localization function (ELF) analysis (Fig. 4g) reveals substantial electron density accumulation, confirming that the extended π-electron delocalization within the PTAP molecular framework lowers the energy barrier for Na+ capture and enhances ion adsorption efficiency.
CDI applications
To maximize the electrochemical capacity generated by redox reactions, we developed an asymmetric design that combines a pseudocapacitive PTAP molecular electrode with excellent Na+ adsorption capability and commercial activated carbon (AC) known for its high surface area and Cl− absorption properties. This innovative pairing enables the creation of a hybrid capacitive deionization (CDI) system with significantly improved total salt removal capacity. For experimental validation, we constructed a CDI device (detailed configuration in Fig. S12†) using PTAP as the anode and AC as the cathode, incorporating an anion exchange membrane adjacent to the AC cathode to block cation adsorption when reversing voltage polarity. The desalination process was continuously monitored using a conductivity measurement system (Fig. 5a), with salt concentrations estimated from a pre-established conductivity–concentration calibration curve (Fig. S13†). The CDI device's performance was systematically assessed under various applied voltages (0.8–1.4 V). The CDI device's desalination performance was assessed across a voltage range of 0.8–1.4 V, well below its theoretical safe operating limit of 1.9 V (Fig. S14†). As shown in Fig. 5b, the current response exhibited distinct, rapid oscillations indicative of sequential ion adsorption and desorption. Concurrently, substantial drops in solution conductivity (Fig. 5c) confirmed the occurrence of electrosorption processes. Our results revealed a strong dependence of desalination efficiency on applied voltage. Higher voltages enhanced ion transport and electrode capture due to stronger electrostatic forces. Quantitative analysis (Fig. 5d and e) showed that increasing the voltage improved salt removal capacity. However, this gain came with a substantial energy cost. Specifically, raising the voltage from 1.2 V to 1.4 V led to a 36.22% increase in energy consumption, while salt removal capacity improved by only 14.15%. This disproportionate trade-off was accompanied by a decline in charge efficiency to 69.53% at 1.4 V (Fig. 5f), suggesting the onset of parasitic electrochemical reactions at higher voltages, which may affect long-term stability and energy efficiency. The specific test results are listed in Table S2.† A comprehensive evaluation of energy consumption versus desalination efficiency identified 1.2 V as the optimal operating voltage. At this voltage, the CDI device achieved an outstanding balance between performance and energy use, delivering a high salt removal capacity of 91.50 mg g−1 with a low energy input of 0.846 W h g−1, and an average removal rate of 3.05 mg g−1 min−1 with a maximum (peak) salt removal rate of 18.83 mg g−1 min−1.56 These values surpass those of state-of-the-art organic electrode-based CDI systems (Fig. 5g)57–66 (refer to Table S3† for details). Long-term operational stability was confirmed through 100 consecutive adsorption–desorption cycles at 1.2 V. The device retained 95.55% of its initial performance (Fig. 5h), with specific energy consumption (SEC) remaining consistently stable between 70.05 and 73.80 kJ mol−1 per cycle. These findings highlight the ability of the system to minimize side reactions and maintain efficiency over extended use. Additional performance data are provided in Fig. S15–S19 and Table S4.† A comparison was made of salt removal capacity and average removal rate with other CDI devices previously reported under the same applied voltage (see Table S5† for details). | ||
| Fig. 5 (a) Schematic diagram of the desalination experiments performed using the PTAP-based CDI device. (b) Current response profiles and (c) corresponding variations in solution conductivity under different applied voltages. (d) Salt removal capacities obtained during the electro-sorption process. (e) Ragone plots and (f) corresponding correlation between salt removal capacities, charge efficiency, and energy consumption at various applied voltages. (g) Comparison of salt removal capacity with previously reported CDI devices utilizing different organic electrodes. (h) Cyclic regeneration at 1.2 V through repeated adsorption–desorption cycles. | ||
Fig. 6a presents a schematic diagram of the hybrid capacitive deionization (HCDI) process using PTAP for the adsorption of metal ions and dye molecules. Beyond its outstanding desalination performance, the CDI device also exhibits excellent potential for treating industrial wastewater containing cationic dyes, with methylene blue (MB) selected as a model contaminant. To simulate textile industry effluents, MB solutions with an initial concentration of 100 mg L−1 were prepared. CDI treatment was performed at an applied voltage of 1.2 V, and dye removal was monitored using UV-vis spectroscopy. As shown in Fig. 6b, the near-complete disappearance of absorption peaks following treatment indicates highly efficient dye removal. This result is visually supported by the dramatic change in solution appearance from deep coloration to complete transparency (Fig. 6c). Moreover, the CDI device maintains exceptional removal rates approaching 100%, even after 50 consecutive adsorption–desorption cycles, with negligible performance degradation (Fig. 6d). Complementary molecular electrostatic potential (MESP) mapping (Fig. 6e) further highlights the electron-rich regions around the CO and CN groups in the PTAP molecule, which serve as active sites for cationic dye adsorption. Conductivity profiles of the saline solution over three HCDI stages (Fig. S20†) confirm stable and efficient ion removal. After three successive HCDI cycles, the Na+ concentration was reduced to 130.1 mg L−1, and Cl− concentration to 200.8 mg L−1 (Fig. 6f), surpassing the safety limits established by China's national standard GB5749-2022. In addition to its purification performance, the CDI device demonstrated superior energy recovery capabilities. Operating within a voltage window of −1.2 V to 1.2 V, the system achieved an energy recovery rate of 36.97% at a current density of 4 A g−1 (Fig. 6g–i), indicating its potential to partially reclaim energy during operation. This combination of high-efficiency salt and dye removal, excellent cycling stability, and notable energy recovery highlights the CDI device as a sustainable and high-performance solution for water purification and environmental remediation.
 | ||
| Fig. 6 (a) Schematic diagram of HCDI of PTAP absorbing metal ions and dye ions. (b) UV-Vis absorption spectra and (c) digital photos of the MB dye-laden solution before and after CDI treatment. (d) Removal efficiencies of MB cationic dye with the CDI device over 50 consecutive adsorption–desorption cycles. (e) MESP mapping illustrating the electron-rich regions (CO and CN sites) responsible for cationic adsorption. (f) Salt ion concentrations profiles. (g) Energy recovery performance of the CDI device during the discharging process. (h) Voltage profiles of the CDI device during charging and discharging. (i) Energy recovery rates at various applied current densities. | ||
Conclusion
In summary, this study successfully engineered and synthesized an electron-delocalized, dual-redox PTAP organic electrode for advanced electrochemical desalination applications. By employing a strategic molecular design approach, multiple CN and CO functional groups were incorporated into a rigid, conjugated backbone, resulting in extensive π-electron delocalization and a notably narrow HOMO–LUMO energy gap of 1.97 eV. This tailored molecular architecture endows the electrode with exceptional electrochemical properties, including a high specific capacitance of 412.73 F g−1 and excellent cycling stability in NaCl electrolyte. The PTAP-based capacitive deionization (CDI) device demonstrated outstanding desalination performance, achieving a high salt removal capacity of 91.50 mg g−1 at an applied voltage of 1.2 V—far exceeding the performance of state-of-the-art organic electrode-based CDI systems (<60 mg g−1)—and an impressive average salt removal rate of 3.05 mg g−1 min−1. Additionally, the device maintained ∼95.55% of its initial salt removal capacity after 100 adsorption–desorption cycles, highlighting its robust long-term stability. Beyond desalination, the PTAP-based CDI device exhibited multifunctional capabilities, including efficient removal of organic dyes and effective energy recovery during operation. This work offers valuable insights into the molecular-level design of high-performance organic electrodes and establishes a versatile platform for the next generation of high-capacity, multifunctional electrochemical water treatment systems. Considering the application prospects of PTAP electrodes in HCDI, future research priorities and challenges primarily include: (1) large-scale production of PTAP electrode materials or development of analogous organic electrode materials; (2) improving the recyclability or efficient decomposition of electrode materials to promote environmental sustainability; (3) integrating in situ characterization with computational modeling to investigate the complex electrochemical mechanisms of electrode materials; (4) expanding the scale of HCDI systems using organic electrodes and discovering broader functional and practical uses.
Data availability
The authors confirm that the data supporting the findings of this study are available within the article [and/or its ESI†]. More detailed data are available on request from the corresponding author, upon reasonable request.Author contributions
The manuscript was collaboratively authored by all contributors. All authors reviewed and approved the final manuscript. The conceptual framework was developed by Jun Yang and Edison Huixiang Ang. Material preparation and theoretical calculations were executed by Xinyue Zhang, Haoyuan Qiu, and Zhiyong Han. Performance evaluation and material characterization studies were performed by Lei Ke, Mugilan Narayanasamy, and Xinyue Zhang. Electrochemical assessment of the integrated device was conducted by Xinyue Zhang, Haoyuan Qiu, Jun Yang, and Can Cui. Experimental data interpretation and manuscript composition were primarily undertaken by Xinyue Zhang and Minjie Shi under the supervision of Jun Yang and Edison Huixiang Ang, with critical feedback and intellectual contributions from all co-authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the funding for this project through the National Natural Science Foundation of China (Grant No. 22409075), Natural Science Foundation of Jiangsu Province (Grant No. BK20241012), the National Institute of Education, Singapore, under its Academic Research Fund (RI 3/23 EAH), and Ministry of Education, under its Academic Research Fund, Tier 1 (RG88/23).Notes and references
- S. Xing, N. Liu, Q. Li, M. Liang, X. Liu, H. Xie, F. Yu and J. Ma, Nat. Commun., 2024, 15, 4951 CrossRef CAS PubMed.
- J. Liu, S. Zhang, Y. Du, C. Wang and J. Yan, Matter, 2024, 7, 4161–4179 CrossRef.
- L. Xiang, X. Xu, Y. Liu, H. Zhang, R. Xu, C. Li, F. Xu, Y. Yamauchi and Y. Mai, Nat. Water, 2024, 2, 1195–1206 CrossRef CAS.
- S. Kumar, N. M. Aldaqqa, E. Alhseinat and D. Shetty, Angew. Chem., Int. Ed., 2023, 62, e2302180 Search PubMed.
- K. Zuo, S. Garcia-Segura, G. A. Cerrón-Calle, F. Y. Chen, X. Tian, X. Wang, X. Huang, H. Wang, P. J. J. Alvarez, J. Lou, M. Elimelech and Q. Li, Nat. Rev. Mater., 2023, 8, 472–490 CrossRef.
- A. Nada, M. G. Ibrahim, M. Elshemy, M. Fujii and M. Sharaan, Desalination, 2024, 586, 117844 CrossRef CAS.
- D. A. De Luca, D. Cocca, E. Egidio and M. Lasagna, Water Reuse and Unconventional Water Resources: A Multidisciplinary Perspective, Springer Book, 2024, vol. 113, pp. 13–38 Search PubMed.
- S. Porada, R. Zhao, A. van der Wal, V. Presser and P. M. Biesheuvel, Prog. Mater. Sci., 2013, 58, 1388–1442 CrossRef CAS.
- Y. Elhenawy, K. Fouad, M. Bassyouni and T. Majozi, Energy Convers. Manage., 2023, 286, 117039 CrossRef CAS.
- Y. Tang, K. Sun, X. Du, J. Zhao, H. Wang and Q. Huang, Sep. Purif. Technol., 2023, 326, 124856 CrossRef CAS.
- R. Feng, X. Zhang, F. Ye, M. A. Götelid and J. Dutta, Nano Energy, 2025, 133, 110444 CrossRef CAS.
- P. Srimuk, F. Kaasik, B. Krüner, A. Tolosa, S. Fleischmann, N. Jäckel, M. C. Tekeli, M. Aslan, M. E. Suss and V. Presser, J. Mater. Chem. A, 2016, 4, 18265–18271 RSC.
- H. Wang, L. Edaño, L. Valentino, Y. J. Lin, V. M. Palakkal, D. L. Hu, B. H. Chen and D. J. Liu, Nano Energy, 2020, 77, 105304 CrossRef CAS.
- R. Kumar, S. S. Gupta, S. Katiyar, V. K. Raman, S. K. Varigala, T. Pradeep and A. Sharma, Carbon, 2016, 99, 375–383 CrossRef CAS.
- J. Yan, M. Qu, Y. Zhao, X. Liu, J. Li, C. L. Sun, X. Zhu and J. He, Desalination, 2024, 575, 117340 CrossRef CAS.
- Y. Lei, S. Wang, L. Zhao, C. Li, G. Wang and J. Qiu, Adv. Sci., 2024, 11, 2402340 CrossRef CAS PubMed.
- Z. Li, B. Song, Z. Wu, Z. Lin, Y. Yao, K. S. Moon and C. P. Wong, Nano Energy, 2015, 11, 711–718 CrossRef CAS.
- S. Porada, L. Borchardt, M. Oschatz, M. Bryjak, J. S. Atchison, K. J. Keesman, S. Kaskel, P. M. Biesheuvel and V. Presser, Energy Environ. Sci., 2013, 6, 3700–3712 RSC.
- W. Shi, X. Liu, T. Deng, S. Huang, M. Ding, X. Miao, C. Zhu, Y. Zhu, W. Liu, F. Wu, C. Gao, S. W. Yang, H. Y. Yang, J. Shen and X. Cao, Adv. Mater., 2020, 32, 1907404 CrossRef CAS PubMed.
- H. Gang, H. Deng, L. Yan, B. Wu, S. I. Alhassan, Y. Cao, D. Wei and H. Wang, J. Colloid Interface Sci., 2023, 638, 252–262 CrossRef CAS PubMed.
- A. Narayanan, A. Siddiqa, N. K. Kodihalli, G. Hegde, D. H. Nagaraju and M. Padaki, ACS Sustainable Chem. Eng., 2023, 11, 3750–3759 CrossRef CAS.
- N. Liu, L. Yu, B. Liu, F. Yu, L. Li, Y. Xiao, J. Yang and J. Ma, Adv. Sci., 2023, 10, 2204041 CrossRef CAS PubMed.
- L. Zhao, H. Zhang, T. Yang, Y. Ma, J. Wang, Z. Guo, Z. Huang, P. Zhang and Z. Ji, ACS Mater. Lett., 2024, 6, 3238–3245 CrossRef CAS.
- T. Ying, Y. Xiong, H. Peng, R. Yang, L. Mei, Z. Zhang, W. Zheng, R. Yan, Y. Zhang, H. Hu, C. Ma, Y. Chen, X. Xu, J. Yang, D. Voiry, C. Y. Tang, J. Fan and Z. Zeng, Adv. Mater., 2024, 36, 2403385 CrossRef CAS PubMed.
- J. Lei, X. Zhang, J. Wang, F. Yu, M. Liang, X. Wang, Z. Bi, G. Shang, H. Xie and J. Ma, Angew. Chem., Int. Ed., 2024, 63, 2401972 Search PubMed.
- Y. Liu, K. Wang, X. Xu, K. Eid, A. M. Abdullah, L. Pan and Y. Yamauchi, ACS Nano, 2021, 15, 13924–13942 CrossRef CAS PubMed.
- Q. Li, Y. Zheng, D. Xiao, T. Or, R. Gao, Z. Li, M. Feng, L. Shui, G. Zhou, X. Wang and Z. Chen, Adv. Sci., 2020, 7, 2002213 CrossRef CAS PubMed.
- J. Guo, X. Xu, J. P. Hill, L. Wang, J. Dang, Y. Kang, Y. Li, W. Guan and Y. Yamauchi, Chem. Sci., 2021, 12, 10334–10340 RSC.
- H. Wang, Y. Liu, Y. Li, X. Xu, X. Liu, Y. Yao, T. Lu and L. Pan, Chem. Eng. J., 2024, 496, 153808 CrossRef CAS.
- S. Wang, Z. Li, G. Wang, Y. Wang, Z. Ling and C. Li, ACS Nano, 2022, 16, 1239–1249 CrossRef CAS PubMed.
- S. Yao, D. Wang, W. Fu, X. Gao, S. Wang, Y. Liu, Z. Hou, J. Wang, K. Nie, J. Xie, Z. Yang and Y. Yan, Chem. Eng. J., 2024, 490, 151608 CrossRef CAS.
- Y. Wang, J. Guo, T. Wang, J. Shao, D. Wang and Y. Yang, Nanomaterials, 2015, 5, 1667–1689 CrossRef CAS PubMed.
- S. Du, S. Wang, Y. Lei, L. Zhao, G. Wang and J. Qiu, Energy Environ. Mater., 2025, 70049 CrossRef.
- S. Wang, Y. Lei, G. Wang, L. Zhao, X. Shen, S. Li, S. Du, C. Yang and J. Qiu, Angew. Chem., Int. Ed., 2025, 64, e202504775 CrossRef CAS PubMed.
- Z. Liu, B. Wei, K. Liu and L. Wang, Sep. Purif. Technol., 2024, 345, 127380 CrossRef CAS.
- J. Lei, X. Zhang, J. Wang, F. Yu, M. Liang, X. Wang, Z. Bi, G. Shang, H. Xie and J. Ma, Angew. Chem., 2024, 136, e202401972 CrossRef.
- X. Xu, A. E. Allah, C. Wang, H. Tan, A. A. Farghali, M. H. Khedr, V. Malgras, T. Yang and Y. Yamauchi, Chem. Eng. J., 2019, 362, 887–896 CrossRef CAS.
- M. Liang, L. Wang, V. Presser, X. Dai, F. Yu and J. Ma, Adv. Sci., 2020, 7, 2000621 CrossRef CAS PubMed.
- F. Yu, Z. Yang, X. Zhang, P. Yang, L. Li and J. Ma, J. Mater. Chem. A, 2022, 10, 23531–23541 RSC.
- D. Jiang, R. Xu, L. Bai, J. P. Hill, J. Henzie, L. Zhu, W. Xia, R. Bu, Y. Zhao, Y. Kang, T. Hamada, R. Ma, N. Torad, J. Wang, T. Asahi, X. Xu and Y. Yamauchi, Adv. Funct. Mater., 2024, 34, 2407479 CrossRef CAS.
- A. Fombona-Pascual, N. Patil, E. García-Quismondo, N. Goujon, D. Mecerreyes, R. Marcilla, J. Palma and J. J. Lado, Chem. Eng. J., 2023, 461, 142001 CrossRef CAS.
- D. Kang, C. Huang, M. Ding and H. Y. Yang, Desalination, 2024, 586, 117884 CrossRef CAS.
- Y. Tao, J. Jin, Y. Cui, H. Wang, Z. Qian and M. Shi, ACS Sustainable Chem. Eng., 2024, 12, 16434–16443 CrossRef CAS.
- H. Zhang, X. Li, C. Xiao, J. Xie, X. Yan, C. Wang, Y. Zhou, J. Qi, Z. Zhu, X. Sun and J. Li, Sep. Purif. Technol., 2022, 302, 122147 CrossRef CAS.
- W. Shi and F. Ren, Phys. Chem. Chem. Phys., 2019, 21, 11871–11882 RSC.
- Y. Kou, Y. Xu, Z. Guo and D. Jiang, Angew. Chem., Int. Ed., 2011, 50, 8753–8757 CrossRef CAS PubMed.
- L. Yang, P. Wang, S. Zhang, Y. Wang, L. Zang, H. Zhu, J. Yin and H. Y. Yang, J. Mater. Chem. A, 2020, 8, 22791–22801 RSC.
- Y. Zhao, J. He, L. Hu, J. Yang, C. Yan and M. Shi, Small, 2023, 19, 2304182 CrossRef CAS PubMed.
- A. Kubaib and P. M. Imran, J. Mater. Sci., 2023, 58, 4005–4019 CrossRef CAS.
- H. Lu, J. Hu, K. Zhang, Y. Zhang, B. Jiang, M. Zhang, S. Deng, J. Zhao, H. Pang and B. Xu, Adv. Mater., 2024, 36, 2408396 CrossRef CAS PubMed.
- Y. Tao, Y. Cui, H. Wang, Z. Li, Z. Qian, P. Zhang, H. Zhou and M. Shi, Adv. Funct. Mater., 2025, 35, 2414805 CrossRef CAS.
- Y. Lin, H. Cui, C. Liu, R. Li, S. Wang, G. Qu, Z. Wei, Y. Yang, Y. Wang, Z. Tang, H. Li, H. Zhang, C. Zhi and H. Lv, Angew. Chem., Int. Ed., 2023, 62, 2218745 Search PubMed.
- H. Liu, J. Zhang, X. Xu and Q. Wang, Chem.–Eur. J., 2020, 26, 4403–4409 CrossRef CAS PubMed.
- Z. Chen, X. Xu, K. Wang, D. Jiang, F. Meng, T. Lu, Y. Yamauchi and L. Pan, Sep. Purif. Technol., 2023, 315, 123628 CrossRef CAS.
- J. Peng, X. Zhao, Y. Huang, C. Zhao, J. Yang and M. Shi, Adv. Funct. Mater., 2025, 2506369 CrossRef.
- Y. Cui, Y. Tao, J. Yang, H. Wang, P. Zhang, G. Li, M. Shi and E. H. Ang, Mater. Horiz., 2025, 12, 2341–2350 RSC.
- J. Yang, Y. Tao, C. Zhao, Y. Cai, P. Xiao and M. Shi, Environ. Sci. Technol., 2025, 59, 10980–10989 CrossRef CAS PubMed.
- P. Zhang, H. Xu, J. Yang, M. Shi and Y. Lin, Water Res., 2025, 282, 123646 CrossRef CAS PubMed.
- D. Jiang, J. P. Hill, J. Henzie, H. N. Nam, Q. M. Phung, L. Zhu, J. Wang, W. Xia, Y. Zhao, Y. Kang, T. Asahi, R. Bu, X. Xu and Y. Yamauchi, J. Am. Chem. Soc., 2025, 147, 12460–12468 CrossRef CAS PubMed.
- Y. Wang, L. Zhang, Y. Wu, S. Xu and J. Wang, Desalination, 2014, 354, 62–67 CrossRef CAS.
- Y. Li, Z. Ding, X. Zhang, J. Li, X. Liu, T. Lu, Y. Yao and L. Pan, J. Mater. Chem. A, 2019, 7, 25305–25313 RSC.
- Q. Li, X. Xu, J. Guo, J. P. Hill, H. Xu, L. Xiang, C. Li, Y. Yamauchi and Y. Mai, Angew. Chem., Int. Ed., 2021, 60, 26528–26534 CrossRef CAS PubMed.
- L. Xu, Y. Liu, Z. Ding, X. Xu, X. Liu, Z. Gong, J. Li, T. Lu and L. Pan, Small, 2024, 20, 2307843 CrossRef CAS PubMed.
- S. Zhang, X. Xu, X. Liu, Q. Yang, N. Shang, X. Zhao, X. Zang, C. Wang, Z. Wang, J. G. Shapter and Y. Yamauchi, Mater. Horiz., 2022, 9, 1708–1716 RSC.
- Y. Li, Z. Ding, J. Li, J. Li, T. Lu and L. Pan, Desalination, 2019, 469, 114098 CrossRef CAS.
- W. Kong, X. Ge, M. Yang, Q. Zhang, J. Lu, H. Wen, H. Wen, D. Kong, M. Zhang, X. Zhu and Y. Feng, Desalination, 2023, 553, 116452 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5sc03868e |
| This journal is © The Royal Society of Chemistry 2025 |