
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUltra-wide time-dependent phosphorescence color of carbon dots via a synergistic strategy of dual confinement structures constructing multiple luminescent centers†
Shiwei Zhang‡
a,
Chenglong Shen‡b,
Jiurong Lia,
Junfei Liaoa,
Peng Miao *c,
Xiujian Zhaoa and
Xiao Gong*a
*c,
Xiujian Zhaoa and
Xiao Gong*a
aState Key Laboratory of Silicate Materials for Architectures, Wuhan University of Technology, Wuhan 430070, P. R. China. E-mail: xgong@whut.edu.cn
bHenan Key Laboratory of Diamond Optoelectronic Materials and Devices, Key Laboratory of Material Physics, Ministry of Education, School of Physics and Microelectronics, Zhengzhou University, Zhengzhou 450052, China
cSuzhou Institute of Biomedical Engineering and Technology, Chinese Academy of Sciences, Suzhou 215163, P. R. China. E-mail: miaopeng@sibet.ac.cn
First published on 30th June 2025
Abstract
Phosphorescent materials with time-dependent phosphorescent color (TDPC) output have significant potential for applications in advanced optical information encryption. However, the effective construction of TDPC composites compatible with multiple phosphorescent emission centers to achieve wider phosphorescent colors evolving over time in a single material system remains a huge challenge. Here, ultra-wide TDPC composites with time-dependent color evolution were realized for the first time in a single system by exploiting the synergistic effect of a doubly confined structure. Utilizing organosilanes to pretreat the reactive precursor, combined with a boric acid (BA) matrix, the composite (Si-CDs@B2O3) was prepared by direct calcination. Our results reveal that the Si-CDs@B2O3 composite has significant ultra-wide TDPC properties, with the phosphorescent emission shifting from red to orange, yellow, green, and cyan blue. Characterization analysis reveals the important role of organosilanes in achieving ultra-wide TDPC properties. It also demonstrates that the red phosphorescence originates from the interaction of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O on the surface of CDs with the BA matrix, while the blue phosphorescence originates from the intrinsic emission of B–O bonds in the BA matrix. The distinctive dynamic room-temperature phosphorescence properties of the Si-CDs@B2O3 composite were leveraged to develop a strategy for its use in information encryption on a precise time scale.
O on the surface of CDs with the BA matrix, while the blue phosphorescence originates from the intrinsic emission of B–O bonds in the BA matrix. The distinctive dynamic room-temperature phosphorescence properties of the Si-CDs@B2O3 composite were leveraged to develop a strategy for its use in information encryption on a precise time scale.
Introduction
Carbon dots (CDs) represent a novel zero-dimensional carbon-based luminescent nanomaterial with several advantages over traditional organic and inorganic materials, including ease of preparation,1 low toxicity,2 good biocompatibility,3 and excellent optical properties.4 It is noteworthy that CDs exhibit a distinctive property known as room-temperature phosphorescence (RTP), which is regarded as one of their most appealing characteristics.5 However, the spin–orbit coupling (SOC) of electrons is typically weak, and the intersystem crossing (ISC) rate is low, which results in the triplet excitons not being filled in a timely manner.6 Furthermore, triple-state excitons are susceptible to deactivation through non-radiative decay processes. Consequently, these drawbacks have become a major obstacle in realizing ultra-long-lasting CD-based RTP materials, limiting their further development and application. In order to overcome these obstacles, enormous efforts have been made to achieve high phosphorescence quantum yields, long phosphorescence lifetime, modulation of multicolor phosphorescence emission, and environmental stability enhancement of CD-based RTP materials. And a series of effective strategies have been developed, such as heteroatom doping, hydrogen-bonding networks, covalent cross-linking, and matrix encapsulation (e.g., silica, boric acid, poly(vinyl alcohol), etc.), which can effectively promote the ISC of CDs, inhibit the nonradiative transitions of triplet excitons, and generate RTP.7–10 Remarkably, these reports present a single emission wavelength, which still cannot satisfy some dynamic information encryption and anti-counterfeiting applications in the multilevel time dimension. Therefore, achieving advanced applications in the time dimension is an important current research direction and a crucial and urgent challenge to be solved.Time-dependent phosphorescence color (TDPC) has the potential to offer a novel avenue for multi-level dynamic information encryption within the temporal domain. It represents a class of multiple dynamic multicolor phosphorescence in a single luminescent system.11 Recently, some researchers have achieved dynamically tunable color-changing phosphorescence and multicolor output by endowing CDs materials with multiple phosphorescence emission centers. For instance, Kang et al.12 achieved bimodal phosphorescence emission of green (550 nm) and red (645 nm) through a straightforward hydrothermal treatment of polyacrylic acid (PAA). This approach successfully confined isolated carboxyl and carboxyl dimer association (CDA) within particles of CDs. A series of time-dependent transient RTP materials exhibiting time-dependent color changes from yellow to green or orange to green were obtained. Similarly, Shi et al.13 demonstrated that CDs with a single emission center can exhibit TDPC properties when doped with metal ions (Mg2+, Ca2+, or Ba2+) owing to the dual role of the inorganic matrix, namely, structural confinement and inducing structural defects. It can be seen that the key to achieving CD-based TDPC properties is how to construct multiple luminescent centers in a single system. Compared with the rich visible spectrum, the investigation of the performance of CD-based TDPCs is still in its infancy, and there are some pressing issues that need to be further investigated. For instance, the majority of CD-based TDPC materials are currently limited to the conversion between two phosphorescence colors, and most of them are focused on short wavelengths (i.e., below yellow light), which have problems such as weak luminescence intensity and short lifetime. To enhance the security of encrypted information in the time dimension, how to construct CD-based ultra-wide time-dependent phosphorescent color (UWTDPC) materials is of great significance in the field of anti-counterfeiting and information security. Typically, UWTDPC materials can usually be utilized to add multi-level encryption functions and thus enhance confidentiality through wide color evolution in the time dimension. However, the realization of UWTDPC materials in a single system is still a current challenge to be solved.14
In this work, we report for the first time a double-constrained structure strategy employing synergistic effects to realize CD-based UWTDPC materials within a single system. Organosilane modification of the reaction precursor for the preparation of CDs was performed in the first stage in order to create more active sites on the surface of CDs and thus facilitate the ISC process. Subsequently, the CDs were immobilized by encapsulating them using BA as an auxiliary matrix so as to stabilize the triplet excitons from deactivation and thus achieve phosphorescent emissions. This strategy was exploited to achieve ultra-wide phosphorescence emission spanning from red to blue with time-dependent properties, which was not shown in previous reports. Through a series of optical and structural characterization studies, the results demonstrate that the red phosphorescence emission can be attributed to the carbonyl (CO) group concentrated within the CDs, while the blue phosphorescence emission originates from the B–O bond present in the BA matrix. Therefore, this unique color output RTP property exhibited by this CD-based UWTDPC material that undergoes color dynamics over time will undoubtedly help to enhance potential applications in the field of anti-counterfeiting and information security. This method also provides a strong technical support for the future synthesis of UWTDPC materials.
Results and discussion
In this work, we designed a distribution synthesis strategy to achieve the double-constrained synergistic effect of CD composites. Currently, carbonyl (CO) has been widely recognized as an important component for RTP generation in CD materials.15,16 Therefore, 1,4,5,8-naphthalenetetracarboxylic acid (1,4,5,8-NTA) was selected as the precursor for the reaction due to its abundance of carboxyl groups, which can be subjected to a silane grafting process. During the formation of CDs, the carbonization of the precursor occurs concurrently with the hydrolysis of the silicone chains on the surface, resulting in the formation of a silicone layer on the surface of the CDs. This layer exhibits high rigidity under high-temperature calcination and effectively promotes the radiative recombination of electron/hole pairs, thereby enhancing phosphorescence.17 The brightness and lifetime of the resulting material are also improved. Furthermore, BA is highly compatible with CDs and can serve as an ideal matrix for protecting the excited state of CDs,2,18 because the high-temperature molten boric acid will act as a secondary coating for the generated CDs, thereby further fixing the phosphorescence-emitting triplet state (Scheme 1).
 | ||
| Scheme 1 Schematic diagram of the synthesis, structure and phosphorescence properties of Si-CDs@B2O3. | ||
The dehydration condensation reaction of the carboxyl group and amino group can be readily achieved by the combination of 1,4,5,8-NTA and APTES (Si-NTA) in a hydrothermal reaction environment characterized by high temperatures and pressures. The alterations in functional groups prior to and following hydrothermal processing can be discerned with precision through the use of Fourier transform infrared spectroscopy (FT-IR) spectra (Fig. S1a†). Compared to APTES, the content of N–H bonds in Si-NTA was significantly reduced,19,20 which is attributed to the fact that the C–O bond of the carboxyl group and the N–H bond of the amino group can be broken down during the dehydration reaction. The carboxyl group and the amino group can be linked together to form the C–N bond, which also explains the enhancement of the C–N characteristic vibration located at 1341 cm−1 in Si-NTA.21–23 Detailed structural information about Si-NTA was obtained using nuclear magnetic resonance (NMR) spectroscopy (1H-NMR, 13C-NMR) and high-resolution mass spectrometry (HR-MS). The 1H-NMR spectrum of Si-NTA in D2O clearly shows the presence of vibrational signals from the protons of the aminosilane chain, as well as from the hydrogen on the benzene ring (Fig. S1b†). The high-density, sharp signals in the range of 2–5 ppm are attributed to the presence of saturated aliphatic hydrocarbons, while the signals in the range of 7–9 ppm are identified as hydrogen atoms of the benzene ring.24 It is noteworthy that the vibrational intensity of hydrogen in the –NH2 group at 1.21 ppm is markedly weak, which aligns with the FT-IR findings and substantiates the involvement of amino groups in silanes in the dehydration condensation reaction. Similarly, from the 13C-NMR spectra of Si-NTA, it can be observed that the vibrational peaks with chemical shifts at 0–60 ppm originate from the silane chain with aliphatic (sp3) carbon atoms (Fig. S1c†).24,25 The vibrational peak located at 59.26 ppm is significantly weaker, which reflects the breaking of the C–O bond during the hydrolysis process of the silanes.
Analysis of the characteristic peaks in the HR-MS spectra of Si-NTA confirmed that the precursor surface had been grafted with numerous silane chains (Fig. 1a and S1d†). The substance resulting from the hydrothermal reaction is a complex mixture of organic compounds, making it unlikely that all of the observed fragment ion peaks originate from the fragmentation of a single organic compound with a mass fraction of 1118. However, the fragment ion peaks demonstrated that the precursors were effectively aminosilanated, resulting in the formation of new organic compounds with an abundance of silane chains. Concurrently, a portion of the silane chain undergoes hydrolysis, resulting in the distribution of numerous Si atoms at the edges of the organic matter. Si-CDs are generated after thermal treatment and are successfully distributed in B2O3 (Si-CDs@B2O3). Transmission electron microscopy (TEM) and high-resolution transmission electron microscopy (HR-TEM) images in Fig. 1b demonstrate that the Si-CDs generated by the thermal treatment process are uniformly distributed in the B2O3 matrix, with an average particle size of approximately 2.33 nm (Fig. 1c). The Si-CDs have a lattice spacing of 0.21 nm, which is attributed to the graphite (100) crystal plane, indicating that the Si-CDs have good crystallinity.26–28 This is corroborated by the X-ray diffraction (XRD) spectrum of Si-CDs@B2O3 (Fig. 1d), which exhibits a distinct broad peak (002) at ≈24°, typical of amorphous carbon.29
 | ||
| Fig. 1 (a) The HR-MS spectrum of the Si-NTA. (b) The TEM and HR-TEM image of Si-CDs@B2O3. (c) The particle size distribution of Si-CDs@B2O3. (d) The XRD spectra of Si-CDs@B2O3 (SiO2, PDF#27-0605; B2O3, PDF#06-0297). (e) The FT-IR spectra of Si-NTA and Si-CDs@B2O3. | ||
The chemical bonding and structure between Si-NTA and Si-CDs@B2O3 were characterized using FT-IR and X-ray photoelectron spectroscopy (XPS). The FT-IR spectra reveal that Si exists as Si–O–C and Si–C bonds in Si-NTA, with Si–O–C dominating, as evidenced by the presence of characteristic vibrational peaks at 1131 and 780 cm−1 (Fig. 1e).30 The asymmetric stretching vibrations of the B–O bond and the vibration of the triangular boron atom connected to the oxygen atom in BA can be observed in the FT-IR of Si-CDs@B2O3, located at 1454 and 1199 cm−1, respectively.6,31 Moreover, the characteristic vibrational peaks corresponding to Si–C bonds are significantly more intense than those of Si–O–C, indicating that at high temperatures, Si–O–C bonds undergo cleavage and reorganize into robust and more stable Si–C bonds. This structural transformation greatly enhances the overall rigidity of the composite materials.32 Furthermore, the appearance of an absorption band at 1059 cm−1 indicates the presence of B–C bonds, demonstrating the successful binding of Si-CDs to BA via a covalent coupling reaction due to the electron-deficient nature of boron and the amphiphilic characteristics of the carbon atoms.33 The aforementioned results indicate that the simultaneous presence of Si–C covalent bonds, B–C covalent bonds, the rigid network structure of the matrix, and the hydrogen bonding interactions between the matrix and the Si-CDs in the system can lock and stabilize the triplet states of the CDs more efficiently by inhibiting the motion of the CDs.
Fig. S2a† illustrates the full-spectrum of XPS, which reveals that Si-NTA is primarily composed of C, N, O, and Si, whereas Si-CDs@B2O3 is predominantly constituted by C, N, O, Si, and B. The high-resolution C 1s spectrum of the Si-NTA was separated into four peaks at 284.7, 285.8, 287.9, and 288.6 eV (Fig. 2a), which correspond to the C–C/CC, C–O/C–N, CO, and OC–O, respectively.34 Notably, the C 1s spectrum of Si-CDs@B2O3 was deconvoluted into a new peak at 283.8 eV, which is attributed to the B–C.35,36 This further suggests that bonding occurs between the host matrix and the Si-CDs, resulting in the formation of a new B–C covalent bond. The high-resolution N 1s spectrum of Si-NTA can be fitted into four peaks (Fig. 2b), which are attributed to pyridinic nitrogen (398.8 eV), aminic nitrogen (399.5 eV), pyrrolic nitrogen (400.7 eV), and graphitic nitrogen (401.7 eV).37,38 Similarly, the high-resolution N 1s spectrum of Si-CDs@B2O3 can be deconvoluted into the aforementioned set of four peaks. The pyridine nitrogen tends to be distributed on the surface of the CDs or defects. It has been established that high-temperature calcination can regulate the defects on the surface and consume the functional groups on the surface of the CDs.39 This may be the primary reason for the decrease in the pyridine nitrogen content. The high-resolution O 1s spectrum of Si-NTA was decomposed into two peaks (Fig. 2c), which are identified as belonging to CO (531.1 eV) and C–O (532.1 eV), respectively.40 It is noteworthy that the high-resolution O 1s spectrum of Si-CDs@B2O3 exhibits an additional new peak, which corresponds to the B–O. This is a consequence of the introduction of a new oxygen atom state, provided by the added boric acid matrix. Furthermore, the high-resolution Si 2p spectra of both Si-NTA and Si-CDs@B2O3 were deconvoluted into two peaks (Fig. 2d), and the two peaks of Si-CDs@B2O3 were found to be located at 102.0 eV and 103.6 eV, respectively, corresponding to Si–C and Si–O.41,42 Notably, the content of Si–C bonds was found to be substantially higher. The deconvoluted high-resolution Si 2p spectra indicate that during the transition from Si-NTA to Si-CDs@B2O3, the high-temperature environment results in a gradual decrease in the Si–O–C content and a gradual increase in stronger Si–C covalent bonds.32 This contributes to a stronger and more stable structure as a whole, which is in agreement with the FT-IR data and analysis results. With respect to the analysis of B 1s, as shown in Fig. S2b,† it could be divided into three bands at 192.0, 192.8, and 193.7 eV, corresponding to BCO2, B2O3, and B–O, respectively. This further demonstrates the presence of a B2O3 matrix and the covalent coupling reaction between Si-CDs and the host matrix to produce B–C bonds.5,43
 | ||
| Fig. 2 (a–d) High-resolution C 1s, N 1s, O 1s and Si 2p spectra of Si-NTA and Si-CDs@B2O3. (a) C 1s, (b) N 1s, (c) O 1s, and (d) Si 2p. (e) Schematic diagram of the Si-CDs@B2O3 composite network structure before and after calcination. | ||
To summarize, a possible formation mechanism is proposed (Fig. 2e). Specifically, this mechanism involves the dehydration and condensation of precursors and organosilane under hydrothermal conditions, resulting in the generation of numerous aminosilanated silicones. These chain-modified silicones can then undergo further calcination and carbonization, leading to the formation of CDs. As the organosilicon atoms are situated in the edge region of Si-NTA, the silicones chains also undergo dehydration reactions during the formation of CDs. Ultimately, this results in the formation of an organosilicon layer or even a SiO2 layer on the surface of the CDs.14 FT-IR and XPS analyses indicate that Si atoms in CDs predominantly exist in the form of Si–C covalent bonds, suggesting that Si-CDs are not merely encapsulated in the SiO2 matrix, but rather stabilized within it through Si–C covalent bonding and hydrogen bonding on the surface. The achievement of a tightly ordered state of the Si elements, which permits effective spatial confinement and the suppression of intramolecular vibrations, is contingent upon the utilization of an appropriate calcination temperature.14 The aforementioned experiments and analyses have confirmed that the Si-CDs were embedded in BA, with new B–C bonds formed between the Si-CDs and the BA matrix. It is proposed that these covalent bonds and glassy states are capable of stabilizing the triplet states (i.e., CO-containing groups) on Si-CDs, promoting the ISC between S1 and T1, and protecting the excited triplet excitons, thereby facilitating RTP emission.31,44,45
As shown in Fig. 3, an investigation was conducted into the photophysical properties of Si-CDs@B2O3 composites. Fig. 3a presents the optical photographs of the naked-eye resolvable phosphorescence color changes for several seconds for the Si-CDs@B2O3 material under different excitation sources at 310 nm and 365 nm, respectively. Particularly, the phosphorescent color change of the Si-CDs@B2O3 material under the illumination of 365 nm light source is very obvious, with a distinctive evolution from red to yellow to green to blue, a typical characteristic of TDPC. It is noteworthy that the CDs@B2O3 material retains the intrinsic phosphorescence emission properties of the BA matrix,46,47 i.e., weak blue phosphorescence emission, which is rarely observed in previous reports. This phenomenon can be attributed to the organosilicon layer on the surface of the CDs, which effectively isolates electron migration between the CDs and the boron oxide matrix, hindering the energy transfer process and thereby protecting the boron oxide triplet state emission. The cooperative interaction between the red phosphorescence of the CDs and the blue phosphorescence of the matrix achieves the UWTDPC effect in CD materials and allows time-resolved visual color RTP output.
 | ||
| Fig. 3 (a) Optical photographs of Si-CDs@B2O3 before and after turning off under different excitation wavelengths. (b) Absorption, fluorescence emission and phosphorescence emission spectra of Si-CDs@B2O3 (Ex: 365 nm). (c) 2D mapping spectra of phosphorescence emission of Si-CDs@B2O3. (d) Time-resolved phosphorescence 2D mapping spectra of Si-CDs@B2O3 after 315 nm irradiation. (e) Time-resolved phosphorescence spectra of Si-CDs@B2O3 after 365 nm irradiation. (f) Time-resolved decay spectra of Si-CDs@B2O3 at room temperature with λex = 365 nm. (g) Schematic illustration of the UWTDPC mechanisms for the Si-CDs@B2O3. | ||
Subsequently, the absorption, fluorescence and phosphorescence spectra of the Si-CDs@B2O3 material were studied (Fig. 3b). The absorption peaks, located at 235 and 378 nm, correspond to the π–π* transitions of CC and n–π* transitions of CO, respectively.48,49 Furthermore, the fluorescence spectra exhibited bimodal emission, with the twin peaks located at 410 and 607 nm, respectively. This explains why Si-CDs@B2O3 exhibits pink fluorescence. Furthermore, the excitation wavelength dependence of the fluorescence of Si-NTA and Si-CDs@B2O3 was examined (Fig. S3†). The fluorescence of both samples exhibited excitation-dependent characteristics, which can be attributed to the presence of rich functional groups on the surface of the CDs. The phosphorescence spectra of Si-CDs@B2O3 exhibited four peaks, located at 450, 610, 668, and 743 nm, respectively. The 2D mapping of the phosphorescence of Si-CDs@B2O3 (Fig. 3c) demonstrates that at different excitation wavelengths, the intensity of the phosphorescence emission is altered without affecting the peak positions. At an excitation wavelength of 316 nm, the phosphorescence emission peak at 450 nm is the most intense, while the optimal excitation wavelength for the phosphorescence emission peaks at 610, 668, and 743 nm corresponds to an emission wavelength of 375 nm. The significant discrepancy between the excitation and emission of the dual emission centers of phosphorescence substantiates the mutually independent property of the dual phosphorescence centers, which is a prerequisite for the successful realization of UWTDPC.
To gain insight into the nature of the TDPC of Si-CDs@B2O3, a detailed analysis of its time-dependent phosphorescence spectrum was conducted (Fig. 3d and e). When the excitation light source is 316 nm, a double peak at 610 nm and 450 nm is evident in the time-dependent phosphorescence spectra. However, with the extension of time, the emission peak at 610 nm decays rapidly, and then almost only the emission peak at 450 nm remains. Fig. S4† illustrates that the lifetime of the phosphorescence emission at 450 nm, following excitation at 310 nm, is as long as 574.64 ms. Consequently, the phosphorescence color of Si-CDs@B2O3 undergoes a gradual transition from red to blue with the passage of time. Similarly, under 365 nm excitation, the red emission peak exhibits a sharp decay, while the weaker intensity blue emission peak decays more slowly. However, when the time is prolonged, the blue phosphorescence emission peak located at 450 nm will become dominant, and blue phosphorescence will be exhibited in the final stage of phosphorescence emission. Moreover, the phosphorescence emission peaks situated in the blue and red regions exhibit broad-peak emission, with a width encompassing the entire 400–800 nm region. This not only demonstrates a transition from red to blue phosphorescence during the double-peak decay but also a dynamic, colored RTP output. The Si-CDs@B2O3 material exhibits ultra-wide TDPC characteristics that not only cover a wide spectrum, but also have diverse color variations during the period, which has never been reported before (Table S1†). Besides, it can be observed that the lifetimes of the phosphorescence emission peaks located at 610, 668 and 743 nm are 45.87, 48.38 and 48.53 ms, respectively (Fig. 3f). However, the lifetime of the emission peak at 450 nm can reach up to 132.88 ms (Fig. S5†), which is considerably longer than that of the red emission peak. This indicates that the Si-CDs@B2O3 composites are capable of completing the colored RTP output in a very short time. Furthermore, the temperature-dependent phosphorescence spectra of Si-CDs@B2O3 composites demonstrate that the entire phosphorescence spectrum displays a pattern of decreasing intensity as the temperature gradually increases from 208 K to 388 K (Fig. S6†), which indicates that they possess notable phosphorescence characteristics, exhibiting dual-emitting phosphorescence centers.30
To verify the synergistic interaction between matrixes, a series of comparison experiments were performed by varying the cladding matrix and distribution synthesis strategy, and the corresponding optical images are presented in Fig. S7.† Compared to Fig. 3a, it is clearly found that the CDs@B2O3 composites synthesized from precursors without organosilane modification exhibit time-dependent changes in the phosphorescence emission color, along with a narrower emission spectrum and shorter phosphorescence lifetime (Fig. S7a and S8†). The results indicate that silane pretreatment facilitates the UWTDPC process. To verify the interaction between the organosilane pretreatment reaction precursor and the assistant matrix, urea was employed to replace the boric acid matrix in the experiments because urea has a similar structure to BA, allowing it to form a framework structure that encapsulates the CDs, and is a commonly used matrix for preparing RTP CDs.50 As demonstrated in Fig. S7b,† Si-CDs@urea did not manifest a discernible TDPC effect, and the phosphorescence perceptible to the naked eye persisted for a mere 0.5 s. However, CDs@urea not only displays a yellow-to-blue TDPC effect but also exhibits phosphorescence for up to 2 s (Fig. S7c†). Furthermore, stabilizing the triple-state excitons with a SiO2 matrix alone is challenging (Fig. S7d†), and this approach does not yield the desired afterglow properties. This result further verifies that the interaction between the organosilane pretreated reactive precursor and the matrix is either promoted or inhibited. Additionally, the stepwise synthesis method is conducive to the formation of layered cladding structures, whereas the one-pot method is challenging to produce ordered structures. As illustrated in Fig. S9,† the afterglow duration of the CD composites synthesized via the one-pot method is markedly shorter, with significantly poor TDPC properties. Subsequently, the phosphorescence spectra of these comparison samples were studied (Fig. S10†). The phosphorescence spectra demonstrated that both urea and BA matrixes, whether distributed or synthesized via a one-pot method, exhibited double-emission characteristics. This is attributed to the triplet state energy level structure formed by the functional groups (CO) on the surface of the CDs and the intrinsic state phosphorescence emission of the matrix.51 Although the phosphorescence emission of these CD composites presents some similarity, the slight difference in lifetime and intensity between the two results in either no TDPC effect or only TDPC in the shorter wavelength range.
To trace the origin of the apparent TDPC performance differences, we further characterized the as-synthesized samples using XRD and FT-IR spectra. The XRD spectrum of the urea-based composite material exhibits characteristic peaks similar to those observed in molten urea (Fig. S11†). The diffraction peaks at 22.2°, 24.5°, and 29.2° correspond to the (110), (101), and (111) crystal planes of urea, respectively, confirming the successful encapsulation of CDs within the urea matrix. Notably, the intense diffraction peak at 28.0° indicates the formation of a melamine acid-like network structure under high-temperature conditions. This cross-linked network plays a crucial role in effectively stabilizing the triplet states of the CDs, thereby enhancing RTP properties. Similarly, the composites capped with either BA or SiO2 as a matrix both exhibited a diffuse broad peak and corresponding characteristic peaks (Fig. S12†), with the former belonging to typical amorphous carbon and the latter indicating that the CDs were encapsulated in the matrix. The FT-IR spectrum demonstrated that the Si-CDs@urea composite exhibited a greater number of functional groups than the Si-CDs@B2O3 composite (Fig. S13†), which is due to the fact that urea is an organic matrix that can provide a greater number of functional groups (e.g., –NH2, CO) than BA. In the XPS spectra (Fig. S14 and S15†) of Si-CDs@urea and CDs@urea, the content of CO bonds increases significantly, indicating that the urea matrix provides a substantial quantity of CO bonds. However, Si-CDs@urea and CDs@urea did not exhibit red phosphorescence emission, suggesting that the addition of CO groups is not always conducive to the phosphorescence emission of CDs. It is hypothesized that the presence of different matrixes engenders divergent chemical environments for CO, as illustrated by the structural diagrams presented in Fig. S16.† Through a comparative analysis of the C 1s spectra of Si-CDs@B2O3 and Si-CDs@urea (Fig. 2a and S14b†), it is proposed that the red phosphorescence is predominantly attributable to the O–CO structure.
Based on the aforementioned results and discussion, we propose a potential mechanism for achieving the distinctive dynamic RTP of UWTDPC and Si-CDs@B2O3. The CDs are generated in situ and embedded into a 3D network of the BA matrix through the action of multiple binding forces. Firstly, the organosilane pretreated reaction precursors provide richer functional groups on the surface of in situ synthesized CDs, providing more active sites for further interactions with the BA matrix. Secondly, the rigid network of the BA matrix serves to isolate the CDs from external quenchers (water and oxygen), thereby enhancing the phosphorescence performance. Thirdly, the stable Si–C and B–C bonds and the abundant hydrogen bonding interactions between the rigid matrix and the CDs can effectively stabilize the triplet excitons of the CDs by suppressing the intramolecular vibrations. It can be found that the Si-CDs@B2O3 composites possess multiple phosphorescence emission centers, where the red phosphorescence emission center originates from CO on the surface of the CDs, and the blue phosphorescence emission belongs to the B–O in the BA matrix. The Jablonski diagrams of the mechanism are demonstrated in Fig. 3g. When exposed to suitable energy light irradiation (e.g., 365 nm), the two phosphorescence centers can be activated simultaneously. The red phosphorescence is generated by the triplet state TCO when returning to the ground state (S0) and exhibits a fast decay. The return of some electrons from the triplet state TB–O to S0 results in the generation of a slowly decaying blue phosphorescence. This process, combined with the large Stokes shift between the two phosphorescence excitations and emissions, ensures that the phosphorescent emission centers are independent of each other and do not interfere with each other. The combination of these features gives rise to the observed UWTDPC phenomenon. Under 365 nm UV irradiation, it is evident that the phosphorescence color undergoes a gradual transition from red to blue, thereby achieving a dynamic and multicolored RTP output.
As a sophisticated anti-counterfeiting material with exceptional time-dependent phosphorescence color, Si-CDs@B2O3 displays considerable promise for deployment in anti-counterfeiting and data encryption applications. As illustrated in Fig. 4a, the specific chameleon pattern model filled with Si-CDs@B2O3 exhibits a time-varying phosphorescence color. The material exhibits pink fluorescence under 365 nm UV light, and then its color takes on a reddish hue immediately after the 365 nm UV light is switched off. Thereafter, it gradually changes to a cyan blue color within 1 s, the phenomenon that can be easily recognized with the naked eye. Fig. 4b illustrates the complete phosphorescence color change process of Si-CDs@B2O3, with the phosphorescence color change encompassing the entire visible spectral range. The distinctive RTP properties of Si-CDs@B2O3 composites offer a potential avenue for multi-level information encryption applications. As illustrated in Fig. 4c, the visual effect of time-dependent information encryption is demonstrated using Si-CDs@B2O3 and N@B2O3. Upon the deactivation of the UV lamp, the flower pattern exhibits a red phosphorescence, rendering the individual parts indistinguishable. However, at a delay of 0.2 s, 0.3 s, and 0.8 s, the central portion of the flower pattern displays orange, yellow, and cyan blue, respectively. The phosphorescence color information is only discernible at a specified time, thereby enabling the encryption of information on a time scale. As illustrated in Fig. 4d, the anti-counterfeiting pattern “WUT” was developed from a CD composite material, with the individual components “W”, “U” and “T” derived from Si-CDs@urea, Si-CDs@B2O3 and N@B2O3, respectively. Upon excitation with UV lamps at varying wavelengths (310 nm, 365 nm), the “WUT” sequence displays a range of fluorescence and phosphorescence colors. The “W” and “T” letters exhibit a single RTP color output, while the “U” letter displays a TDPC phenomenon at different excitation wavelengths. The “U” can undergo a change in color from pink to blue phosphorescence when the 310 nm excitation source is removed. In particular, the UWTDPC phenomenon is exhibited by “U” when the excitation source at 365 nm is removed, whereby the color changes from red to cyan blue. Moreover, this process is not merely a transition from red to cyan-blue, it involves a series of intermediate color changes, gradually shifting from long to short wavelengths (Video S1†). The combination of the excitation dependence of Si-CDs@B2O3 and the TDPC properties results in an intrinsic improvement in the information carrying capacity and security factor of the CD-based phosphorescence material. Consequently, the above time-varying dynamic multilayer information encryption properties provide a new research avenue for advanced information based on precise time scales.
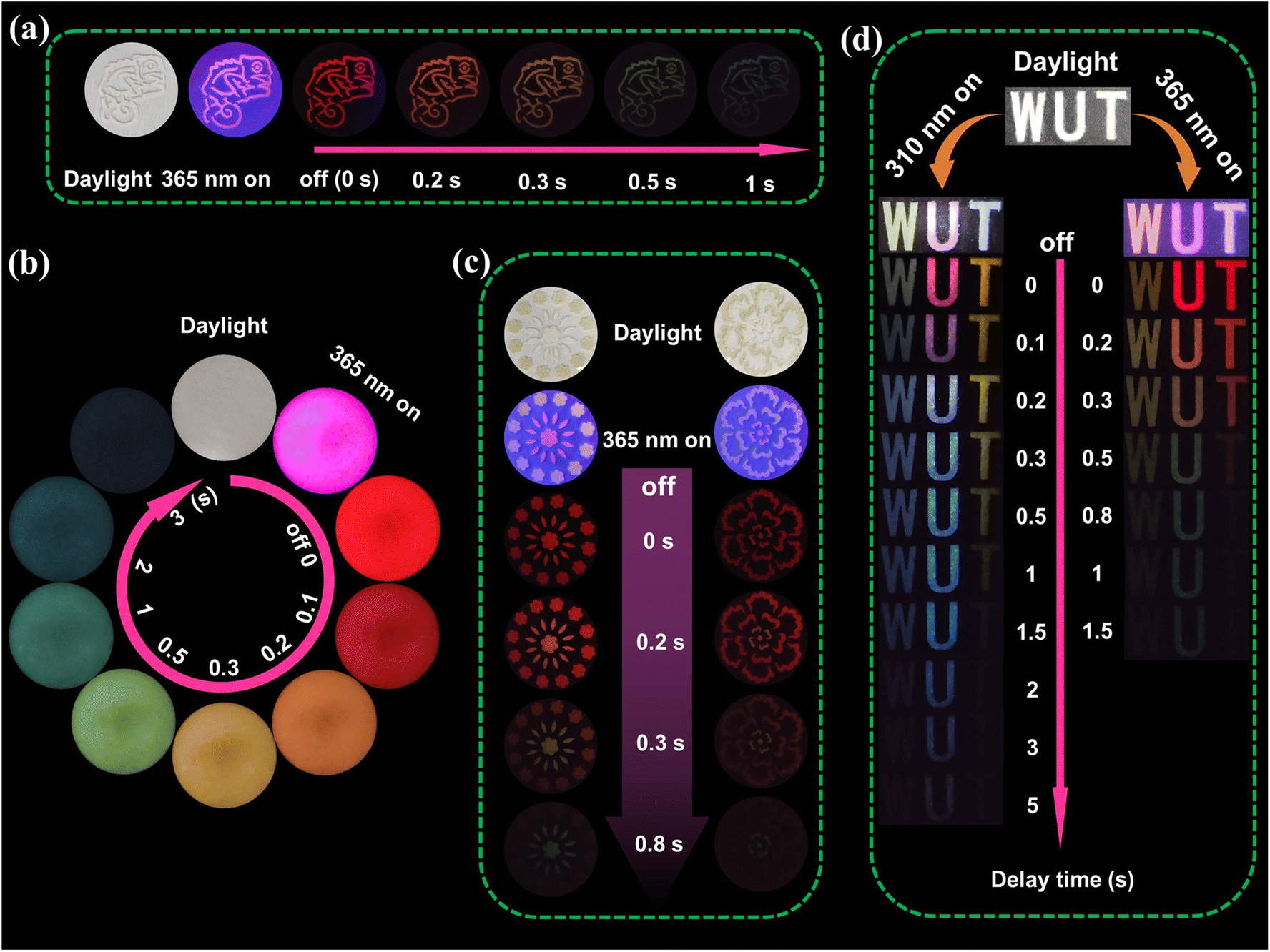 | ||
| Fig. 4 (a) Optical photographs of the chameleon pattern after 365 nm irradiation. (b) Optical photographs of Si-CDs@B2O3 composites after 365 nm irradiation. (c) Optical photographs of the flower pattern after 365 nm irradiation. (d) Images of the “WUT” letter under 310 nm and 365 nm irradiation on and off. | ||
Conclusions
In summary, we designed a novel CD composite that exhibits distinctive UWTDPC properties through organosilane modification of the precursor and its subsequent embedding within the rigid network structure of a BA matrix. The results of the structural studies indicate that the phosphorescence is caused by the formation of Si–C bonds between the organosilicon layer present on the surface of the CDs and the CDs themselves, as well as by the formation of B–C bonds between the boric acid and the CDs and the domain-limited structure. Upon excitation with a wavelength of 365 nm, Si-CDs@B2O3 exhibits distinctive UWTDPC properties, spanning from red to cyan-blue. This is attributed to the red phosphorescence exhibited by the interaction between the CO on the surface of the CDs and the substrate, as well as the blue phosphorescence emitted by the B–O in the BA. There is a notable difference in the phosphorescence intensity and lifetime between the two. The excellent optical properties of Si-CDs@B2O3 have been demonstrated to have potential for application in spatiotemporal multidimensional dynamic afterglow anti-counterfeiting systems for advanced information encryption. However, CD materials exhibiting TDPC characteristics generally suffer from relatively short phosphorescence lifetimes and low intensity, highlighting the urgent need for further optimization. Moreover, enhancing the resolution of multicolor RTP output remains a critical challenge for the practical application of TDPC CD materials. Despite these limitations, the design concepts presented in this work are expected to facilitate the development of TDPC properties based on CD materials and open up new opportunities for the preparation of dynamic luminescent materials with more colors.
Data availability
All data including experimental and analytical details are in the ESI.†Author contributions
X. Gong conceived and designed the project; S. Zhang, C. Shen and P. Miao contributed to the characterization of products; S. Zhang performed the experiments, interpreted the data and wrote the original manuscript; J. Li, C. Shen and J. Liao performed the analysis; X. Gong directed the whole project; X. Gong, X. Zhao and P. Miao acquired the funding. All authors have approved the final version of the manuscript.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 21774098).References
- B. Wang, Y. Yu, H. Zhang, Y. Xuan, G. Chen, W. Ma, J. Li and J. Yu, Carbon dots in a matrix: energy-transfer-enhanced room-temperature red phosphorescence, Angew. Chem., Int. Ed., 2019, 58, 18443–18448 CrossRef CAS PubMed.
- W. Li, W. Zhou, Z. Zhou, H. Zhang, X. Zhang, J. Zhuang, Y. Liu, B. Lei and C. Hu, A universal strategy for activating the multicolor room-temperature afterglow of carbon dots in a boric acid matrix, Angew. Chem., Int. Ed., 2019, 58, 7278–7283 CrossRef CAS PubMed.
- C. Hu, M. Li, J. Qiu and Y. Sun, Design and fabrication of carbon dots for energy conversion and storage, Chem. Soc. Rev., 2019, 48, 2315–2337 RSC.
- S. Zong, J. Zhang, X. Yin, J. Li and S. Qu, Time-dependent and excitation-dependent afterglow color evolution from the assembly of dual carbon dots in zeolite, Nano Lett., 2024, 24, 1859–1866 CrossRef CAS PubMed.
- J. Hu, M. Lei, L. Yan, L. Chen, Y. Yang, J. Zheng, X. Liu and B. Xu, Carbon dot-based afterglow composite with tunable room temperature phosphorescence and thermally activated delayed fluorescence and their anti-counterfeiting and encryption application, Chem. Eng. J., 2024, 489, 151245 Search PubMed.
- S. Cui, B. Wang, Y. Zan, Z. Shen, S. Liu, W. Fang, X. Yan, Y. Li and L. Chen, Colorful, time-dependent carbon dot-based afterglow with ultralong lifetime, Chem. Eng. J., 2021, 431, 133373 CrossRef.
- Y. Liu, D. Cheng, B. Wang, J. Yang, Y. Hao, J. Tan, Q. Li and S. Qu, Carbon dots-inked paper with single/two-photon excited dual-mode thermochromic afterglow for advanced dynamic information encryption, Adv. Mater., 2024, 36, 2403775 CrossRef CAS.
- L. Ai, W. Xiang, J. Xiao, H. Liu, J. Yu, L. Zhang, X. Wu, X. Qu and S. Lu, Tailored fabrication of full-color ultrastable room-temperature phosphorescence carbon dots composites with unexpected thermally activated delayed fluorescence, Adv. Mater., 2024, 36, 2401220 CrossRef CAS PubMed.
- Z. Song, Y. Shang, Q. Lou, J. Zhu, J. Hu, W. Xu, C. Li, X. Chen, K. Liu, C. Shan and X. Bai, A molecular engineering strategy for achieving blue phosphorescent carbon dots with outstanding efficiency above 50%, Adv. Mater., 2022, 35, 2207970 Search PubMed.
- T. Zhang, M. Wang, L. Liu, Z. Li and H. Bi, Visible-light manipulated reversible and ultralong phosphorescence of carbon dots through dynamic cross-linking, Adv. Funct. Mater., 2024, 34, 2406672 CrossRef CAS.
- L. Zhang, X. Chen and Y. Hu, Pyrolysis of Al-based metal-organic frameworks to carbon dot-porous Al2O3 composites with time-dependent phosphorescence colors for advanced information encryption, Small, 2023, 20, 2305185 CrossRef PubMed.
- C. Kang, S. Tao, F. Yang, C. Zheng, Z. Qu and B. Yang, Enabling carbonized polymer dots with color-tunable time-dependent room temperature phosphorescence through confining carboxyl dimer association, Angew. Chem., Int. Ed., 2023, 136, e202316527 CrossRef.
- W. Shi, R. Wang, J. Liu, F. Peng, R. Tian and C. Lu, Time-dependent phosphorescence color of carbon dots in binary salt matrices through activations by structural confinement and defects for dynamic information encryption, Angew. Chem., Int. Ed., 2023, 135, e202303063 CrossRef.
- J. Chen, J. Tan, P. Liang, C. Wu, Z. Hou, K. Shen, B. Lei, C. Hu, X. Zhang, J. Zhuang, L. Sun, Y. Liu and M. Zheng, Dynamic room temperature phosphorescence of silane-functionalized carbon dots confining within silica for anti-counterfeiting applications, Small, 2023, 20, 2306323 CrossRef.
- B. Zhao, R. Yu, K. Xu, C. Zou, H. Ma, S. Qu and Z. a. Tan, Highly efficient carbon dot-based room-temperature fluorescence–phosphorescence dual emitter, J. Mater. Chem. C, 2021, 9, 15577–15582 RSC.
- H. Shi, Z. Niu, H. Wang, W. Ye, K. Xi, X. Huang, H. Wang, Y. Liu, H. Lin, H. Shi and Z. An, Endowing matrix-free carbon dots with color-tunable ultralong phosphorescence by self-doping, Chem. Sci., 2022, 13, 4406–4412 Search PubMed.
- Y. Sun, S. Liu, L. Sun, S. Wu, G. Hu, X. Pang, A. T. Smith, C. Hu, S. Zeng, W. Wang, Y. Liu and M. Zheng, Ultralong lifetime and efficient room temperature phosphorescent carbon dots through multi-confinement structure design, Nat. Commun., 2020, 11, 5591 CrossRef CAS.
- Q. Li, Z. Zhao, S. Meng, Y. Li, Y. Zhao, B. Zhang, Z. Tang, J. Tan and S. Qu, Ultra-strong phosphorescence with 48% quantum yield from grinding treated thermal annealed carbon dots and boric acid composite, SmartMat, 2021, 3, 260–268 CrossRef.
- Y. Xu, W. Hou, K. Huang, H. Guo, Z. Wang, C. Lian, J. Zhang, D. Wu, Z. Lei, Z. Liu and L. Wang, Engineering built-in electric field microenvironment of CQDs/g-C3N4 heterojunction for efficient photocatalytic CO2 reduction, Adv. Sci., 2024, 11, 2403607 Search PubMed.
- L. Xie, C. Liang, Y. Wu, K. Wang, W. Hou, H. Guo, Z. Wang, Y. M. Lam, Z. Liu and L. Wang, Isomerization engineering of oxygen-enriched carbon quantum dots for efficient electrochemical hydrogen peroxide production, Small, 2024, 20, 2401253 Search PubMed.
- L. Vallan, E. P. Urriolabeitia, F. Ruipérez, J. M. Matxain, R. Canton-Vitoria, N. Tagmatarchis, A. M. Benito and W. K. Maser, Supramolecular-enhanced charge-transfer within entangled polyamide chains as origin of the universal blue fluorescence of polymer carbon dots, J. Am. Chem. Soc., 2018, 140, 12862–12869 CrossRef CAS PubMed.
- S. Tao, S. Lu, Y. Geng, S. Zhu, S. A. T. Redfern, Y. Song, T. Feng, W. Xu and B. Yang, Design of metal-free polymer carbon dots: a new class of room-temperature phosphorescent materials, Angew. Chem., Int. Ed., 2018, 57, 2393–2398 Search PubMed.
- M. Shi, Q. Gao, J. Rao, Z. Lv, M. Chen, G. Chen, J. Bian, J. Ren, B. Lü and F. Peng, Confinement-modulated clusterization-triggered time-dependent phosphorescence color from xylan-carbonized polymer dots, J. Am. Chem. Soc., 2023, 146, 1294–1304 CrossRef PubMed.
- S.-Y. Song, L.-Z. Sui, K.-K. Liu, Q. Cao, W.-B. Zhao, Y.-C. Liang, C.-F. Lv, J.-H. Zang, Y. Shang, Q. Lou, X.-G. Yang, L. Dong, K.-J. Yuan and C.-X. Shan, Self-exothermic reaction driven large-scale synthesis of phosphorescent carbon nanodots, Nano Res., 2021, 14, 2231–2240 CrossRef CAS.
- D. Zhou, P. Jing, Y. Wang, Y. Zhai, D. Li, Y. Xiong, A. V. Baranov, S. Qu and A. L. Rogach, Carbon dots produced via space-confined vacuum heating: maintaining efficient luminescence in both dispersed and aggregated states, Nanoscale Horiz., 2018, 4, 388–395 RSC.
- H. Guo, J. Raj, Z. Wang, T. Zhang, K. Wang, L. Lin, W. Hou, J. Zhang, M. Wu, J. Wu and L. Wang, Synergistic effects of amine functional groups and enriched-atomic-iron sites in carbon dots for industrial-current-density CO2 electroreduction, Small, 2024, 20, 2311132 CrossRef CAS PubMed.
- H. Guo, Y. Lu, Z. Lei, H. Bao, M. Zhang, Z. Wang, C. Guan, B. Tang, Z. Liu and L. Wang, Machine learning-guided realization of full-color high-quantum-yield carbon quantum dots, Nat. Commun., 2024, 15, 4843 CrossRef CAS PubMed.
- J. Li, H. Zhao, X. Zhao and X. Gong, High-efficiency luminescent solar concentrators based on carbon dots with simultaneously ultrabright solid-state and liquid-state luminescence, Adv. Funct. Mater., 2024, 34, 2404473 CrossRef CAS.
- F. Yuan, T. Yuan, L. Sui, Z. Wang, Z. Xi, Y. Li, X. Li, L. Fan, Z. a. Tan, A. Chen, M. Jin and S. Yang, Engineering triangular carbon quantum dots with unprecedented narrow bandwidth emission for multicolored LEDs, Nat. Commun., 2018, 9, 2249 CrossRef PubMed.
- X. Wang, Y. Han, W. Li, J. Li, S. Ren, M. Wang, G. Han, J. Yu, Y. Zhang and H. Zhao, Doped carbon dots enable highly efficient multiple-color room temperature phosphorescence, Adv. Opt. Mater., 2023, 12, 2301962 CrossRef.
- Y. Huang and P. Li, Visible-light-excited high efficiency multicolor room temperature phosphorescence of carbon dots in boric acid matrix, Chem. Eng. J., 2023, 480, 148157 CrossRef.
- Y. Zhang, M. Li and S. Lu, Rational design of covalent bond engineered encapsulation structure toward efficient, long-lived multicolored phosphorescent carbon dots, Small, 2022, 19, 2206080 CrossRef PubMed.
- Y. Ding, X. Wang, M. Tang and H. Qiu, Tailored fabrication of carbon dot composites with full-color ultralong room-temperature phosphorescence for multidimensional encryption, Adv. Sci., 2021, 9, 2103833 CrossRef PubMed.
- L. Ai, H. Wang, B. Wang, S. Liu, H. Song and S. Lu, Concentration-switchable assembly of carbon dots for circularly polarized luminescent amplification in chiral logic gates and deep-red light-emitting diodes, Adv. Mater., 2024, 36, 2410094 CrossRef CAS PubMed.
- K.-B. Cai, H.-Y. Huang, M.-L. Hsieh, P.-W. Chen, S.-E. Chiang, S. H. Chang, J.-L. Shen, W.-R. Liu and C.-T. Yuan, Two-dimensional self-assembly of boric acid-functionalized graphene quantum dots: tunable and superior optical properties for efficient eco-friendly luminescent solar concentrators, ACS Nano, 2022, 16, 3994–4003 CrossRef CAS PubMed.
- Q. Li, D. Cheng, H. Gu, D. Yang, Y. Li, S. Meng, Y. Zhao, Z. Tang, Y. Zhang, J. Tan and S. Qu, Aggregation-induced color fine-tunable carbon dot phosphorescence covering from green to near-infrared for advanced information encryption, Chem. Eng. J., 2023, 462, 142339 CrossRef CAS.
- Y. Liu, X. Kang, Y. Xu, Y. Li, S. Wang, C. Wang, W. Hu, R. Wang and J. Liu, Modulating the carbonization degree of carbon dots for multicolor afterglow emission, ACS Appl. Mater. Interfaces, 2022, 14, 22363–22371 CrossRef CAS.
- Z. Li, H. Cheng, B. Wang, L. Wang, J. Wu, B. Zhang, Z. Tang and S. Qu, Polylysine-modified near-infrared-emitting carbon dots assemblies: amplification of tumor accumulation for enhanced tumor photothermal therapy, J. Colloid Interface Sci., 2024, 668, 132–141 CrossRef CAS.
- J. Liu, Y. Luo, Z. Ran, F. Wang, M. Sun, Y. Luo, J. Zhuang, X. Zhang, B. Lei, Y. Liu and C. Hu, Calcination temperature tuning of RTP and TADF with wide range of emission color from carbon dots confined in Al2O3, Chem. Eng. J., 2023, 474, 145597 Search PubMed.
- K. Jiang, Y. Wang, C. Lin, L. Zheng, J. Du, Y. Zhuang, R. Xie, Z. Li and H. Lin, Enabling robust and hour-level organic long persistent luminescence from carbon dots by covalent fixation, Light: Sci. Appl., 2022, 11, 80 CrossRef CAS PubMed.
- H. Hu, J. Li and X. Gong, Hour-level persistent multicolor phosphorescence enabled by carbon dot-based nanocomposites through a multi-confinement-based approach, Small, 2023, 20, 2308457 CrossRef PubMed.
- Y. Luo, Q. Jiang, J. Liu, H. Yang, X. Liao, F. Huang, J. Zhuang, C. Hu, B. Lei, Y. Liu and J. He, Mesoporous collapsing encapsulated carbon dots: direct evidence for the effect of multiple-confined interactions of silica on efficient long-lived phosphorescence, Chem. Eng. J., 2024, 486, 150436 CrossRef CAS.
- Z. Guan, Z. Tang, J. Zeng, J. Deng, Y. Zheng, H. Li and X. Liu, Molecular engineering enables multi-color room temperature phosphorescence of carbon dots composites derived in situ, facilitating their utilization for advanced information encryption, Adv. Opt. Mater., 2024, 12, 2302820 CrossRef CAS.
- R. B. Pricilla, P. Urbanek, J. Sevcik, D. Skoda, J. Antos, L. Munster, E. Domincova-Bergerova and I. Kuritka, Exploring the ultralong lifetime of self-matrix 1,10 phenanthroline and boron-based room temperature phosphorescence carbon dots for multiple applications, Adv. Opt. Mater., 2024, 12, 2400753 Search PubMed.
- W. He, X. Sun and X. Cao, Construction and multifunctional applications of visible-light-excited multicolor long afterglow carbon dots/boron oxide composites, ACS Sustainable Chem. Eng., 2021, 9, 4477–4486 CrossRef CAS.
- H. Zheng, P. Cao, Y. Wang, X. Lu and P. Wu, Ultralong room-temperature phosphorescence from boric acid, Angew. Chem., Int. Ed., 2021, 133, 9586–9592 Search PubMed.
- Z. Zhang, Z. Wang, X. Liu, Y.-E. Shi, Z. Li and Y. Zhao, Modulating emission of boric acid into highly efficient and color-tunable afterglow via dehydration-induced through-space conjugation, Adv. Sci., 2023, 10, 2300139 CrossRef CAS.
- Z. Guo, C. Wang, F. Qi, J. Dong, J. Xue, Y. Zhang, B. Xu, G. N. Liu, Y. Sun and C. Li, Sunlight-activated room-temperature phosphorescent carbon dots, Adv. Funct. Mater., 2024, 35, 2414178 CrossRef.
- X. Chen, J. Li, W. Zou and X. Gong, Regulating the surface state of carbon dots as ultrahigh-capacity adsorbents for water treatment, Small, 2024, 20, 2404407 CrossRef CAS PubMed.
- Q. Zhao, C. Fan, H. Bu, J. Gao, L. Li, X. Yu, X. Yang, Z. Lu and X. Zhang, Electrochemical synthesis of multicolor carbon dots with room temperature phosphorescence to thermally activated delayed fluorescence via surface state modulation, Chem. Eng. J., 2024, 500, 156704 CrossRef CAS.
- Z. Zhou, Z. Song, J. Liu, B. Lei, J. Zhuang, X. Zhang, Y. Liu and C. Hu, Energy transfer mediated enhancement of room-temperature phosphorescence of carbon dots embedded in matrixes, Adv. Opt. Mater., 2021, 10, 2100704 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5sc02963e |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2025 |