
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCluster versus coordination: the chemistry of cyclopentadienyl titanium and vanadium complexes with B- and C-functionalized carborane-thiols, [C2B10H12−n(SH)n] (n = 2 or 3)†
Subhash
Bairagi‡
 a,
Deepak Kumar
Patel‡
abc,
Debipada
Chatterjee
a,
Monika
Kučeráková
d,
Jan
Macháček
c,
Tomas
Base
*c,
Thalappil
Pradeep
*ab and
Sundargopal
Ghosh
*a
a,
Deepak Kumar
Patel‡
abc,
Debipada
Chatterjee
a,
Monika
Kučeráková
d,
Jan
Macháček
c,
Tomas
Base
*c,
Thalappil
Pradeep
*ab and
Sundargopal
Ghosh
*a
aDepartment of Chemistry, Indian Institute of Technology Madras, Chennai 600036, India. E-mail: sghosh@iitm.ac.in
bDST Unit of Nanoscience (DST UNS) and Thematic Unit of Excellence (TUE), Department of Chemistry, Indian Institute of Technology Madras, Chennai 600036, India. E-mail: pradeep@iitm.ac.in
cInstitute of Inorganic Chemistry, The Czech Academy of Science, 25068 Rez, Czech Republic. E-mail: tbase@iic.cas.cz
dInstitute of Physics, The Czech Academy of Science, 182 21 Prague 8, Czech Republic
First published on 10th July 2025
Abstract
A series of B- and C-functionalized di- and trithiol chelating o-carborane ligands have been employed to explore the coordination chemistry with cyclopentadienyl titanium and vanadium complexes. Treatment of [Cp*TiCl3] with [LiBH4·THF], followed by thermolysis with a C-functionalized carborane-dithiol ligand [1,2-(SH)2-1,2-C2B10H10], yielded octacapped octahedral [(Cp*Ti)4{Ti(1,2-(S)2-1,2-C2B10H10)}2(μ3-S)6(μ3-O)2] (1) and hexacapped trigonal bipyramidal [(Cp*Ti)4{Ti(1,2-(S)2-1,2-C2B10H10)}(μ3-S)6] (2) clusters. One of the driving forces of these reactions is the cleavage of C–S bonds of carborane-dithiols that resulted in sulfide ligands and subsequently generated clusters 1 and 2. In contrast, a similar reaction with a B-functionalized carborane-dithiol [9,12-(SH)2-1,2-C2B10H10] led to B–B bond formation that yielded a κ2-hydridoborato complex, [(Cp*Ti){κ2-BH3(9,12-(S)2-1,2-C2B10H10)}] (3). To the best of our knowledge, complex 3 is the first example of a carborane-dithiol functionalized hydridoborato complex. Interestingly, when the reactions of [Cp*TiCl3] or [Cp2TiCl2] were carried out with a B-functionalized carborane-trithiol, [8,9,12-(SH)3-1,2-C2B10H9], they led to coordination complexes, [(Cp/Cp*Ti){8,9,12-(S)3-1,2-C2B10H9}] (Cp* (4a) and Cp (4b)). Similarly, when [(Cp*VCl2)3] was employed as a metal precursor, deboronation was observed at the icosahedral cage that resulted in a zwitterionic complex, [(Cp*V){1,5,6-(S)3-nido-7,8-C2B9H9}] (5). All the clusters have been characterized by NMR, IR, mass spectrometry, and X-ray diffraction analysis. Furthermore, the theoretical analyses provided valuable insights into the electronic structures of these unusual clusters.
Introduction
Since the inception of coordination chemistry from the work of Alfred Werner,1 the field of inorganic chemistry has expanded into various branches, such as organometallics, polymers, bioinorganics, and materials chemistry. Central to these developments and their comprehensive understanding is the specific design of ligands exhibiting specific geometry and enabling site-specific functionalization. These play a critical role in shaping the properties of metal complexes.2 Ligand design not only influences the formation of new structures but also accelerates reaction rates, alters mechanisms, and fine-tunes the topology and functionality of metal–organic frameworks.3 One of the biggest challenges in ligand design, which often involves modifying the chemical properties (such as electron-donating or withdrawing behaviour) as well as changing the steric requirements of ligands, is represented by modification of one whilst keeping the other identical.4 In this regard, icosahedral carboranes represent a fascinating class of highly stable, boron-rich clusters that can be readily modified at various vertices through simple chemical reactions while preserving their steric demands.5Despite significant progress in the coordination chemistry of carboranes, systematic investigation focusing on the effect of direct positional attachment of exo-substituents to carboranes has not been much explored.5–7 This is surprising given that the distinct chemical reactivity of the CH and BH vertices in these clusters has been well established.5 In this regard, Mirkin and Spokoyny showed that the coordination strength of a carborane-functionalized thioether to Pt(II) (complex II, Chart 1) can be significantly influenced by the positional attachment of the sulfur-containing moiety.8 This means that icosahedral carboranes can function as strong electron withdrawing or donating ligands, a unique behaviour in coordination chemistry that could regulate ligand electronic properties.
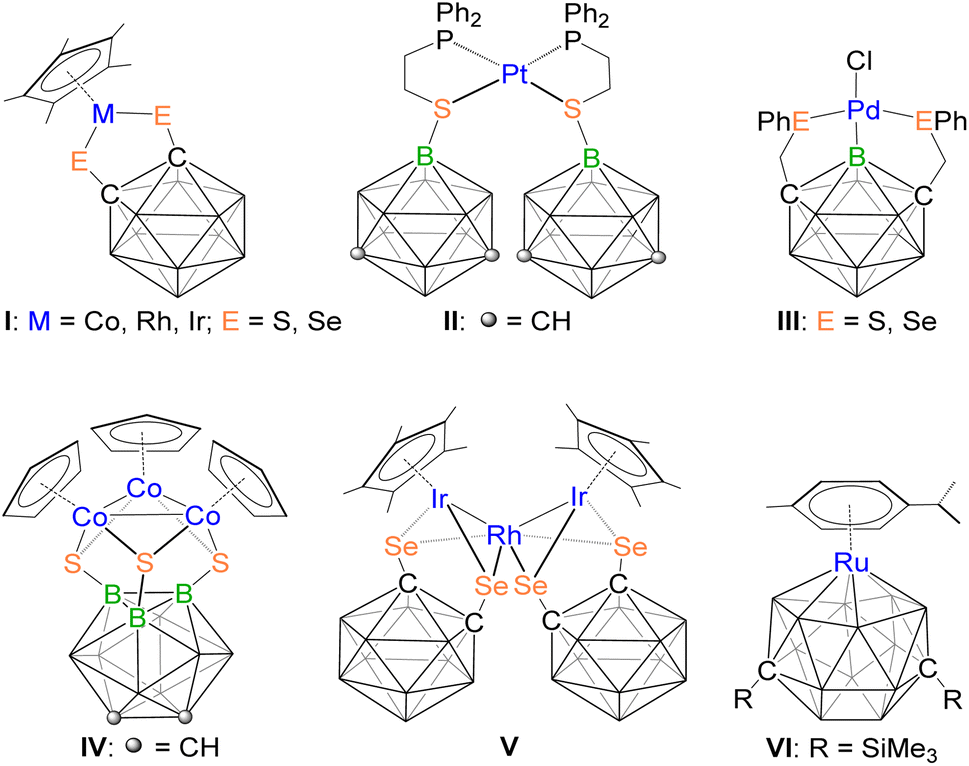 | ||
| Chart 1 Various examples of functionalized-carborane-protected transition metal compounds. | ||
Over several decades, the research on o-carborane derivatives has garnered significant attention due to their unique structures, fundamental properties, and potential applications in material synthesis, medicine, optics, and microelectronics.7b,9,10 In this regard, the monometallic 16e half-sandwich o-carborane species I, such as, [Cp*M{1,2-(E)2-1,2-C2B10H10}] (M = Co, Rh, Ir; E = S, Se) have been well explored.11,12 In these complexes, the thiolate ligands in the metalladithiolene ring [MS2C2] often modulate carbon-based substituent effects, allowing for the tuning of the steric and electronic properties of the compounds. These sterically congested, mononuclear coordination compounds serve as stable starting materials and have demonstrated rich reaction chemistries.11–14 For instance, these complexes can undergo alkyne insertion into metal–chalcogen bonds, which can subsequently lead to B–H activation, the formation of metal–boron bonds, and the functionalization of the carborane cage at B(3)/B(6) positions.12 Additionally, the construction of novel polycarborane molecular architecture using two or more [1,2-(E)2-1,2-C2B10H10]2− units has been reported.15 One notable metalation product, [1-(σ-S)-2-(η5-C5H4CH(Ph))-1,2-C2B10H10]Ti(NMe2)2, was synthesized, where the appended carborane-thiol unit functions as both a linking and η1-bonding group.16
Jin group has nicely established the chemistry of bimetallic rhodium complexes utilizing the [1,2-(E)2-1,2-C2B10H10]2− ligand, such as [(Cp*Rh)2{1,2-(S)2-1,2-C2B10H10}]17a and [(Cp'Rh)2{1,2-(E)2-1,2-C2B10H10}]17b (Cp′ = η5-1,3-tBu2C5H3). Subsequently, Jin and Herberhold demonstrated that type I complexes can serve as models for further transformations due to their quasi-aromaticity and electronic unsaturation.7b,12c,17,18 These complexes are particularly useful for the synthesis of heterometallic clusters with metal–metal bonds, stabilized by carborane-dichalcogenolato ligands. Interestingly, Jin group reported the first trimetallic cluster [{Cp*Ir(1,2-(Se)2-1,2-C2B10H10)}2Rh] (V), synthesized from the reaction of [Cp*Ir{1,2-(Se)2-1,2-C2B10H10}] with [RhCl(cod)2].19a Using a similar method, they have prepared the tetrametallic clusters [{Cp*Ir(1,2-(E)2-1,2-C2B10H9)}2Rh2(cod)] (E = S or Se).19b It is noteworthy that 1,2-dithiol-o-carborane has been extensively used as a strong electron-withdrawing group in the construction of both mononuclear and multinuclear complexes. Furthermore, Mirkin and Spokoyny reported the first carborane-based pincer ligand family and the respective Pd complexes of these ligands (complex III).7c The structure represents a new class of compounds with a Pd–B σ-coordination bond pincer motif.
Although the chemistry of C-functionalized carborane-thiols are well established by several groups, the chemistry of B-functionalized carborane has not been explored much.7c,8,20 Yan and co-workers reported a monometallic 16-electron half sandwich complex of cobalt containing a 9,12-dithiol-o-carborane ligand, [CpCo{9,12-(S)2-1,2-C2B10H10}], and demonstrated its utility in the synthesis of boron-fused 1,4-dithiin compounds.20b Furthermore, Yan and Bregadze synthesized a new B-functionalized 8,9,12-trithiol-o-carborane and used it as a strongly electron-donating ligand to construct an unusual, isolable trinuclear cobalt cluster IV with 51 cluster valence electrons.20c They also showed that boron-based substituents significantly influence the stability and reactivity of these trimeric cobalt clusters.
The chemistry of metallacarborane has progressed impressively from the isolation of small cages to 16-vertex clusters.7a,21,22 Xie and co-workers recently isolated and structurally characterized the 16-vertex metallacarborane VI by reduction of a 15-vertex carborane [1,14-(SiMe3)2-1,14-C2B13H13] with excess of sodium, followed by reaction with 0.5 equiv. [Ru(p-cym)Cl2]2.22a They have also developed the chemistry of metallacarboranes of early transition metals. Many metallacarboranes of group 4 were synthesized utilizing closo-12 or nido-11 vertex carboranes.22b–d These metallacarboranes exhibit reactivity with unsaturated organic substrates such as alkynes, alkyl nitriles, diaryl ketones, and carbodiimides, with reactions primarily occurring at the metal centers. In parallel, our group has also focused on transition metal–boron compounds with diverse structures and properties. These include higher-vertex metallaborane clusters,23 electron-precise transition metal–boron complexes,24 and metallachalcogenaboranes25 containing group 4–9 metals. The synthetic methodologies to generate metallachalcogenaborane clusters involve treating chalcogens, diaryl dichalcogenides, or carbon disulfide with Cp*-based transition metal chlorides ([Cp*MClx]) in the presence of [LiBH4·THF].23–25 These synthetic strategies have enabled us to isolate complexes in which small borane fragments are stabilized within the coordination sphere of bimetallic or trimetallic frameworks. Inspired by these results, we have attempted to synthesize metallachalcogenaboranes using carborane-thiols as the chalcogen source. In this article, we have explored the chemistry of B- and C-functionalized o-carborane-thiol ligands with in situ generated intermediates, generated from the Cp*-based metal chlorides and [LiBH4·THF]. These reactions led to the formation of clusters and coordination complexes featuring o-carborane-thiol ligands. We have further demonstrated how the C- or B-functionalized carboranethiol influences the formation of clusters or coordination complexes, respectively, without altering the steric properties of the ligands.
Results and discussion
Reactivity of [Cp*TiCl3] with the C-functionalized carborane-dithiol [1,2-(SH)2-1,2-C2B10H10]
Reaction of [Cp*TiCl3] with [LiBH4·THF] at low temperature generated an unstable and sensitive intermediate, which, on treatment with [1,2-(SH)2-1,2-C2B10H10] at elevated temperature, led to the formation of an octacapped octahedral cluster [(Cp*Ti)4{Ti(1,2-(S)2-1,2-C2B10H10)}2(μ3-S)6(μ3-O)2] (1) and a hexacapped trigonal bipyramidal cluster [(Cp*Ti)4{Ti(1,2-(S)2-1,2-C2B10H10)}(μ3-S)6] (2; Scheme 1). Note that this reaction also yielded other air and moisture sensitive products in low yields. The reaction mixture was separated using thin layer chromatography (TLC) on aluminium-supported silica gel plates. Although elution with hexane and dichloromethane solvent (50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 v/v) allowed us to isolate 1 and 2 as brown solids, all of our attempts to isolate other species were unsuccessful. Detailed characterization of Ti-clusters 1 and 2 is discussed in the sections below.
:50 v/v) allowed us to isolate 1 and 2 as brown solids, all of our attempts to isolate other species were unsuccessful. Detailed characterization of Ti-clusters 1 and 2 is discussed in the sections below.
 | ||
| Scheme 1 Synthesis of 1,2-dithiol-o-carborane protected Ti-clusters 1 and 2. | ||
:1:2 ratio which corresponded to the Cp* protons. In addition, the 1H{11B} chemical shifts at δ = 2.52–2.98 ppm are attributed to the BHt protons of the carborane ligand. The 11B NMR spectrum of 1 displayed very broad resonances between δ = −13.0 and −1.0 ppm, consistent with the presence of the [1,2-(S)2-1,2-C2B10H10]2− ligand in cluster 1. In the IR spectrum of 1, an absorption band at 2580 cm−1 suggested the presence of B–H bonds. The mass spectrum of 1 exhibited a molecular-ion peak at m/z 1504.2403 ([M + K]+). However, these spectroscopic data were not sufficient to identify the core geometry of 1. A definitive structural explanation was achieved through single-crystal X-ray analysis of a suitable crystal of 1, grown from an n-hexane/CH2Cl2 solution at −5 °C.
The solid-state X-ray structure of 1 revealed a distorted octahedral metallic framework consisting of six titanium atoms, where the eight triangular faces of the octahedron are capped by six triply bridging sulfur atoms and two triply bridging oxygen atoms, forming a distorted cube {S6O2} (Fig. 1). All the sulfur atoms in 1 must have originated from the de-thiolation of the ligand 1,2-dithiol-o-carborane, as this ligand is the only source of sulfur in the reaction. This agrees with previous studies, as the C–S bond is weaker than the B–S bond.26 Furthermore, we believe that the trace amount of water bound to the silica gel led to the incorporation of oxygen that resulted in the formation of 1 during chromatographic work-up. Despite our best efforts, we were unable to isolate the unstable intermediate involved in this conversion. Although we are unable to provide any direct evidence to support this, a similar phenomenon has previously been observed in transition metal boron complexes.27 Interestingly, at two of six Ti atoms (Ti2 and Ti3), the Cp* ligand is replaced by the bidentate [1,2-(S)2-1,2-C2B10H10] ligand. To the best of our knowledge, cluster 1 is the first example of an [M6] cluster containing a carborane ligand. The [Ti6S6O2] core in 1 is isostructural with [Ti6O8] in [Cp6Ti6O8].28
 | ||
| Fig. 1 Molecular structure and labelling diagram of 1. Cp* ligands attached to Ti4 and Ti5 atoms are omitted for clarity. Selected bond lengths (Å) and bond angles (°): Ti⋯Ti 3.130–3.598, Ti1–S5 2.4274(12), Ti1–O1 1.919(2), Ti2–S5 2.4835(12), Ti2–S1 1.3963(13), C1–S1 1.785(4), B1–C1 1.703(7), Ti3–Ti2–Ti5 93.913, Ti1–S5–Ti2 82.52(4). | ||
The coordination geometry around four Ti centers (Ti1, Ti4, Ti5, and Ti6) in 1 is square pyramidal, with the basal plane defined by μ3-chalcogen atoms and the apex occupied by the Cp* ligand. The average displacement of titanium atoms from the basal plane is 0.951 Å, which is slightly higher compared to the reported complexes [Cp6Ti6O8]28a (0.762 Å) and [Cp*6Ti6S8]28b (0.857 Å). The coordination geometry around the other two Ti centers (Ti2 and Ti3) is octahedral, with the equatorial plane defined by one μ3-S, one μ3-O, and two S atoms from one of the 1,2-dithiol-o-carborane ligands, while the axial positions are occupied by one μ3-S and one μ3-O ligand. The Ti–Ti bond distances in 1 range from 3.130 Å to 3.598 Å, which are comparable to those in [Cp*6Ti6S8] (av. 3.503 Å) and [(CpTi)5(μ3-S)5(μ3-S2)(μ4-O)]28c (3.207(3)–3.597(3) Å) but considerably longer than those in [Cp5Ti5S6] (av. 3.156 Å).28b,29 Furthermore, due to the replacement of Cp* by the 1,2-dithiol-o-carborane ligand, which reduces steric crowding at the titanium centers, the Ti2–Ti3 bond distance (3.130 Å) is significantly shorter compared to other Ti–Ti bond distances (3.230–3.598 Å). The average Ti–S–Ti bond angle in 1 is 89.68°, similar to the value observed in [Cp*6Ti6S8] (91.29°). The two 1,2-dithiol-o-carborane ligands attached to two equatorial titanium atoms are nearly coplanar with each other. Additionally, the Ti–S–C–C–S plane is not coplanar with the Ti4 equatorial plane but is tilted by an average angle of 42.27°.
The cluster valence electron (CVE) count for an M6 octahedral cluster is expected to have 86 valence electrons (VEs).30 However, the total number of VEs available in 1 is 80 [9 (Cp*Ti) × 4 + 6 {Ti(1,2-(S)2-1,2-C2B10H10)} × 2 + 4 (μ3-S) × 8]. For comparison, the clusters [Cp6Ti6S8] and [Cp6Ti6Cl8] possess 86 and 94 VEs, respectively. Therefore, while [Cp6Ti6S8] and [Cp6Ti6Cl8] are electronically saturated and hyper-electronic clusters, respectively, cluster 1 is hypo-electronic. On the other hand, if we consider 1 as an organometallic aggregate, one of the important aspects of this chemistry is the electronic configuration of [M6X8] (X = O, S, Cl) clusters. According to the inert gas formalism, clusters [Cp6M6X8] (X = O, S, Cl) and 1 require 84 and 80 electrons, respectively.28e,f However, [Cp6Ti6S8], [Cp6Ti6Cl8], and 1 possess 86, 94, and 80 electrons, respectively. The number of electrons in excess of those required for Ti–Cp and Ti–X (X = O, S, Cl) bonding increases precisely with the formal number of Ti(III) d1 ions present in each cluster: ten in [Cp6Ti6Cl8], two in [Cp6Ti6S8], and zero in 1. Therefore, in terms of electronic configuration, cluster 1 is a new addition to these [M6X8] cluster series. Based on valence-bond arguments, Gillespie has suggested that the two excess electrons in [Cp6Ti6S8] reside at the center of the octahedron.31a Mealli et al. have reported a qualitative molecular orbital (MO) analysis of such octahedral metal clusters.31b They have shown that the excess electron pair occupies the a1g bonding MO, composed of metal d orbitals, resulting in 1/12 M–M bond order at each edge. In 1, although, the a1g remains unoccupied, there are other factors that cement the Ti6 unit together, which include mixing of other orbitals and second-order perturbation effects. Notably, the electron count in clusters of the early transition metals can be as low as 80, and cluster 1 is an example of an 80 VE octahedral cluster.31b The MO calculations and presence of all Ti(+4) ions (resulting in zero VE) explain the diamagnetism of 1, which is consistent with the intensity and low energy of the visible absorption; cluster 1 is brown.
Density functional theory (DFT) studies at the BP86/def2-SVP level were performed to understand the electronic structure and bonding situation in 1. The optimized structural parameters are closely aligned with those obtained from X-ray crystallography. MO analysis of 1 revealed that the HOMO is predominantly localized on the p orbitals of the thiolato groups, with a minor contribution from the d orbitals of the Ti2 and Ti3 atoms (Fig. 2a). On the other hand, the LUMO is entirely localized on the d orbitals of the six Ti atoms (Fig. 2b). MO analysis further showed that the bonding of the Ti atoms (Ti2 and Ti3) with the thiolato groups of the 1,2-dithiol-o-carborane ligand involved substantial overlap of d orbitals of Ti with p orbitals of the sulfur atoms as observed in HOMO−13 (Fig. 2c). The coordination of the 1,2-dithiol-o-carborane ligand to the Ti atom through the thiolato groups was also depicted by a contour line diagram of the Laplacian of electron density drawn along the Ti2–S2–C1–C2–S1 ring (Fig. 2d). Additionally, the natural charge analysis revealed that the thiolato groups carry positive natural charges, while the μ3-S atoms bear negative natural charges (Table S3†).
 | ||
| Fig. 2 (a) HOMO, (b) LUMO, and (c) HOMO−13 of 1 (isocontour values: ±0.045 [e bohr−3]1/2). (d) Contour-line diagram of the Laplacian of the electron density along the Ti3–S3–S4 plane in 1. | ||
The solid-state X-ray diffraction study revealed the identity of 2 as [(Cp*Ti)4{Ti(1,2-(S)2-1,2-C2B10H10)}(μ3-S)6], which showed that 2 contains sulfur atoms bridging the triangular faces of a distorted trigonal bipyramid of titanium (Fig. 3). The [1,2-(S)2-1,2-C2B10H10] ligand replaced a Cp* ligand on one of the Ti atoms (Ti2) of the equatorial plane. The axial titanium (Ti1, Ti5) and one of the equatorial titanium centers (Ti2) have a coordination number of six. On the other hand, the titanium atoms (Ti3, Ti4) have a coordination number of seven. A crystallographic mirror plane lies in the vertical direction containing Ti1, Ti2, and Ti5 atoms. Cluster 2 represents another metal-sulfide cluster with a carborane ligand, which is isostructural with [Cp5Ti5S6].29 The Ti–Ti bond distances (av. 3.274 Å) in 2 are shorter compared to those in cluster 1 (av. 3.384 Å) but are slightly longer than the Ti–Ti distance in [Cp5Ti5S6] (av. 3.156 Å). The Ti–S bond distances in 2 (av. 2.436 Å) are comparable to those in cluster 1 and [Cp5Ti5S6]. Notably, the Ti3–Ti4 bond distance (3.201 Å), opposite to the Ti atom attached to the 1,2-dithiol-o-carborane ligand, is shorter than the other Ti– Ti bond distances (3.243–3.306 Å). The axial Ti–S distances (av. 2.290 Å) are significantly shorter than the equatorial Ti–S distances (av. 2.513 Å). The differing axial and equatorial Ti–S distances are due to the different formal oxidation states of the Ti atoms. The two axial Ti atoms adopt a +4 oxidation state, whereas among the equatorial Ti centers, two coordinated to Cp* are formally Ti(+3), while the remaining Ti, bound to the dithiol-o-carborane ligand, exhibits +4 oxidation state. In this regard, a 3c–2e bonding interaction was observed along the equatorial trititanium (Ti2–Ti3–Ti4) framework in the HOMO of 2 (Fig. 4). This kind of interaction in trimetallic frameworks has recently been well-studied due to their aromatic and magnetic properties.32 Perhaps the most persuasive evidence for Ti–Ti interactions is the acute Ti–S–Ti angles, averaging 83.27°. Since the μ3-sulfur bridge could hold the cluster together without any Ti–Ti interaction, one would expect that the Ti–S–Ti angles would approach the idealized tetrahedral angle without interaction. Therefore, the observed angles and distances indicate the presence of Ti–Ti interactions. The CVE count for an M5 trigonal bipyramidal cluster is expected to be 72 VEs.30 However, cluster 2 contains only 66 valence electrons, confirming that it is hypo-electronic.
 | ||
| Fig. 3 Molecular structure and labelling diagram of 2. The Cp* ligand attached to Ti3 atom is omitted for clarity. Selected bond lengths (Å) and bond angles (°): Ti⋯Ti 3.201–3.305, Ti1–S1 2.2839(17), Ti2–S1 1.5004(16), Ti2–S7 1.446(4), Ti3–S1 2.5089(17), C2–S8 1.782(9), Ti3–Ti2–Ti4 58.270, S7–Ti2–S8 81.09(12), Ti1–S1–Ti2 82.24(5). | ||
 | ||
| Fig. 4 HOMO of 2 showing 3c–2e equatorial trititanium interactions (isocontour values: ±0.045 [e bohr−3]1/2). | ||
The 1,2-dithiol-o-carborane ligand in 2 is attached to the equatorial Ti-atom in such an orientation that the Ti–S–C–C–S plane makes an angle of 43.28° with the Ti3 equatorial plane. The incorporation of the 1,2-dithiol-o-carborane ligand has measurable effects on the bond lengths, angles, and overall geometry of the Ti-clusters 1 and 2. The ligand appears to enhance structural stability by balancing electronic and steric factors, influencing both intra-cluster interactions and resistance to external perturbations. These findings suggest that carborane-based ligands are promising tools for stabilizing high-nuclearity metal-sulfide clusters with potential applications in catalysis and materials science.
Reactivity of [Cp*TiCl3] with the B-functionalized carborane-dithiol [9,12-(SH)2-1,2-C2B10H10]
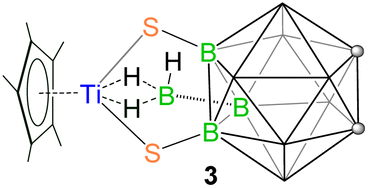
The successful isolation of clusters 1 and 2 interested us to investigate the role of ligand connectivity in cluster formation. Note that in these systems, the SH groups are influenced by the strong electron-donating effect of the carborane cage, causing them to act as weak acids.33 The presence of these changes in B-functionalized carborane-dithiol is likely to modify the course of the reaction as compared to the C-functionalized ligand. As a result, we have explored the reactivity of B-functionalized carborane-dithiol [9,12-(SH)2-1,2-C2B10H10]. The low-temperature reaction of [Cp*TiCl3] with [LiBH4·THF] resulted in an unstable, sensitive intermediate, just like in the previous case, which on treatment with [9,12-(SH)2-1,2-C2B10H10] at elevated temperature yielded the yellow complex 3 with a relatively higher yield of 64% (Scheme 2). Unlike clusters 1 and 2, the 1H NMR spectrum of 3 displayed a single peak at δ = 2.24 ppm, which can be assigned to the chemically equivalent methyl protons of the Cp* ligand. Interestingly, the high-field region showed a broad resonance at δ = −0.86 ppm that suggested the presence of the Ti–H–B proton. The 11B{1H} NMR showed six resonances at δ = −15.8, −11.2, −10.1, −6.8, −3.1 and 8.4 ppm. The IR spectrum of 3 exhibits an absorption band at 2602 cm−1 that suggests the presence of terminal B–Ht stretching vibrations originating from the 9,12-dithiol-o-carborane ligand. The 13C NMR spectrum confirms the presence of Cp* (δ = 13.9 and 127.8 ppm) and 9,12-dithiol-o-carborane (δ = 44.3 ppm) ligands. Furthermore, the ESI-MS spectrum of 3 displayed a molecular ion peak at m/z 403.2129 with the typical isotopic distribution pattern corresponding to the [M+H]+ fragment. | ||
| Scheme 2 Synthesis of the 9,12-dithiol-o-carborane funcionalized hydridoborato complex of titanium, 3. | ||
The single-crystal X-ray diffraction analysis of 3 revealed a structure that is completely different from that of 1 and 2, a 9,12-dithiol-o-carborane-funcionalized κ2-hydridoborato complex [(Cp*Ti){κ2-BH3(9,12-(S)2-1,2-C2B10H10)}] (Fig. 5). In complex 3, one Cp* ligand and one {κ2-BH3(9,12-(S)2-1,2-C2B10H10)} ligand are coordinated to the central titanium atom opposite to each other. This 9,12-dithiol-o-carborane-funcionalized borate ligand is connected to the metal through four atoms: two are thiol substituents of the carborane cage, and two are hydrogens of the exo-BH3 unit. Therefore, this structure can be described as a four-legged piano-stool geometry, where the coordination number around the titanium center is seven, assuming the Cp* ligand as three-coordinate. The structure possesses a σ-plane of symmetry through the plane containing [Ti1–B11–B1–B10–B9–B4] atoms. Unlike 1 and 2, the metallacycle, [Ti1–S1–B2–B3–S2] in 3 is puckered at Ti caused by the newly formed Ti–dihydridoborato interaction, while the remaining four atoms [S1–B2–B3–S2] form an almost perfect plane. Consequently, the [TiS2B2] metallacycle in 3 is bent, and the titanium atom is out of the plane of the 9,12-dithiol-o-carborane ligand, with the dihedral angles between the planes defined by [Ti–S–S] and [S–B–B–S] being 66.29°. The Ti–S bond lengths in 3 (av. 2.327 Å) are shorter than those in clusters 1 (av. 2.47 Å) and 2 (av. 2.45 Å) but are in good agreement with the existing values in the literature for a wide variety of Ti–S σ-bonds.
 | ||
| Fig. 5 Molecular structure and labelling diagram of 3 (left) and 4a (right). Selected bond lengths (Å) and bond angles (°): 3: Ti1–S1 2.3321(9), Ti1–S2 2.3214(9), Ti1⋯B11 2.259(4), Ti1–H1 2.00(3), Ti–H2 1.93(3), B11–H1 0.90(3), B11–H2 1.03(4), B1–B11 1.750(5), B2–S1 1.851(4), C1–C2 1.626(6), Ti1–S1–B2 81.17(11), B2–B1–B11 121.9(3). 4a: Ti1–S1 2.3095(12), Ti1–S3 2.3231(12), B1–S3 1.849(5), B1–B2 1.829(5), C1–C2 1.635(7), C1–B10 1.713(7), Ti1–S1–B2 80.72(14). | ||
One of the interesting features of complex 3 is the presence of the 9,12-dithiol-o-carborane functionalized hydridoborato ligand. The isolation of the Ti-hydridoborato unit suggests the formation of an intermediate [(Cp*Ti)(κ2-BH4){9,12-(S)2-1,2-C2B10H10}] followed by the formation of a new B–B bond via a dehydrocoupling reaction between one terminal hydrogen of (κ2-BH4) and one B(8)–H hydrogen from the [9,12-(SH)2-1,2-C2B10H10] ligand. The κ2-coordinated BH3R group exhibits a Ti⋯B distance of 2.259(4) in 3, which is shorter than those for the κ2-BH3Me ligand in [(Cp*Ti)(κ2-BH3Me)(κ3-BH3Me)]32a (2.377(2) Å), but longer than those in κ3-hydridoborato ligands in [(Cp*Ti)(κ3-BH4)(thf)2](BPh4)34a (2.178(6) Å) and [(CO)4Ti(κ3-BH4)]34b (2.158(7) Å). A qualitative electron counting scheme suggests 3 to be a 14 electron system ((Cp*− (6e) + {κ2-BH3(9,12-(S)2-1,2-C2B10H10)}3− (6e)) + 2 Ti–H–B (2e)), and Ti4+ (0e), where Ti is in +4 oxidation state.
To get insight into the bonding scenario in dihydridoborate coordination, we have performed DFT calculations on 3. The natural bond orbital (NBO) and MO analyses indicated the existence of strong 3c–2e bonding interactions along the Ti1–H– B11 units (Fig. 6a and S62d†). The Laplacian of electron density revealed the presence of BCP along the bond paths of Ti1–Hbridging and B11–Hbridging, respectively (Fig. 6c). The bond paths appear to be curved inwards, confirming these interactions to be of 3c–2e type. Furthermore, an extended delocalized bonding interaction was observed in HOMO−6 that shed light on the coordination of the Ti-borate unit with the carborane cage through B1 boron (Fig. S62c†). In this regard, NBO analysis suggested the presence of a 2c–2e bond between B11–B1 (Fig. 6b), which was further supported by a BCP along the corresponding bond path in the electron density contour plot (Fig. 6d). Additionally, MO analysis illustrated the bonding of the two thiolato groups with the metal center aided by the overlap of the d orbital of Ti with p orbitals of S (Fig. S62b†).
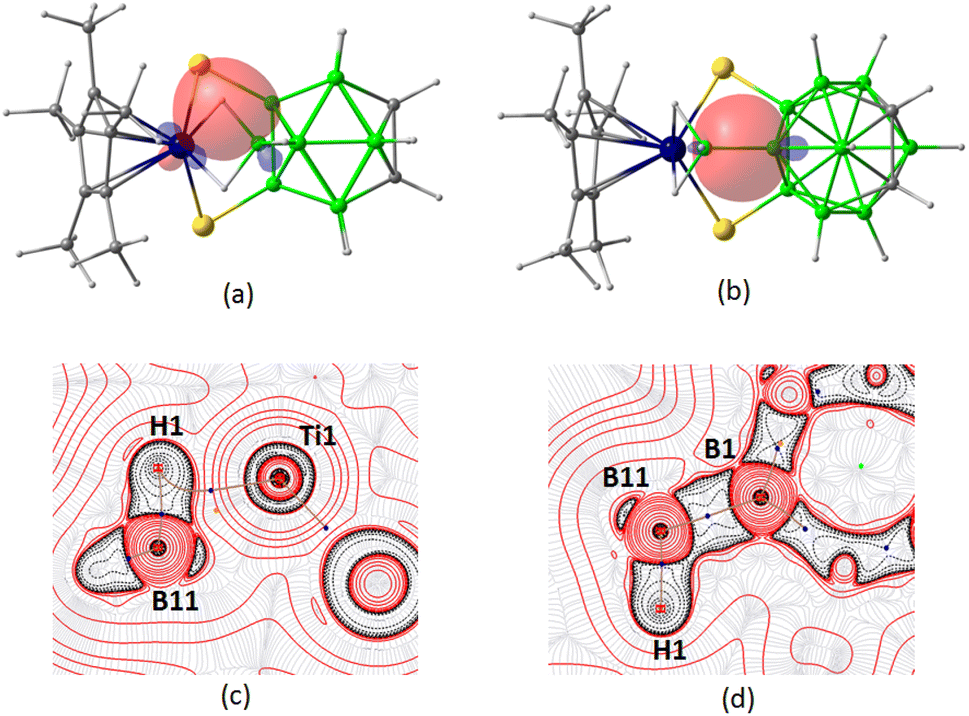 | ||
| Fig. 6 Selected NBO of 3 showing (a) 3c–2e interaction of Ti1–H1–B11 and (b) 2c–2e interaction of B1–B11 (isocontour values: ±0.045 [e bohr−3]1/2). Contour-line diagram of the Laplacian of electron density along (c) Ti1–H1–B11 and (d) B1–B11–H1 planes in 3. | ||
Reactivity of [Cp*TiCl3] and [Cp2TiCl2] with the B-functionalized carborane-trithiol [8,9,12-(SH)3-1,2-C2B10H9]
To explore the coordination chemistry of carborane-thiol ligands, we have now explored the chemistry of the B-functionalized carborane-trithiol ligand [8,9,12-(SH)3-1,2-C2B10H9] under similar reaction conditions. An unstable intermediate was formed through a low-temperature reaction of [Cp*TiCl3] with [LiBH4·THF], which on treatment with [8,9,12-(SH)3-1,2-C2B10H9] yielded, [(Cp*Ti){8,9,12-(S)3-1,2-C2B10H9}] (4a; Scheme 3). With an objective to isolate a trimetallic Ti3 cluster, [(Cp2Ti)3{8,9,12-(S)3-1,2-C2B10H9}2], we carried out the reaction of the trithiol ligand with [Cp2TiCl2]. Unexpectedly, this led to the formation of [(CpTi){8,9,12-(S)3-1,2-C2B10H9}] (4b; Scheme 3), analogous to 4a. Complexes 4a and 4b were isolated as pale-yellow solids in 72% and 57% yield, respectively. The 1H NMR spectra of 4a and 4b showed single intense signals at δ = 2.24 and 6.65 ppm, corresponding to the Cp* and Cp ligands, respectively. The 13C NMR spectra are consistent with one type of Cp* or Cp ligand.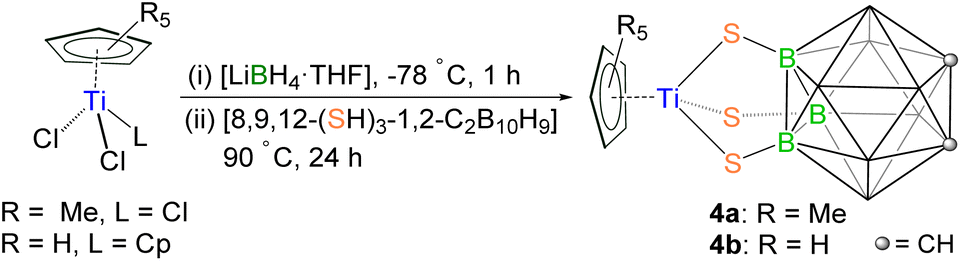 | ||
| Scheme 3 Synthesis of 8,9,12-trithiol-o-carborane protected Ti-thiolate complexes 4a and 4b. | ||
Besides the Cp* or Cp signals, 1H NMR spectra displayed additional peaks at δ = 3.49 ppm for 4a and δ = 3.59 ppm for 4b attributed to carborane-CH protons. The 13C NMR peaks for 4a and 4b are also observed at δ = 39.7 and 39.8 ppm, respectively, which confirmed the CH group of [8,9,12-(SH)3-1,2-C2B10H9] ligand. The 11B chemical shifts of the boron bonded to sulfur in 4a and 4b shifted to the downfield region compared to the free ligands, suggesting an electronic influence from the Cp*/CpTi-fragment on the carborane ligands. The IR spectra exhibited absorption bands at 2569 and 2605 cm−1 for 4a and 2598 and 2623 cm−1 for 4b, attributed to the terminal B–H stretches, respectively. Furthermore, the mass spectra of 4a and 4b showed molecular-ion peaks at m/z = 421.1511 and m/z = 723.1343, respectively, consistent with (M + H) and (2M + H). Finally, single-crystal X-ray diffraction analyses were conducted on suitable crystals of 4a and 4b.
As shown in Fig. 5 and S5,† the solid-state X-ray structures of 4a and 4b can be described as three-legged piano-stool geometry with a coordination number of six around the titanium center. In both the complexes, Cp*/Cp and [8,9,12-(S)3-1,2-C2B10H9] ligands are coordinated to titanium atom in opposing positions. The dihedral angle between the Cp* plane and [B1–B2–B3] plane of the carborane cage is 1.05° (4a) and 0.91° (4b), which are closer to planar and less deviated as compared to 3 (2.15°). This may be due to the symmetrical nature of the [8,9,12-(S)3-1,2-C2B10H9] ligand. In 4a and 4b, the three metalladithiolene [TiS2B2] rings are formed to fix the monometallic Cp*Ti fragment into the carborane scaffold through three Ti–S bonds. The dihedral angle between [B2S2] and [TiS2] in 4a (av. 64.12°) and 4b (64.86°) indicates that these metallacycles [TiS2B2] are non-planar. The Ti–S bond lengths in 4a (av. 2.313 Å) and 4b (2.318 Å) are significantly shorter than in clusters 1 and 2 but are in good agreement with 3. The overall structure possesses a σ-plane of symmetry, perpendicular to the C1–C2 bond and containing the [Ti1–S3–B1–B10–B9–B4] atoms. A qualitative electron counting scheme suggests 4a and 4b to be the 12 electron systems, where Ti(d0) is in +4 oxidation state.
Reactivity of [Cp*VCl2]3 with the B-functionalized carborane-trithiol [8,9,12-(SH)3-1,2-C2B10H10]
Intrigued by the formation of the BH3-R group coordinated to Ti and relatively higher yields, we have done further experiments aimed at investigating the role of the metal in the formation of metal carborane-thiolate complexes. As a result, we have now changed the metal from Ti to V. The low-temperature reaction of [Cp*VCl2]3 with [LiBH4·THF] resulted in an unstable and sensitive intermediate, which on treatment with the [8,9,12-(SH)3-1,2-C2B10H9] ligand at elevated temperature yielded a green solid 5 with a yield of 26% (Scheme 4). The 1H and 13C NMR spectra of 5 confirmed the presence of both Cp* and carborane ligands. Interestingly, an alluring signal at δ = −0.43 ppm in the 1H NMR spectrum suggests the formation of a B–H–B bridged bond. Unlike 4a and 4b, the 11B NMR spectrum of 5 displayed five peaks at δ = −30.3, −25.8, −22.0, −6.4, and −4.6 ppm, indicative of a different boron environment. The IR spectrum of 5 showed absorption bands at 2619 and 2683 cm−1 associated with terminal B–H stretches. To validate the spectroscopic data and elucidate the solid-state X-ray structure of 5, the single-crystal X-ray diffraction analysis was carried out. | ||
| Scheme 4 Synthesis of 1,5,6-trithiol-nido-carborane protected V-thiolate complex 5. | ||
The molecular structure of 5, shown in Fig. 7, revealed a six-coordinate vanadium atom, bonded to three S atoms from the nido-carborane ligand [1,5,6-(S)3-nido-7,8-C2B9H9] and Cp* ligand, forming a three-legged piano-stool geometry. The C2B3 open face of the nido-carborane ligand is positioned opposite to the V–S coordination. The dihedral angle between the Cp* plane and the [B1–B2–B3] plane of the carborane cage measured 3.07°, indicating a near-planar orientation but with greater deviation compared to complexes 4a and 4b, likely due to the asymmetry of the nido-carborane ligand. The V–S bond lengths (2.213 Å) are typical and comparable with those observed in [(iPrCp)2V2S4] (2.236 Å).35 The C–C distance (1.555(8) Å) within the cup-shaped carborane cage is similar to the C–C bond lengths in closo-carborane cage complexes 4a (1.629(3) Å) and 4b (1.635(7) Å).
 | ||
| Fig. 7 Molecular structure and labelling diagram of 5. Selected bond lengths (Å) and bond angles (°): V1–S1 2.2194(16), V1–S3 2.2099(15), B3–S3 1.859(6), B1–B8 1.780(9), C1–C2 1.555(8), C2–B7 1.596(8), B8-Hb 1.15(2), B9-Hb 1.16(2), V1–S1–B1 81.01(19), V1–S3–B3 79.42(17), B7–B8–B9 101.7(5), C1–C2–B7 114.3(5). | ||
Interestingly, complex 5 can be considered as a zwitterionic complex, arising from the separation of the cationic metal fragment (Cp*V) and the nido-carborane anion, which was formed by deboronation of one of the B(3)/B(6) vertices.36 The vanadium atom in 5 possesses 12 valence electrons, which accounts for the observed diamagnetism of the complex. During the reaction, the removal of a B(3)–H unit from the parent closo-carborane cage transformed it into a nido-carborane (C2B9) anion, with the anionic charge delocalized onto the pendant [C2B3] ring and consequently, 5 became a zwitterion with the [Cp*V(κ3-S,S,S)]+ fragment. Note that the conversion of closo-carboranes to nido-carboranes through deboronation is a well-known reaction, typically induced by nucleophilic attack.11e,37 In this case, the employed borohydride anion (BH4−) can act as a nucleophile.38 Additionally, deboronation reactions generally require higher energy.5 Therefore, this transformation may be attributed to the combined effect of the nucleophile and the elevated temperature. In contrast to 4a and 4b, the isolation of 5 highlights the significant influence of both the ligand coordination and the metal center in directing the formation of transition-metal carborane-thiolate complexes.
The MO analysis of 4a and 5 showed that the coordination of three thiolato groups with the metal in both the complexes involves similar bonding interactions of the metal d orbitals with the p orbitals of sulfur (Fig. S65 and S68†). The NBO analysis further revealed a 3c–2e interaction in 5, involving bridging hydrogen (H1) and boron atoms B8 and B9 on the open face of the nido-carborane cage (Fig. 8a). This interaction was illustrated by the contour line diagram of Laplacian of electron density along the B8–H1–B9 unit (Fig. 8b). Additionally, a contour plot along the plane of the open face of the carborane cage showed the bonding situation within the five-membered ring (Fig. 8c).
 | ||
| Fig. 8 (a) Selected NBO of 5 showing the 3c–2e interaction of B8–H1–B9 (isocontour values: ±0.045 [e bohr−3]1/2). Contour-line diagram of the Laplacian of the electron density along (b) B8–H1–B9 and (c) B7–B8–B9 planes in 5. | ||
Molecular structure, spectroscopic, and frontier molecular orbital analysis of 1–5
Having a series of carborane-thiol stabilized clusters and coordination complexes of titanium and vanadium, it became of interest to examine their structural and spectroscopic data (Table 1).11b–d,20c The Ti–Scarborane bond lengths in 1 (av. 2.385 Å) and 2 (av. 2.452 Å) are significantly longer than those observed in I (Co: av. 2.125 Å and Ir: av. 2.263 Å). This difference can be attributed to π-type interactions in the latter, where electron donation from the sulfur pπ orbitals stabilizes the coordinatively unsaturated metal center, leading to shorter M–S bonds.11b–d In contrast, the longer Ti–S bond suggests the absence of these π-type interactions. Interestingly, the Ti–S bond lengths in 3–5 are shorter than those in 1 and 2, likely due to different attachment sites of the thiol substituents of the carborane cage. While the S is attached to the more electropositive B rather than C, there is more electron donation from the S atoms to form a bond with the electron-deficient Ti atom, which makes the Ti–S bond stronger in 3. Consequently, the Ti–S bond length decreased in 3–5 compared to 1 and 2. The π-type interaction in complexes I makes the metallacycle, [M–S–C–C–S] planar. Although this π-type interaction is absent in 1 and 2, the [Ti–S–C–C–S] rings in 1 and 2 are nearly planar, similar to I. However, unlike 1 and 2, the [M–S–B–B–S] rings in 3–5 are puckered at the metal center resulting from the tridentate coordination of the ligand; the remaining four atoms [S–B–B–S] form an almost perfect plane. Thus, the [MS2B2] metallacycles in 3–5 are bent, and the metal atom is positioned out of the plane of the carborane ligand, with the dihedral angles between the planes defined by [M–S–S] and [S–B–B–S] being 64.12–66.29°.| Complexes | Structural parameters | Spectroscopic details | TEC | Oxidation state of M | |||
|---|---|---|---|---|---|---|---|
| d av.M–S (Å) | ϴ av.S–M–S (°) | ϴ av.S–S vector (°) | 11B NMR | IR | |||
| a 11B NMR: range of 11B chemical shifts; IR (ῡ in cm−1) corresponding to terminal B–H stretches of the carborane ring is shown; in dav.M–S, ϴav.S–M–S, and ϴav.S–S vector S atoms refer to the sulfur atoms attached to the carborane ligand only; ϴav.S–S vector represents the angle between the planes of S–M–S and S–C/B–C/B–S. b Data not available. | |||||||

|
2.385 | 84.445 | 5.24 | −13.0 to −1.0 | 2580 | 80 | +4 |

|
2.452 | 81.09 | 1.94 | −12.4 to −0.8 | 2589 | 66 | +3 and +4 |

|
2.125 (Co) | 96.609 (Co) | 3.00 (Co) | (Co)b | 2554 (Co) | 16 | +3 |
| (Rh)b | (Rh)b | (Rh)b | −10.7 to −5.2 (Rh) | 2590, 2561 (Rh) | |||
| 2.263 (Ir) | 92.69 (Ir) | 1.93 (Ir) | (Ir)b | 2600, 2560 (Ir) | |||

|
2.203 | 97.983 | 11.91 | −13.6 to 13.0 | 2601 | 51 | +2 |
|
2.327 | 101.497 | 66.29 | −15.8 to 8.4 | 2602 | 14 | +4 |

|
2.313 (4a) | 103.555 (4a) | 64.12 (4a) | −22.3 to 6.3 (4a) | 2605, 2569 (4a) | 12 | +4 |
| 2.318 (4b) | 103.203 (4b) | 64.86 (4b) | −22.2 to 7.1 (4b) | 2623, 2598 (4b) | |||

|
2.213 | 106.293 | 67.94 | −30.3 to −4.6 | 2683, 2619 | 14 | +5 |
On the other hand, in terms of spectroscopic data, if we look into the 11B NMR spectra of the complexes 1–5, the changes of 11B chemical shifts in complexes 3 and 5 compared to free carborane-thiol ligands are significantly larger than those in 1, 2, 4a, and 4b, which is due to the B–B bond formation (in 3) or deboronation of the carborane cage (in 5). In the IR spectra, in addition to the terminal B–H stretches of the carborane ring, a distinct B–H stretch at 2489 cm−1 is observed in 3, corresponding to the presence of an exo-BH3 moiety. In UV-vis absorption spectra, the absorptions with λ > 300 nm (330–652 nm) primarily display two to three absorption bands, which can be assigned to charge transfer transitions (Fig. S54 and S55†). Time-dependent DFT studies indicate that the number of possible electronic transitions is higher in the hexa- and pentametallic clusters 1 and 2 than in the monometallic complexes 3–5 (Fig. S71–S82 and Tables S6–S11†). In complexes 1 and 2, the low-intensity absorption bands near 365–490 nm can be assigned to intramolecular LMCT transitions, where electron density flows from the sulfur or carborane ring to the metal centers. In contrast, the absorption bands in the 330–388 nm region for complexes 3, 4a, and 4b are mainly due to the electron density transfer from the Cp* ligand or sulfur atoms to the metal center. Notably, the absorption bands at 335 and 652 nm for 5 correspond to LMCT transition, where electron density flows from the carborane ring to the metal center. This is mainly due to the zwitterionic nature of the complex 5, wherein the anionic charge is delocalized onto the pendant [C2B3] ring of the nido-carborane (C2B9). Furthermore, variation in the electronic configuration and oxidation state of metals across complexes in Table 1 is observed with the variation of the transition metal.
A comparative theoretical analysis of 3, 4a, and 5 was carried out. MO analysis revealed that 3 (2.65 eV) and 4a (2.86 eV) have similar HOMO–LUMO gaps, both significantly larger than that of 5 (1.59 eV) (Fig. 9). The smaller HOMO–LUMO gap and energetically higher-lying HOMO of 5, as compared to 3 and 4a, suggest that 5 may exhibit a greater reactivity. Although the LUMOs of all three complexes are identical (localized on Ti or V center and two S atoms), the HOMOs feature different interactions. The HOMO of 4a is completely localized on the p orbitals of the three sulfur atoms, while that of 3 has an additional small contribution from the borate boron (B11) center. In contrast, the HOMO of 5 is largely delocalized over the open [C2B3] face of the nido-carborane cage, along with little contribution from the p orbitals of sulfur atoms. This delocalization suggests the donating ability or reactive nature of the open face to generate a more stable closo-structure. Furthermore, it is evident that the thiolato groups in 3 and 4a are more nucleophilic than those in 5. This observation is also supported by natural population analysis that revealed negative natural charges on sulfur atoms of 3 and 4a and positive charges on those of 5 (Table S3†).
 | ||
| Fig. 9 Presentation of the energy levels, HOMO–LUMO gap, and orbital composition distribution of the HOMO and LUMO for 3, 4a, and 5 (isocontour values: ±0.045 [e bohr−3]1/2). | ||
Conclusions
In conclusion, we have demonstrated how the thiol functionalization site on carborane cages influences the formation of either cluster or coordination complexes when treated with cyclopentadienyl titanium and vanadium chlorides. The C-functionalized carborane-thiol, due to its lower C–S bond stability, resulted in the formation of penta- and hexa-metallic TiS clusters, whereas the B-functionalized variant, with its more stable B–S bond, gave rise to a coordination complex featuring an exo-BH3 moiety. The formation of a new B–B bond in complex 3 offers strong evidence of metal-induced B–H activation. The B-functionalized di- and tri-thiol ligands are crucial in stabilizing the electron-deficient monometallic coordination complexes 3–5. Interestingly, changing the metal from Ti to V facilitated the deboronation of the neutral closo-carborane, leading to the formation of 14-electron zwitterionic nido-5. This highlights the crucial role of ligand variation and emphasizes the significant impact of metal choice on the formation of transition metal–carborane thiolate coordination complexes. Our findings highlight the higher thermodynamic stability of B–S bonds over C–S bonds in carboranes, providing valuable insights into the nature of these bonding interactions. This work offers new opportunities for the strategic design of carborane-based complexes with specific properties, and potential applications in organometallic chemistry and materials science.Data availability
The data supporting this article have been included as part of the ESI.† Crystallographic data have been deposited at the Cambridge Crystallographic Data Center under 2403346 (1), 2451038 (2), 2311787 (3), 2358616 (4a), 2311786 (4b), and 2358617 (5).Author contributions
S. B., D. K. P., D. C., and J. M. executed the experimental synthesis, characterization, and data analysis. D. C. and S. B. carried out the theoretical calculations. M. K. carried out single-crystal X-ray diffraction data collection, structure refinement, and analysis. S. B. prepared the first draft of the manuscript and all authors contributed to the preparation of the manuscript. S. G., T. P., and T. B. supervised the project.Conflicts of interest
There are no conflicts to declare.Acknowledgements
S. G. thanks the support of the SERB, India, Grant No. CRG/2023/000189. T. B. acknowledges the support of the Ministry of Education, Youth, and Sports, Grant No. MSMT LTAIN 19152. T. P. thanks SERB for funding through Grant No. SPR/2021/000439 and a JC Bose Fellowship. We gratefully acknowledge funding through the Center of Excellence (CoE) on Molecular Materials and Functions as part of the Institute of Eminence (IoE) scheme of IIT Madras. S. B. and D. C. thank IIT Madras, and D. K. P. thanks PMRF, Grant No. SB22230356CYPMRF008224 for fellowships. We thank Ms Swetashree Acharya for performing the ESI-MS experiment for complexes 1 and 2. We also extend our gratitude to Dr B. Varghese and Dr Elias Jesu Packiam, IIT Madras, for Single Crystal X-ray diffraction data collection and structure refinement. We gratefully acknowledge DST-FIST, India, for HRMS and IIT Madras for providing computational facilities.Notes and references
-
Coordination Chemistry: A Century of Progress, ed. G. B. Kauffman, American Chemical Society, Washington, DC, 1994, vol. 565 Search PubMed
.
-
(a) T. M. Trnka and R. H. Grubbs, Acc. Chem. Res., 2001, 34, 18–29 CrossRef CAS PubMed
-
(a) J. D. Atwood, M. J. Wovkulich and D. C. Sonnenberger, Acc. Chem. Res., 1983, 16, 350–355 CrossRef CAS
-
(a)
R. H. Crabtree, The Organometallic Chemistry of the Transition Metals, Wiley, 3rd edn, 2001 Search PubMed
-
R. N. Grimes, Carboranes, Elsevier, Oxford, 3rd edn, 2016 Search PubMed
-
(a) M. F. Hawthorne, J. I. Zink, J. M. Skelton, M. J. Bayer, C. Liu, E. Livshits, R. Baer and D. Neuhauser, Science, 2004, 303, 1849–1851 CrossRef CAS PubMed
-
(a) A. K. Saxena and N. S. Hosmane, Chem. Rev., 1993, 93, 1081–1124 CrossRef CAS
- A. M. Spokoyny, C. W. Machan, D. J. Clingerman, M. S. Rosen, M. J. Wiester, R. D. Kennedy, C. L. Stern, A. A. Sarjeant and C. A. Mirkin, Nat. Chem., 2011, 3, 590–596 CrossRef CAS PubMed
-
(a) J. Plesek, Chem. Rev., 1992, 92, 269–278 CrossRef CAS
-
(a) G. L. Moxham, T. M. Douglas, S. K. Brayshaw, G. Kociok-Köhn, J. P. Lowe and A. S. Weller, Dalton Trans., 2006, 5492–5505 RSC
-
(a) M. Herberhold, G.-X. Jin, H. Yan, W. Milius and B. Wrackmeyer, Eur. J. Inorg. Chem., 1999, 873–875 CrossRef CAS
-
(a) M. Herberhold, H. Yan, W. Milius and B. Wrackmeyer, Angew. Chem., Int. Ed., 1999, 38, 3689–3691 CrossRef
-
(a) M. Herberhold, H. Yan, W. Milius and B. Wrackmeyer, Organometallics, 2000, 19, 4289–4294 CrossRef CAS
-
(a) S. Liu, Y.-F. Han and G.-X. Jin, Chem. Soc. Rev., 2007, 36, 1543–1560 RSC
-
(a) J.-Q. Wang, C.-X. Ren and G.-X. Jin, Chem. Commun., 2006, 162–164 RSC
- J.-H. Wang, C. Zheng, J. A. Maguire and N. S. Hosmane, Organometallics, 2003, 22, 4839–4841 CrossRef CAS
-
(a) S. Cai and G.-X. Jin, Organometallics, 2005, 24, 5280–5286 CrossRef CAS
-
(a) M. Herberhold, G.-X. Jin, H. Yan, W. Milius and B. Wrackmeyer, J. Organomet. Chem., 1999, 587, 252–257 CrossRef CAS
-
(a) G.-X. Jin, J.-Q. Wang, Z. Zhang, L. H. Weng and M. Herberhold, Angew. Chem., Int. Ed., 2005, 44, 259–262 CrossRef CAS
-
(a) D. Olid, R. Núñez, C. Viñas and F. Teixidor, Chem. Soc. Rev., 2013, 42, 3318–3336 RSC
-
(a) A. J. Mannix, X.-F. Zhou, B. Kiraly, J. D. Wood, D. Alducin, B. D. Myers, X. Liu, B. L. Fisher, U. Santiago, J. R. Guest, M. J. Yacaman, A. Ponce, A. R. Oganov, M. C. Hersam and N. P. Guisinger, Science, 2015, 350, 1513–1516 CrossRef CAS PubMed
-
(a) F. Zheng, T. H. Yui, J. Zhang and Z. Xie, Nat. Chem., 2020, 11, 5943–5947 CAS
-
(a)
S. Kar and S. Ghosh, Borane Polyhedra beyond Icosahedron, in 50th Anniversary of Electron Counting Paradigms for Polyhedral Molecules. Structure and Bonding, ed. D. M. P. Mingos, Springer, Berlin, 2021, vol. 187, pp. 109–138 Search PubMed
-
(a) K. Saha, D. K. Roy, R. D. Dewhurst, S. Ghosh and H. Braunschweig, Acc. Chem. Res., 2021, 54, 1260–1273 CrossRef CAS PubMed
-
(a) K. Pathak, C. Nandi and S. Ghosh, Coord. Chem. Rev., 2022, 453, 214303 CrossRef CAS
- T. Baše, J. Macháček, Z. Hájková, J. Langecker, J. D. Kennedy and M. J. Carr, J. Organomet. Chem., 2015, 798, 132–140 CrossRef
-
(a) S. Gayen, F. Assanar, S. Shymal, D. P. Dorairaj and S. Ghosh, Chem. Sci., 2024, 15, 15913–15924 RSC
-
(a) J. C. Huffman, J. G. Stone, W. C. Krussel and K. G. Caulton, J. Am. Chem. Soc., 1977, 99, 5829–5831 CrossRef CAS
- F. Bottomley, G. O. Egharevba and P. S. White, J. Am. Chem. Soc., 1985, 107, 4353–4354 CrossRef CAS
-
(a) K. Wade, Inorg. Nucl. Chem. Lett., 1972, 8, 559–562 CrossRef CAS
-
(a) R. Gillespie, Chem. Soc. Rev., 1979, 8, 315–352 RSC
-
(a) X.-W. Li, W. T. Pennington and G. H. Robinson, J. Am. Chem. Soc., 1995, 117, 7578–7579 CrossRef CAS
- J. C. Thomas, D. P. Goronzy, A. C. Serino, H. S. Auluck, O. R. Irving, E. Jimenez-Izal, J. M. Deirmenjian, J. Macháček, P. Sautet, A. N. Alexandrova, T. Baše and P. S. Weiss, ACS Nano, 2018, 12, 2211–2221 CrossRef CAS PubMed
-
(a) A. C. Molina, E. Horno, J. Jover, A. P. -Redondo, C. Yélamos and R. Zapata, Organometallics, 2023, 42, 1360–1372 CrossRef
- C. M. Bolinger, T. B. Rauchfuss and A. L. Rheingold, J. Am. Chem. Soc., 1983, 105, 6321–6323 CrossRef CAS
- X.-K. Huo, G. Su and G.-X. Jin, Dalton Trans., 2010, 39, 1954–1961 RSC
-
(a) R. A. Wiesboeck and M. F. Hawthorne, J. Am. Chem. Soc., 1964, 86, 1642–1643 CrossRef CAS
-
(a) H. C. Brown and S. Krishnamurthy, J. Am. Chem. Soc., 1972, 94, 7159–7161 CrossRef CAS
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2403346, 2451038, 2311787, 2358616, 2311786, and 2358617. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5sc03562g |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |