
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe prospect of substrate-based kinase inhibitors to improve target selectivity and overcome drug resistance
Biswajit
Biswas
 ,
Yen-Hua
Huang
,
David J.
Craik
* and
Conan K.
Wang
*
,
Yen-Hua
Huang
,
David J.
Craik
* and
Conan K.
Wang
*
Institute for Molecular Bioscience, Australian Research Council Centre of Excellence for Innovations in Peptide and Protein Science, The University of Queensland, Brisbane QLD 4072, Australia 4072. E-mail: c.wang@imb.uq.edu.au; d.craik@imb.uq.edu.au
First published on 13th July 2024
Abstract
Human kinases are recognized as one of the most important drug targets associated with cancer. There are >80 FDA-approved kinase inhibitors to date, most of which work by inhibiting ATP binding to the kinase. However, the frequent development of single-point mutations within the kinase domain has made overcoming drug resistance a major challenge in drug discovery today. Targeting the substrate site of kinases can offer a more selective and resistance-resilient solution compared to ATP inhibition but has traditionally been challenging. However, emerging technologies for the discovery of drug leads using recombinant display and stabilization of lead compounds have increased interest in targeting the substrate site of kinases. This review discusses recent advances in the substrate-based inhibition of protein kinases and the potential of such approaches for overcoming the emergence of resistance.
 Biswajit Biswas | Biswajit Biswas is a PhD candidate at the Institute for Molecular Bioscience, the University of Queensland. His research focuses on targeting kinases for anti-cancer drug development, employing diverse high-throughput screening techniques and peptide/protein engineering tools. |
 Yen-Hua Huang | Yen-Hua Huang was awarded her PhD from the University of Queensland (UQ) in 2010 and has >14 years of post-PhD experience in synthetic peptide chemistry, analytical chemistry and assay development. Her career focus is on methods for peptide drug optimisation, particularly relating to efficacy towards intracellular protein targets, in vivo stability, and cell permeability and penetration. |
 David J. Craik | David Craik, AO, FAA, FRS, is a Group Leader at the Institute for Molecular Bioscience at the University of Queensland, and Director of the Australian Research Council Centre of Excellence for Innovations in Peptide and Protein Science. His group focusses on the discovery and applications of disulfide-rich macrocyclic peptides from plants and animals. |
 Conan K. Wang | Conan Wang obtained his PhD from the University of Queensland (UQ) in 2009. He now leads the Technology-Driven Drug Discovery (Tech3D) group at the Institute for Molecular Bioscience, UQ. His group is interested in computational and molecular approaches for peptide and protein engineering. |
Introduction
Protein kinases catalyze one of the most fundamental biochemical reactions of life. They transfer the γ-phosphate of a purine nucleotide triphosphate (ATP/GTP) to the hydroxyl group of their substrate proteins.1 Over 500 human kinases carry out this type of reaction to regulate key cellular processes that range from cell growth to cell cycle progression and cell differentiation, proliferation, metabolism, and apoptosis. However, when kinases become dysregulated, they transition into drivers of disease, commonly cancer.2,3 The first cellular proto-oncogene, identified over 40 years ago, was found to encode c-Src kinase.4 Kinases are amongst the most common cancer gene-encoded protein domains and are attractive drug targets.5Inhibiting the ATP binding site of kinases was initially viewed as an unsurmountable challenge because of the high intracellular ATP concentration.6 However, with the FDA approval in 2001 and the clinical success of BCR-Abl inhibitor imatinib, interest in developing oral ATP-competitive kinase inhibitors skyrocketed.7,8 As of April 2024, 81 protein kinase inhibitors have been approved by the FDA, most of which work by inhibiting ATP binding to the kinase domain.9 Moreover, six additional inhibitors have been approved for lipid kinases and a staggering 600 kinase-targeting agents are under clinical trials according to a report in 2021.6 The major challenge in drug discovery today, however, is the rapid emergence of drug resistance, typically developing from such acquired mutations within the kinase domain that prevent ATP-inhibitor binding.8,10 Another, now pressing challenge, relates to polypharmacology, i.e., ATP-competitive inhibitors can act on more than one kinase. Interestingly, targeting multiple kinases can have favourable efficacy effects but also lead to adverse patient outcomes in other cases,11,12 pointing to the need in general of being able to fine-tune inhibitor activity.
To enable greater selectivity and combat drug resistance, there are increasing calls to explore non-ATP-mediated kinase inhibition.6,13 Substrate binding is another molecular interaction essential to kinase function. Unlike the ATP-binding site, the substrate-binding site of protein kinases is less conserved and thus offers better selectivity.14 Should mutations be acquired within the site to reduce inhibitor binding, the coincident effect would be reduced kinase activity. However, the substrate binding site has a shallow and open surface, which has made the design of small molecule inhibitors difficult.14,15 Furthermore, the molecular details of kinase–substrate interactions have been sparse until recently, which has further hindered the development of substrate-site inhibitors.15 Despite these difficulties, the field has progressed, and a multitude of kinase substrate-site inhibitors have been reported.
In this review, we aim to provide an up-to-date discussion of substrate-site inhibitors and their potential in overcoming drug resistance. As background for the need for such inhibitors, we summarize the FDA-approved ATP-competitive kinase inhibitors and highlight mutations to the ATP-binding region associated with their drug resistance. For comprehensive discussions of ATP-competitive inhibitors, we refer readers to several recent reports.6,8,9,12 Substrate-site inhibitors, on the other hand, have been reviewed in very few articles, each focusing on specific modalities without providing a complete overview and not including recent studies.14,16,17 This article will cover both small molecule and peptide-based substrate-site inhibitors, but with a focus on the latter. This focus on peptides and their design technologies is because most reported substrate-site inhibitors have been peptides. Peptides naturally mimic the substrates of protein kinases and therefore present as promising leads in drug development. Finally, we discuss the evidence that explores the potential of substrate-site inhibitors in overcoming the emergence of drug resistance.
Structural and mechanistic features of kinases
Human protein kinases can be classified based on their substrate specificity and/or sequence similarity. According to the amino acid they phosphorylate, most are named either serine/threonine kinases (STKs) or tyrosine kinases (TKs), with STKs (>300 reported) being more prevalent than TKs (>50).18 Sequence analyses of these kinases have borne a separate and more granular classification scheme that begins with their division into eukaryotic (ePKs, 478 kinases) and atypical protein kinases (aPKs, 40 kinases), with the former, but not latter, having the ‘kinase catalytic domain’.19 The ePKs are further divided into 9 groups, and these are, in order of abundance: TK (tyrosine kinase), CAMK (Ca2+/calmodulin-dependent kinase),TKL (tyrosine kinase-like), AGC (protein kinase A, G and C related), CMGC (Cdk, GSK, MAPK, Cdk-like related), STE (STE20, STE11, and STE7 related), CK1 (casein kinase 1), RGC (receptor guanylyl cyclase), and “others”.20,21 PKA (c-AMP dependent protein kinase) is a prototypical example of both an STK and an AGC, and its crystal structure was the first to reveal the bi-lobal fold of the kinase catalytic domain.22 Another example, Abl, is prototypical of TKs, and its kinase domain was the first to be successfully drugged for the treatment of cancer.8Fig. 1a shows a typical catalytic cycle carried out by the protein kinase domain once activated, usually by being itself phosphorylated. The ATP binding, magnesium complexation and substrate recognition and positioning steps at the catalytic site are followed by phosphoryl transfer, and finally release of the substrate (phosphorylated) and ADP products.23 In some cases the order of these steps varies, for instance substrate binding can precede ATP binding, and ADP can be released before substrate dissociation.24 Regardless, when the catalytic cycle becomes dysregulated, it results in aberrant phosphorylation and disease.25 As an example, a constitutive active mutant of Abl is the oncogenic cause of chronic myeloid leukaemia.26
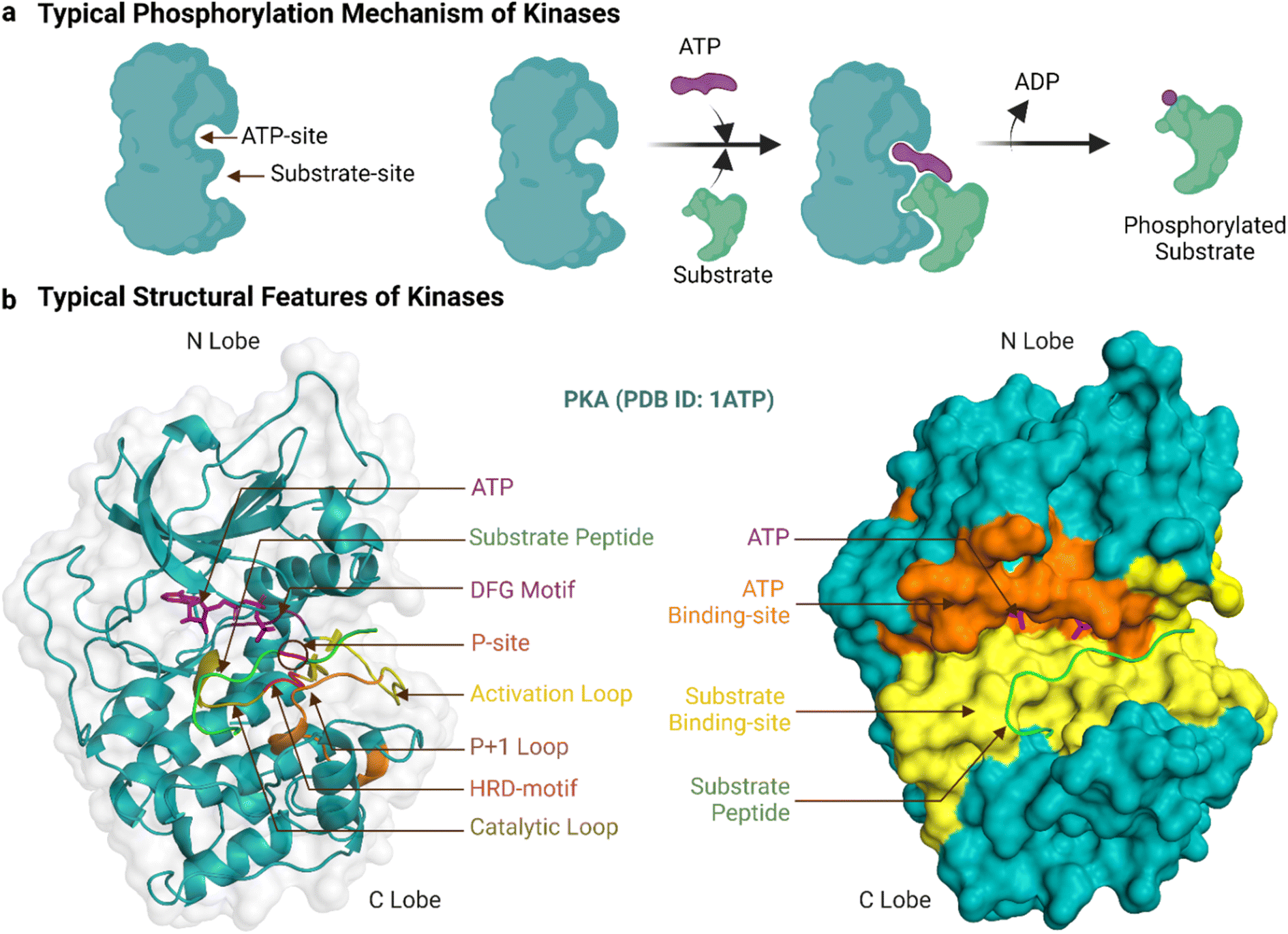 | ||
| Fig. 1 Typical phosphorylation mechanism and structural features of protein kinases. ‘a’ shows the typical catalytic reaction carried out by kinases and ‘b’ shows the essential structural features of the enzyme's kinase domain. Created with https://BioRender.com. | ||
Kinase binding to either ATP or substrates is governed by different binding properties. Generally, ATP binding is driven by moderate affinity in the 10–100 μM range combined with high intracellular ATP concentrations of 1–10 mM.27 By contrast, the substrate binding affinity is substrate and kinase-dependent and can involve regions outside the substrate sequence motif (a contiguous ∼10 amino acid region around the acceptor residue) and catalytic site. For example, an N-terminal SH2 domain often aids the positioning of protein substrates for catalysis by Src TKs.28 Abl utilizes the SH2 and SH3 domains adjacent to its kinase domain to mediate substrate recognition.29 Substrate binding is thus likely stronger than the μM affinity for a peptide representing the sequence motif.30 It is also difficult to generalise for the substrate concentrations inside cells. While there are ∼700![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 potential intracellular phosphorylatable sites, kinases vary greatly in the number of sites they phosphorylate and their substrate recognition motifs.24
000 potential intracellular phosphorylatable sites, kinases vary greatly in the number of sites they phosphorylate and their substrate recognition motifs.24
The ePK catalytic domain has approximately 250 amino acid residues and contains the essential structural features for catalysing substrate phosphorylation (Fig. 1b). The domain has an N-terminal and a C-terminal lobe connected through a hinge region.31 The N-terminal lobe is made up of five β-strands (β1-β5) and one α-helix (αC-helix), while the C-terminal lobe comprises four short β-strands (β6-β9) and seven α-helices (αD-αI).32 The C-terminal lobe also contains a flexible polypeptide segment, which is divided into the catalytic, activation and ‘P+1’ loops, and is important for catalysis and coordinating kinase binding to magnesium, ATP and substrates.32 The conformation of the activation loop can change between activated and inactivated kinase states to facilitate or inhibit/block binding of ATP and substrates.33
The ATP-binding site is a deep pocket formed between the two lobes of the kinase domain.15 Once bound, ATP resides near 23 residues in PKA (PDB ID: 3X2V) and 17 residues in Abl (PDB ID: 2 G2I, residues within 5 Å proximity). In general, the adenine of ATP is surrounded by conserved hydrophobic residues and forms hydrogen bonds to the hinge region.34 The remainder of ATP binds to a hydrophilic channel that extends towards the substrate binding site, usually interacting with the N-terminal lobe through an AxK motif (Ala, x, Lys) and a glycine-rich loop (GxGxxG motif) that binds with the triphosphate group and the ribose moiety.35,36 This phosphate binding region of the N-terminal lobe between β1 and β2 containing the AxK motif and glycine-rich loop is also known as the ‘P-loop’.37,38 In the activated kinase state, the DFG motif (Asp, Phe, Gly) of the activation loop positions the ATP for phosphotransfer.39
The majority of the approved kinase inhibitors target this ATP-binding site for inhibiting phosphorylation. Some inhibitors, as elaborated on later, engage adjacent regions outside the ATP pocket that are not occupied by ATP in attempts to increase inhibitor potency and/or selectivity. The ‘entrance’ and ‘buried’ regions are two such regions and have structural and sequence diversity among different kinases.40 The entrance region resembles a solvent-exposed hydrophobic slot and access to it is controlled by the conformation of the DFG motif.41 Access to the buried region is controlled by a single amino acid residue in the hinge region - known as the ‘gatekeeper residue’.42 Mutation of the gatekeeper residue is a predominant cause of drug resistance for ATP competitive inhibitors.43
The substrate-binding site is a shallow cleft adjacent to the ATP-binding site in the C-terminal lobe.15 The co-crystal structure of PKA bound to a 20-amino acid peptide substrate, identified 32 residues that constitute this site (PDB ID: 3X2V, within 5 Å proximity). Compared to those of STKs, the substrate-binding sites of TKs are deeper to accommodate the larger tyrosine acceptor residue.24 Generally, peptide substrates bind the substrate-binding site in an extended conformation.30 In this canonical binding mode, the substrate phosphorylation site is secured by the ‘P+1’ loop, which in turn is anchored to the αF-helix. The HRD-arginine of the catalytic loop anchors the primary phosphate.36 Residues upstream of the phosphorylation site form multiple bonds to the αC-helix and activation loop while residues downstream lie in a groove formed by the αF, αD and αG-helices.32 These residues in contact with the substrate help determine substrate specificity. For PKA and many other AGC subfamily kinases, the His of the HRD motif is replaced by Tyr.44 Moreover, two Glu residues of PKA in the β6 and αF helix select for positively charged residues at P-2 and P-5 substrate positions and a hydrophobic pocket formed by residues from the P+1 loop favours a hydrophobic residue at the P+1 position of the substrate.32,45
Protein kinases phosphorylate substrates with specific sequence motifs, typically described using short linear peptides (∼10 amino acids). Multiple studies have revealed the phosphorylation site motifs for specific kinases.18,30 For instance, c-Src and Abl show distinct preferences for consensus substrate sequence motifs: c-Src favors Ile at −1 and Phe at +3 of its substrate whereas Abl prefers Ala at +1 and Pro at +3 (positions relative to the central Tyr).46 However, overlaps in sequence specificity have also been reported.47 Kinases with close homology from the same subgroups can share identical or similar phosphorylation motifs, as seen in the consensus sequences reported for three Pim kinases.48 Additionally, considerable overlaps also exist between different kinase groups; for instance, both MAPKs and CDKs share the same minimal phosphorylation sequence motifs (Ser-Thr-Pro).47
Frequent mutations within and around the substrate-binding site are often observed in cancer and congenital diseases. Such cancer-associated mutations can cause substantial changes in substrate phosphorylation site specificity, rewire signalling networks (by impairing recognition of the kinase to the specificity determining residues of the substrate for example), and result in large decreases in the catalytic activity.30,49 Even a single amino acid substitution can cause marked changes in specificity and catalytic activity. For example, mutation within the activation loop of PKA from Phe to Val altered the substrate selectivity.50 As expected, multiple point mutations tend to have greater effects and can render the enzyme inactive.30 For instance, mutations of four residues within the substrate binding site of Pim-1 STK converted it into a nonfunctional kinase. Interestingly, introduction of two compensating mutations into the substrate restored its phosphorylating ability.51
Activity and selectivity of FDA-approved small molecule inhibitors of kinases
As noted earlier, 81 protein kinase inhibitors have been approved by the US FDA as of April 2024, and most of which are for use against cancer. The approved cancer indications include, but are not limited to, chronic myeloid leukaemia, acute lymphoblastic leukaemia, HER2-positive breast cancer, non-small cell lung carcinoma, renal cell carcinoma, metastatic melanoma, squamous-cell carcinoma, hepatocellular carcinoma, pancreatic cancer, systemic mastocytosis, gastrointestinal stromal tumors, anaplastic thyroid cancers.9 Amongst the 81 drugs, 38 have been approved for only one cancer indication so far, whereas 34 have been approved for multiple cancer types. Kinase inhibitors can be used for non-cancerous applications as well. Three drugs are approved for use in both cancerous and non-cancerous diseases, and nine drugs for non-cancerous disorders only. Altogether, these therapeutic activities have been achieved by targeting mostly TKs (Fig. 2b, generated with KinMap52), which indicates that untapped drug development opportunities remain as STKs are also associated with human diseases.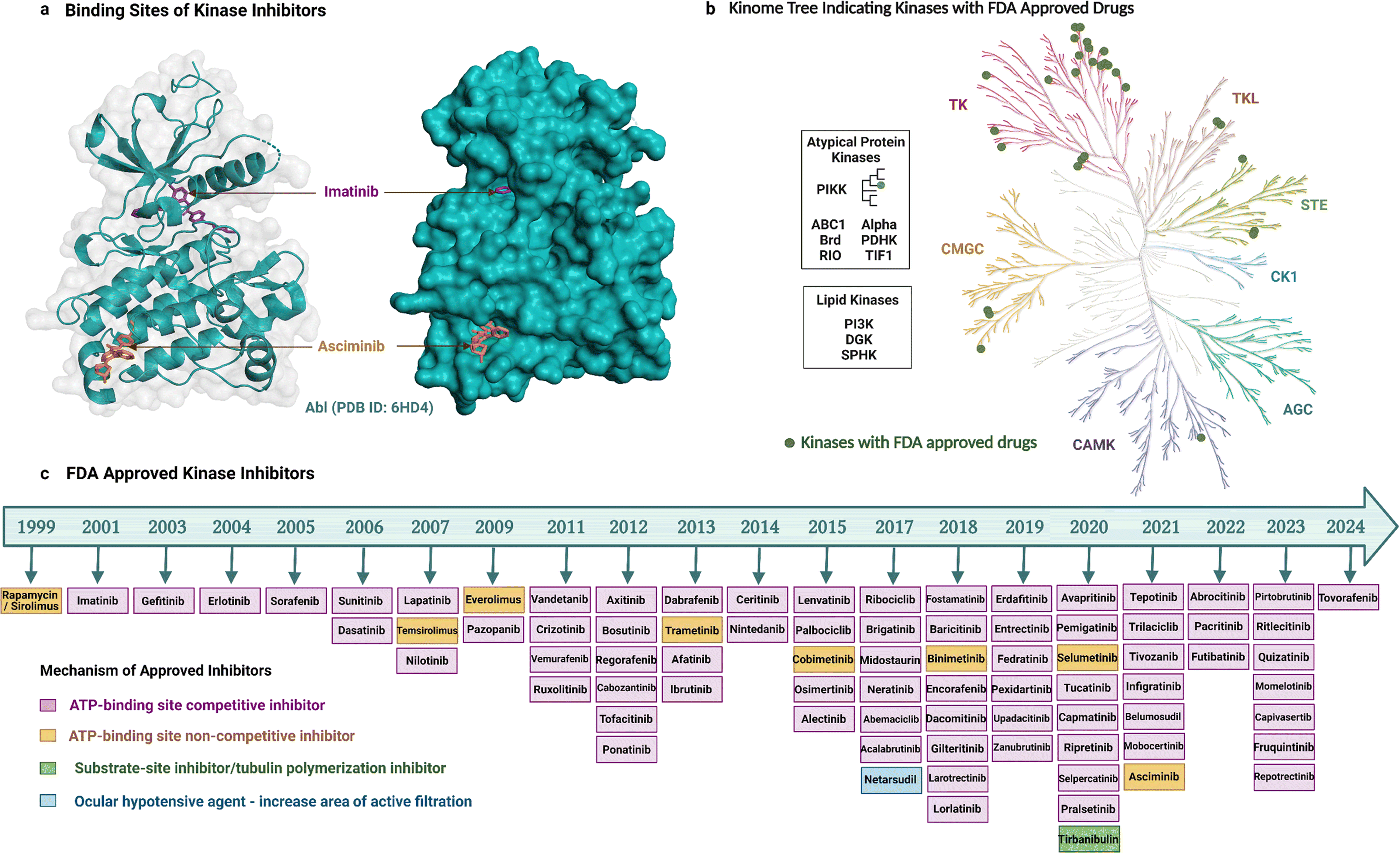 | ||
| Fig. 2 FDA approved kinase inhibitors. ‘a’ shows the binding sites of imatinib and asciminib to the ATP-binding site and an allosteric site of Abl TK, ‘b’ shows the Kinome tree highlighting the targets of the FDA-approved drugs (generated using the KinMap), ‘c’ shows the timeline of FDA-approved drugs and their mechanism of kinase inhibition. Created with https://BioRender.com. | ||
Most of the inhibitors approved by the FDA are small molecules and have resulted from over two decades of drug development (Fig. 2). The first kinase inhibitor approved by the FDA was rapamycin (also known as sirolimus) as an immunosuppressive agent in 1999.53 The next inhibitor approved was imatinib two years later in 2001, and it was the first anticancer kinase inhibitor.54 The per-year approval number has increased significantly since then, with the greatest number of kinase inhibitors (nine) approved in 2020. Apart from the approved drugs, over 600 kinase inhibitors are in clinical trials, including both small molecules and other biological agents,6 which highlights the ongoing importance of kinases as drug targets and the current interest in modalities beyond small molecules.
Small-molecule kinase inhibitors have been categorized into ‘types’ to distinguish their mechanisms of action.6,55 Type I and II inhibitors bind within the ATP-binding pocket but target different activation and conformational states. Type I inhibitors target the active DFG-in kinase conformation. Conversely, type II inhibitors target the inactive DFG-out conformation to access additional sites occluded in the active state. An intermediate binding mode between the two types, referred to as type I1/2, targets the inactive DFG-in conformation. Type III and IV inhibitors do not compete with ATP but differ in their binding proximity to the ATP-binding site.13 In addition, bivalent inhibitors (type V), covalent inhibitors (type VI), and macrocycles have been reported. Bivalent inhibitors usually link a type 1 inhibitor with another binder, i.e., a substrate-site targeting ligand or SH2 domain ligand.56 Covalent inhibitors usually attach to the ATP binding region, thereby working as ATP-competitive inhibitors.57 Macrocycles are generated by macrocyclization of a previously approved kinase inhibitor.58
Most of the FDA-approved kinase drugs, i.e., 71 of the inhibitors, show their activity by binding to the ATP-binding site of target kinases (Fig. 2). These ATP-competitive inhibitors comprise 60 inhibitors of type I, II and/or I1/2 (some have multiple binding modes); nine covalent inhibitors of type VI; and two that have a yet undefined mode of binding. By contrast, only 10 inhibitors do not compete with ATP (Fig. 2). This minority group comprises four inhibitors of type III; four of type IV; and two that are unclassified. The much smaller number of non-ATP-competitive compared to ATP-competitive inhibitors could potentially reflect the underlying difficulty in their development. For instance, targeting allosteric sites can be challenging because these sites are shallower, broader, more solvent-exposed, and less well-defined than the ATP-binding site.15,59 Despite this, asciminib represents a type IV drug that was recently developed to target BCR-Abl. It perturbs substrate binding by interacting with the C-terminal myristate-binding site, which is distal to the ATP-binding site.60 Tirbanibulin is another recently approved drug, interesting because of its dual mechanism of action, acting as an inhibitor of tubulin polymerization and reportedly also as a substrate-site inhibitor of c-Src kinase.61
ATP-competitive kinase inhibitors need to be sufficiently potent to out-compete intracellular ATP concentrations. Inhibitor potency has commonly been expressed as IC50 values (concentration of inhibitor required to inhibit 50% of activity). This indicator of therapeutic efficacy is useful to understand the culminative effect of the intrinsic affinity of inhibitors, the kinase and ATP concentration, and the affinity between ATP and the kinase,62 with the corresponding caveat of it being difficult to replicate precisely between independent experimental setups. Accordingly, reported IC50 values for the same inhibitor have varied widely, but generally range between the sub-nanomolar to micromolar levels.9,63 To provide some indication of the magnitude of these values, imatinib showed an IC50 value of 436 nM in ELISA and 682 nM in ATP-depletion assay against BCR-Abl1 expressed in BA/F3 cells64 while asciminib had an IC50 value of 0.5 nM in a Caliper assay against Abl1 expressed in E. coli.65 The high potency of ATP-competitive inhibitors is important as it increases their selectivity.62
Nevertheless, many kinase inhibitors have low selectivity and are effective against more than one kinase. Non-selective kinase inhibition is often linked to unwanted side effects. For example, sorafenib has been reported for severe side effects including desquamation, alopecia, pruritus, hand/foot–skin reaction, and sublingual hemorrhage.66 Three of the FDA-approved drugs are effective against multiple kinase groups and 26 against multiple subgroups within the same kinase group. For example, imatinib is effective against BCR-Abl, c-KIT, and PDGFRα; and sorafenib against c-RAF, B-RAF, VEGFR1-3, and PDGFRβ. In total, 49 of the approved drugs have activity against one subgroup of kinase. Some of the drugs, however, are extremely selective and have activity against only one subgroup type. For example, abrocitinib is only effective against JAK167 while fedratinib is only selective to JAK2.68 It is possible that the number of multi-kinase inhibitors is much higher than reported as many of the drugs have not been examined thoroughly against the complete panel of human kinases.9
The need to develop selective kinase inhibitors has motivated wider exploration into determinants of kinase selectivity. Within all inhibitor types and ATP-competitive inhibitors, type I inhibitors are generally thought to be less selective than type II inhibitors because they occupy only the conserved ATP-binding pocket. However, several type I inhibitors are more selective than some type II inhibitors,69 indicating difficulty in predicting selectivity based solely on the type of ATP-competitive inhibitor or the current classification scheme is insufficiently nuanced at the molecular level. The irreversible ATP-competitive inhibitor ibrutinib gained attention for having high selectivity,70 but along with other type VI inhibitors, it has only been successfully developed for selectivity targeting BTK (Bruton's tyrosine kinase). The most selective drugs are thought to be inhibitors that do not compete with ATP binding, such as type III and IV inhibitors because they occupy less conserved regions.13,70 For instance, cobimetinib is a carboxamide-based type III MEK1/2 inhibitor that is positioned to form an H-bond with amino acid residues of β3 and catalytic loop as well as γ-phosphoryl oxygen of ATP.63 Similar to cobimetinib, other type III inhibitors like binimetinib, selumetinib have selective activity against MEK1/2.71 Asciminib is a type IV BCR-Abl1 inhibitor that binds to the myristate binding pocket in the N-terminal lobe distant from the ATP or substrate binding site (Fig. 2) and is selective for Abl kinase.60
Emergence of resistance to approved small molecule kinase inhibitors
Although an increasing number of ATP-competitive kinase inhibitors are being approved, they are not curative, and patients risk disease relapse due to drug resistance.72,73 Resistance to kinase inhibitors can be pre-existing (innate) or develop after treatment (acquired).74 The causes of resistance are complex and diverse and include mutation of the target kinase, acquisition of bypass signalling pathways, and histological transformation.72,75 Amongst these, point mutations within the kinase domain are the predominant cause of acquired resistance76 and can develop within a short timeframe, at times occurring within months to years of treatment.77 These types of mutations are best characterized for Abl and EGFR TKs, which were the earliest to be drugged by ATP-competitive inhibitors, i.e., imatinib54 and gefitinib,78 respectively. Most resistance-causing mutations have been found within the hinge region (specifically the gatekeeper residue), the entrance region, and the DFG-motif.79All six Abl kinase inhibitors approved by the FDA have already been reported as developing resistance in CML (chronic myeloid leukaemia) patients.80 Studies with imatinib-resistant CML and Philadelphia chromosome-positive acute lymphoblastic leukaemia patients detected mutations in 30% to 83% cases.81 Mutations giving rise to drug resistance are shown in Fig. 3. Such mutations either reduce the inhibitors' affinity towards the target kinase while maintaining catalytic activity and sometimes increase the affinity of the kinase for ATP over the inhibitor.76 The mutation at the gatekeeper residue T315 is one of the most common and causes resistance to five of the FDA-approved ATP-competitive Abl kinase inhibitors – imatinib, nilotinib, dasatinib, bosutinib, and natinib (Fig. 3). Therefore, although Abl kinase inhibitors have developed since imatinib, in some cases to address drug resistance challenges of the previously approved drugs, there remain acquired mutations that persistently subvert drug activity. Apart from the gatekeeper T315, the other mutations associated with resistance usually centre within the P-loop at positions M244, G250, Q252, Y253, and E255; hinge region at position F317; activation loop at position H396; and the C-lobe at position M351 and F359.80 The allosteric inhibitor asciminib is not affected by some of these mutations around the ATP-binding pocket at T315, G250, Y253, E255, and H396; however, resistance still occurs due to mutations in the C-lobe at A337, W464, P465, V468, and I502.80,82
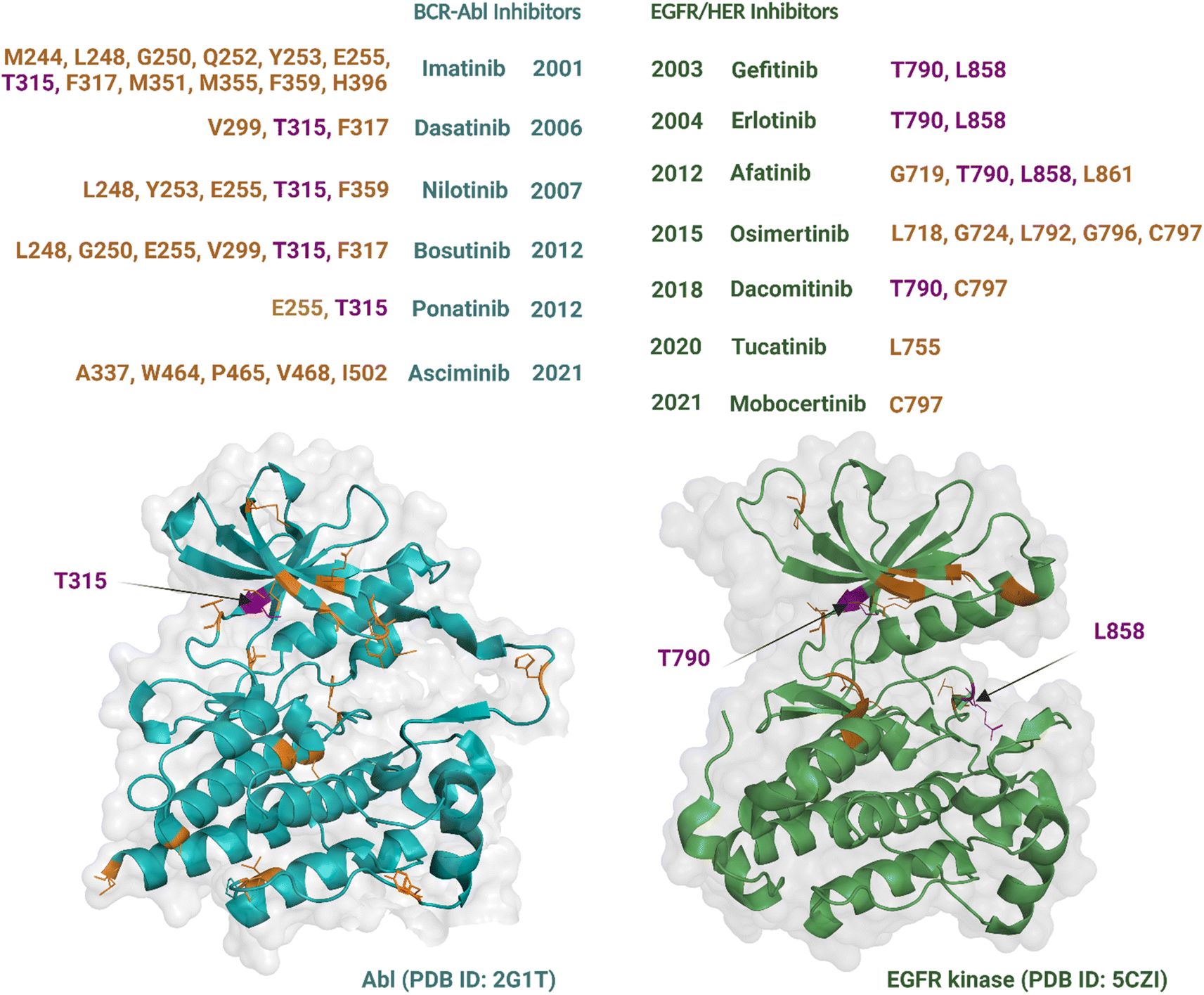 | ||
| Fig. 3 Single point mutations conferring resistance to the FDA-approved Abl and EGFR TK inhibitors over the years. Residues with the most frequent mutations are indicated by purple colour. Created with https://BioRender.com. | ||
Nine EGFR/HER kinase inhibitors are approved by the FDA. Mutation of the gatekeeper T790 residue can be found in 50% of NSCLC patients83 and mutation of the activation loop at L858 is associated with 90% of NSCLC patients treated with EGFR inhibitors (Fig. 3).84 Resistance to the first two EGFR inhibitors gefitinib and erlotinib is associated with these two mutations.85 In the case of afatinib resistance, mutations at T790 and L858 are also the most common, with mutations in the activation loop residue L861 and P-loop G719 being less frequent.86,87 Osimertinib, a third generation TKI, has been developed to target NCSLC patients with T790 and L858 mutations.88 However, resistance to the drug has been linked to mutations at the entrance region of the ATP-binding pocket of residues L792, G796, C797 and in the P-loop of residues L718, G724.89,90 For dacomitinib, mutations of the gatekeeper T790 and ATP-entrance region C797 residues have been reported as the mechanism of acquired resistance to the drug.91 The acquired resistance to tucatinib has been linked to the mutation in the αC-helix of residue L755.92 Finally, resistance to mobocertinib has been linked to a single-point mutation at C797.93
Like TKs and their inhibitors, single-point mutations to STKs have been linked to resistance to their respective inhibitors. Such emergence of resistance from mutations is often observed in drugs that target the RAS/RAF/MEK/ERK pathway. For instance, mutations to allosteric pocket gatekeeper residue V211 of MEK1 have been reported for creating resistance to binimetinib and other allosteric inhibitors.94 Another study reported mutation in K57 of MEK1, Q61 of NRAS, and Q61 of KRAS for developing resistance to BRAF/MEK inhibitors dabrafenib and trametinib.95 These mutations increased the catalytic activity of the kinases, reduced their sensitivity for the drug and/or amplified the activation of the targeted pathway.
Apart from acquired mutations, a major mechanism of mutation-independent resistance is the reactivation of signalling pathways downstream of the targeted kinase, resulting again in dysregulated cell proliferation and disease relapse. In CML patients with the BCR-Abl as the oncogenic driver, increased downstream signalling of pathways involving the kinases PI3K, MAPK, SRC, JAK/STAT can overcome the inhibitory effects of Abl-targeted drugs.80 The activity of EGFR-targeted inhibitors can be bypassed through downstream reactivation of pathways including RAF–MEK–ERK and PI3K–PDK1–AKT.96 The other mutation-independent mechanisms involve plasticity-mediated resistance with epigenetic and transcriptional changes, mutations in the epigenetic regulators like, DNMT3A, ASXL1, SETBP1, and IDH1 as well as downstream signalling activation by stromal cytokines.97–99 For CDK4/6 STKs, the mutation-independent resistance mechanisms include alterations of the controlling factors for cell cycle progression, amplification or overexpression of CDK4/6, cyclin D1, cyclin E, p16, and E2F, epigenetic alterations, aberrant PI3K/AKT/mTOR signaling, immune evasion as well as autophagy.100 The most common approach considered for targeting mutation independent resistance is through combination therapies. These include the use of BCR-Abl inhibitors combined with drugs that target JAK2/STAT;101 EGFR inhibitors combined with drugs that target IGF1R;102 and CDK4/6 STK inhibitors combined with drugs that target PI3K, mTOR, AKT.103
Substrate-site inhibitors of kinases
Substrate-site inhibitors have yet to reach the level of clinical impact of ATP-competitive drugs, and thus we highlight below studies on their discovery and activities to exemplify the early stages of drug development. We also summarise trends relating to their selectivity and activity against drug-resistant kinases so that comparison with approved drugs can be made. These properties are reviewed separately for small molecules and peptides – the two modalities on which many studies have been investigated.Small molecule inhibitors
Small molecule substrate-site TK and STK inhibitors have been sourced from nature and synthetic chemical libraries (Table 1 and Fig. 4). One of the earliest reports refined an active extract from the flowering plant Euphorbia lagascae to isolate the secondary metabolite picearannol, which inhibited the p40 TK in a substrate-competitive and non-ATP-competitive manner.110 In a more focussed approach, Reddy et al., chemically elaborated chemotypes unrelated to ATP and any other kinase-inhibitory purine or pyrimidine analogues to search for non-ATP-competitive inhibitors.121 They found substrate-site inhibitors in the form of the α-benzoyl styryl benzyl sulfides ON012380 and ON044580 as BCR-Abl inhibitors and the unsaturated sulfone ON01910 as a Plk1 inhibitor.104,120,122 One challenge for discovery of novel chemotypes from synthetic chemical libraries is the requirement for a custom-built assay because the substrate can vary between kinases. Stebbins et al. developed a lanthanide-based immunoassay to detect the interaction of JNK1 with its substrate JNK interacting protein 1 and used it to screen 30000 compounds, discovering potential candidates including the JNK inhibitor BI-78D3.116 Finally, chemical libraries have been screened in silico through computational docking into the substrate-binding site followed by validation through activity assays. The PI4KIIα inhibitor PI-273119 and ERK inhibitor compound 76114 are examples of molecules discovered in such an approach.
| Class | Origin and development approach | Name | Target kinase | Assay | Potency (nM) | Reference | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a Substrate Competition (SC), Kinase Inhibition (KI), ATP Competition (AC), Computer Aided Molecular Docking (CAMD), Surface Plasmon Resonance (SPR), X-ray Crystallography (XRC), Deuterium Exchange Mass Spectrometry (DXMS), * IC50, ∼ Ki, aIC50/Ki value determined using SC, bIC50 value determined using KI. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| TK inhibitors | Screening of styryl benzylsulfone library | ON012380 | BCR-Abl | SC | 9*a | 104 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ON044580 | BCR-Abl, JAK2 | SC | 7940*a, 1230*a | 105 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Natural product isolated from Streptomyces sp. | Erbstatin | EGFR | SC | 3070*a | 106 and 107 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Derivatives of erbstatin | ST638 (Tyrphostin) | EGFR | SC | 1100*a | 108 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| AG 538 | IGF-1R | SC | 400*a | 109 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Secondary metabolite from Euphorbia lagascae | Piceatannol | p56lck | SC | 66000*a |
110 and 111 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Screening of low molecular-weight phenols | MEB-SCI (compound 12) | c-Src | KI | 16000*b |
112 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Screening of hydroxynaphthalene and hydroxyindole methyl esters and amides | Compound 2f | pp60c-src | KI, AC | 16000*b |
113 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Modification of tyrosine | Tirbanibulin (KX-01/KX2-391) | c-Src | KI, CAMD | 46000*b |
61 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| STK inhibitors | In silico screening of a compound database | Compound 76 | ERK | SC, CAMD | 5000*a | 114 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Screening of compounds from corn silks | Compound 30 | GSK-3β | SC, CAMD | 590*a | 115 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Screening of a compound database with a lanthanide-based immunoassay | BI-78D3 | JNK | KI, CAMD | 280*b | 116 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| High throughput screening of a compound library | CMPD1 | p38α | SC, SPR, XRC, DXMS | 330∼a | 117 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Structure-based design | PS210 | PDK1 | KI, XRC, CAMD | 2000*b | 118 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| In silico screening of a SPECS compound database | PI-273 | PI4KIIα | SC, SPR | 470*a | 119 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Screening of a styryl benzylsulfone library | ON01910 | Plk1 | SC | 10*a | 120 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
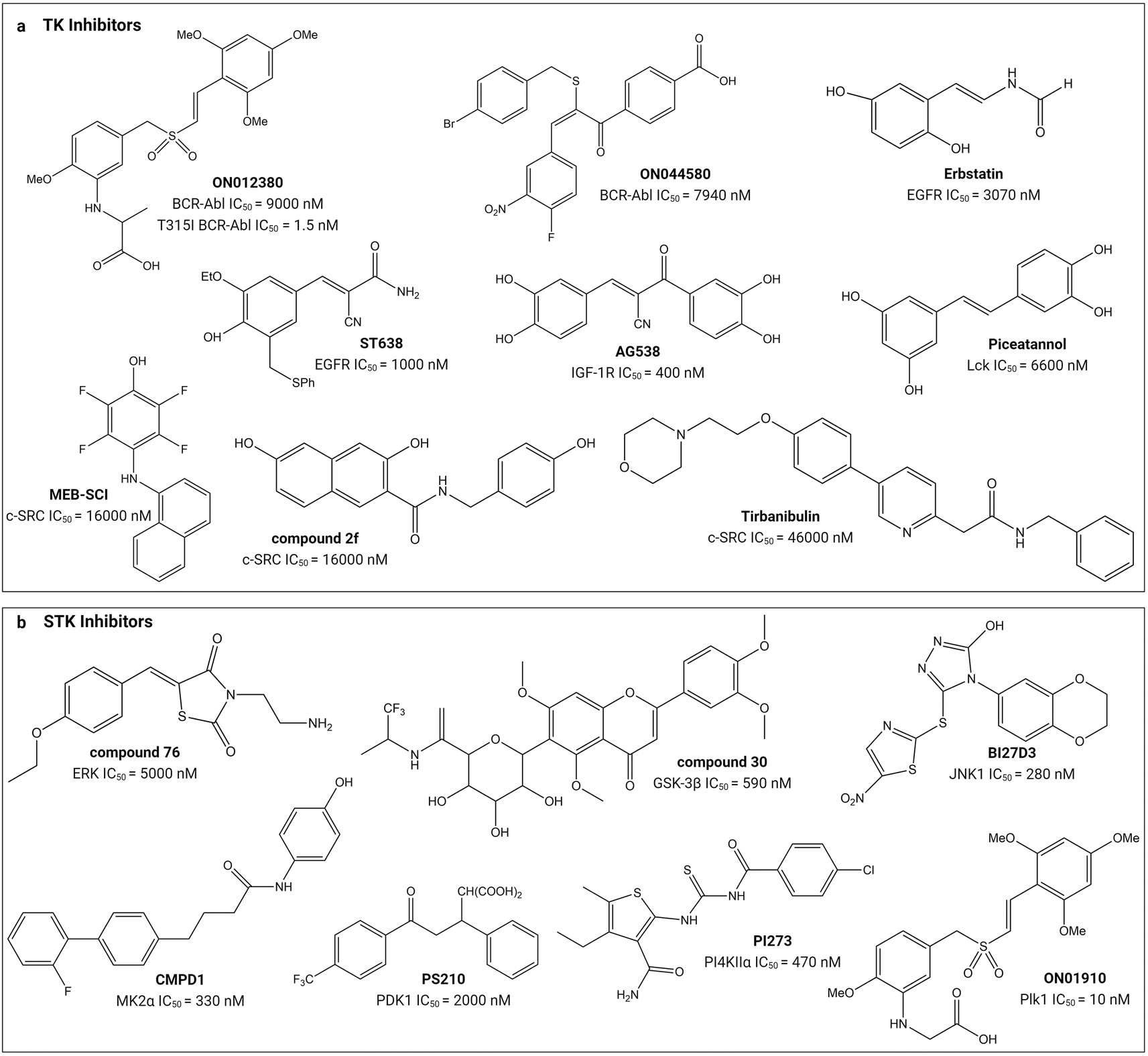 | ||
| Fig. 4 Structures of small-molecule substrate-site inhibitors of protein kinases and their IC50 values. ‘a’ shows the small-molecule inhibitors for TKs and ‘b’ shows the small-molecule inhibitors for STKs. Refer to Table 1 for the inhibitory assays. Created with https://BioRender.com. | ||
Validation of substrate-site inhibitory mechanisms has mainly been done using competition assays, and in some cases, with additional structural data. Under the assumption of the substrate and ATP-binding sites being distinct, one expects that true substrate-site inhibitors are strictly affected by competing substrates and not ATP molecules. Thus, competitive assays have provided seemingly compelling evidence of substrate-competitive modes of action, such as for ON012380, ON044580, CMPD1, PI-273, ON01910 because their inhibitory curves and IC50 values shift only upon varying of the substrate but not the ATP concentrations.105,117,119,120,123 However, directly linking substrate-site inhibition to interaction with the kinase requires binding experiments, such as was done using surface plasmon resonance (SPR) to evaluate the PI4KIIα inhibitor PI-273,119 and fluorescence binding assays to assess the p38α inhibitor CMPD1.117 For the FDA-approved tirbanibulin, kinase binding was unaffected by an ATP-site binder, suggesting binding to a non-ATP site.61 Higher resolution data is essential to show that inhibition happens specifically at the substrate-binding site. Computer-aided docking is useful for this purpose and has been performed for the ERK inhibitor (compound 76), c-Src inhibitor tirbanibulin, and MEB-SCI (compound 12),61,112,114 but experimental evidence is more definitive. Davidson et al. applied deuterium exchange mass spectrometry to map the binding interactions of CMPD1.117 The most concrete evidence of substrate-site binding was provided for the PDK1 inhibitor PS210 which comprises competition assays, differential scanning fluorimetry, cell-based experiments, and crystal structures of the inhibitors engaging the substrate-binding site of PDK1.118
There is some uncertainty regarding the true mechanism of action of some reported molecules. For instance, the apoptotic action of ON012380 on CML cells might be unrelated to its kinase inhibition based on cellular substrate phosphorylation levels,123 contrary to earlier suggestions based primarily on competitive assays.104 In addition, tirbanibulin has also been linked to the inhibition of tubulin polymerization as a second mechanism of action.61
Substrate-site inhibitors have IC50 values that lie within the low nM to μM range (Table 1). As mentioned above, IC50 values are difficult to compare between studies and they are illustrated in Fig. 4 simply for indicative purposes of potency. As illustrated, the styryl benzyl sulfones ON012380 and ON01910 have IC50 values of 9 and 10 nM against BCR-Abl TK and Plk1 STK, respectively, whereas another styryl benzyl sulfide, ON044580, has an IC50 value of 7.94 μM against BCR-Abl. The FDA approved tirbanibulin has a IC50 value of 46 μM against c-Src TK. Though tempting to speculate that the substrate competitive inhibitors are less potent than FDA-approved inhibitors, it is difficult to conclude this because of their opposing mechanisms of action and variable ATP and unknown substrate concentrations inside cells.24,27
Selectivity has been reported as a key feature of substrate-site inhibitors. For instance, the PDK1 inhibitor PS210 was tested against a panel of 121 kinases at 10 μM concentration and did not alter the activity of any other kinase except its target, including downstream signalling components such as S6K, PKB/Akt or GSK3.118 Other molecules have also exhibited single target selectivity, including compound 30 against 41 kinases (at 5 μM concentration), and ON012380 against 23 kinases (and demonstrated substantially higher IC50 values than for BCR-Abl).104,115 In some cases, target specificity has been reported after testing against a smaller number of 5 to 11 kinases. AG538, compound 12, PI-273, ON01910 are examples of such inhibitors. In those studies, some of the inhibitors like AG538 and ON01910 inhibited other kinases but have much higher IC50 values than their targets.120,124 To our knowledge, no selectivity studies have been reported for compound 2f, BI-78D3 and tirbanibulin. In general, the small-molecule substrate site kinase inhibitors are more selective than the ATP competitive inhibitors, but more investigations are needed to definitively confirm this observation.
A few small molecule substrate-site inhibitors have been reported to be effective against resistant mutants of kinases. For example, the BCR-Abl kinase inhibitors ON012380104 and ON044580105 are effective against the imatinib-resistant T315I mutant variant. ON044580 is effective against the V617F mutant variant of JAK2.105 It can be argued that the conserved mutable residues within the ATP binding site make the ATP-competitive inhibitors more susceptible to resistance and the flexible substrate-site inhibitors have the potential to be advantageous in this regard. However, the effectiveness of the majority of the substrate-site inhibitors against mutant kinases has yet to be explored.
| Origin | Development approach | Name | Target kinase | Assay | Potency (nM) | Reference | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a Substrate Competition (SC), Substrate Phosphorylation (SP), Kinase Inhibition (KI), X-ray Crystallography (XRC), Computer Aided Molecular Modelling (CAMM), Computer Aided Molecular Docking (CAMD), Surface Plasmon Resonance (SPR), Laser Scanning Microscopy (LSM); * IC50, ∼ Ki, aIC50/Ki value determined using SC, bIC50 value determined using KI, cIC50 value determined using SP. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Endogenous substrates | Alteration of optimum substrate sequence | Peptide 29 | p60c-src | SC | 130*a | 126 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Peptide 4 | Akt1 | SC, CAMM | 95∼a | 127 and 128 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| N-myristoyl-RKRTLRRL | PKC | SC | 6900*a | 129 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Optimization of consensus sequence from downstream signaling substrate | Peptide 6 | PKCα | SC | 1.9*a | 130 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Grafting of optimum substrates with other scaffolds | MTAbl13 | T315BCR-Abl | KI, CAMM | 1300*b | 131 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Replacement of substrate amino acids with non-canonical ones | PTR 6164 | Akt/PKB | SC, CAMM | 450*a | 132 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Conformational constraints of peptide substrate | Peptide 31 | p60c-src | SC | 280*a | 133 and 134 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Conformational constraints of consensus sequence | Peptide 13 | c-Src | SC | 40*a | 135 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Unconventional peptide substrate | Peptide 5 and 10 | EGFR | CAMD | — | 136 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Prephosphorylation of peptide substrate | L803 | GSK-3β | SC, CAMM | 40000*a |
137 and 138 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Optimization of pseudosubstrate sequence | PKI | PKA | SC | 2.3∼a | 139 and 140 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| [K17] PKC | PKC | SP | 75*c | 141 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| GSK3β-N (3–12) | GSK3β | SC, XRC | 700000∼a |
142 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Exogenous peptide | Optimization of HIV-1 Tat peptide | Tat-peptide | PKCα, PKA | SC, LSM | 22*a, 1200*a | 143 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Optimization of cationic cell-penetrating peptides | C[RW]5 | c-Src | SC | 2800*a | 144 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Bioengineering of lactazole-like thiopeptides | TP15 | TNIK | SC, SPR, XRC | 14*a | 145 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Allosteric inhibitors | Optimization of downstream target (MAPKAPK2) sequence | Peptide 6 | p38α | SC, CAMD | 1.3*a | 146 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Optimization of JNK scaffolding protein (JIP-1) sequence | TI-JIP | JNK | SC, SPR | 1100∼a | 147 and 148 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Retro-inverso form of yeast-screened peptide | D-PYC98 | JNK | SC, SPR | — | 149 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
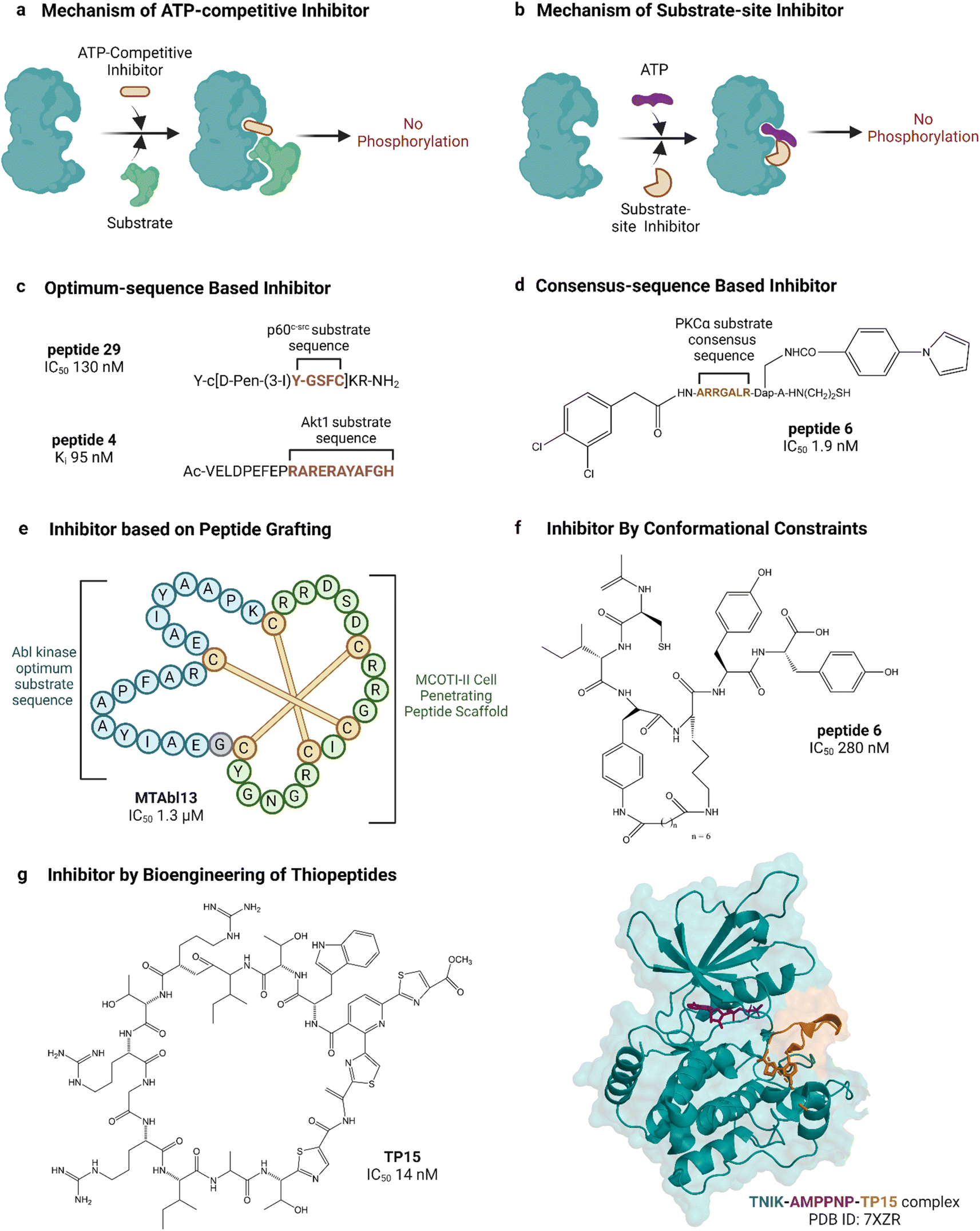 | ||
| Fig. 5 Peptide inhibitors of protein kinases. ‘a’ and ‘b’ show the functional difference between ATP-competitive and substrate site kinase inhibitors. ‘c’, ‘d’, ‘e’ and ‘f’ show different approaches for designing endogenous substrate-sequence based peptide inhibitors. ‘g’ shows an example of a pseudo-natural thiopeptide kinase inhibitor. Refer to Table 2 for inhibitory assays associated with the IC50s. Created with https://BioRender.com. | ||
Peptide substrates are typically limited by their low activity, poor stability and poor cell penetration. Litman et al. developed an AKT inhibitor by using a substrate sequence from GSK3, a downstream signaling protein.132 They altered the substrate amino acids using non-canonical amino acids to enhance cell permeability as well as stability and reported PTR 6164 as a potent AKT inhibitor (IC50 0.45 μM). Huang et al. grafted the optimum peptide substrate of Abl kinase into a cyclic cystine knotted scaffold (MCoTI-II) reported to have high stability and cell-penetrating properties (Fig. 5).131 Kumar et al. reported the introduction of conformational constraints as well, i.e., head-to-tail and terminus-to-side chain bridging of synthetic c-Src substrate-based peptide inhibitor for increased activity.133 Hah et al. developed c-Src inhibitor based on a strategy to convert weak consensus sequences into higher affinity ligands by tethering to a sequence that binds the SH2 domain adjacent to the catalytic domain.135
Unconventional peptide substrates have also been developed into inhibitors. Mitogen-inducible gene 6 (MIG6) binds and inhibits EGFR, involving a region of MIG6 that is primed by an upstream kinase and becomes phosphorylated again during inhibition.150 This substrate binding region of MIG6 has been used for targeting the EGFR-resistant mutant L858R.136 Plotkin et al. reported the use of a pre-phosphorylated substrate of GSK-3 called SXXXS(p) for synthesizing an inhibitor in the μM range.138 Another interesting source of substrates found naturally are pseudosubstrates, which occupy the substrate site but are not phosphorylated. An example is the protein kinase inhibitor peptide, also named as PKI, discovered in the 1970s, which was the first lead in designing peptide inhibitors of protein kinases.151 PKI is an endogenous thermostable peptide that specifically binds to and inhibits the catalytic subunit of PKA.140,152
Substrate-site inhibitors have been developed without using known substrates as leads. For example, HIV-1 Tat, a cationic cell-penetrating peptide was used by Ekokoshi et al. to develop substrate-competitive inhibitors of PKCα in the nM range and PKA in the μM range.143 Although only evidence for a non-ATP-competitive mechanism was shown, it is intriguing that Shirazi et al. also reported cationic cell-penetrating peptides to be kinase inhibitors, albeit for a different type of peptide and for c-Src kinase, because those types of peptides were not originally designed for kinase inhibitory activity.144 Vinogradov et al. reported the bioengineering of thiopeptides based on lactazole A.145 They built a pseudo-natural product library of over 1012 using mRNA display and screened it against TNIK, discovering TP15 as the most potent and selective TNIK substrate-site inhibitor with an IC50 in the low nM range (14 nM, Fig. 5).
We note that other studies have also considered peptides and proteins that bind outside of the catalytic cleft to be substrates. These binders therefore potentially and/or partially involve allosteric mechanisms to inhibit substrate binding at the catalytic site. For example, Nagao et al. developed an inhibitor of a mitogen-activated protein kinase (MAPK) p38α based on binding to its docking site by MAPK-activated protein kinase 2 (MAPKAPK2).146 Niu et al. reported a peptide inhibitor of JNK derived from the amino acid residue of JIP-1 (JNK-interacting protein 1).147 Ngoei et al. discovered a peptide binder of JNK from a yeast two-hybrid screen of a gene fragment library, and surprisingly (because specific binding is assumed to require stereospecific interactions) found that its retro-inverso form, D-PYC98, was more potent.149 The peptide had a non-ATP-competitive mechanism and was suggested to bind the docking site of JNK.
The substrate-competitive inhibitory effect of most of the studied peptides has been demonstrated in assays where substrate concentrations were varied to observe the inhibition of phosphorylation. For instance, endogenous PKI,139 PKC pseudo-substrate-based inhibitors,141 c-Src optimum substrate-based inhibitors,126 analogues of CIYKYY as c-Src inhibitors,134 AKT peptide inhibitor,132 PKCα peptide inhibitor130 and HIV-1 Tat-peptide inhibitors143 have been described as substrate-site inhibitors based on such substrate-competitive assays. Sometimes the substrate-competitive assay has been supported by molecular docking as evidenced in the case of BCR-Abl inhibitor MTAbl13131 and p38αMAPK inhibitor peptides.146 For some of the reported peptide inhibitors, direct binding to the substrate site of target kinases has been proven by SPR analysis. For instance, the binding of JNK inhibitor TI-JIP and D-PYC98-TAT to the kinase was studied by SPR.148,149 In addition to substrate-competitive assays and SPR analyses, the substrate-site binding of the TNIK inhibitor TP15 was validated by determining the crystal structure of the inhibitor-bound protein.145
Like small molecules, most peptide substrate-site inhibitors offer specificity and selectivity for their target kinases. The AKT inhibitors AKTide-2T and PTR6164 were tested against a panel of structurally related kinases (IC50 was measured),127,132 GSK-3β inhibitor L803 was tested against five other kinases (at 200 μM concentration),138 and c-Src inhibitor peptides were tested against two other TKs (IC50 was measured).144 The inhibitors showed substantially high potency for their target kinase compared to a pool of other kinases. All these mentioned peptide inhibitors were designed based on their substrate sequence. On the other hand, the PKCα inhibitor HIV-1 Tat exogenous peptide was tested against 70 kinases at 1 and 10 μM concentrations. It showed inhibitory activity against other AGC kinase groups (PKB, SGK1, S6K1, MSK1), two CAMK-kinases (CAMK1 and MELK), and one STE kinase (MKK1).143 Another non-substrate-based TNIK inhibitor, TP15, was tested against 67 kinases at 1 and 10 μM concentrations and selectively inhibited its target over other kinases, inhibiting two other Ste20 family kinases, mammalian sterile twenty-like 1 and, to a lesser extent, lymphocyte-oriented kinase.145 In short, although most peptide substrate-site kinase inhibitors have efficient target selectivity, more investigations are necessary to establish the generality of this specificity.
The potency of peptide inhibitors is mostly in the lower μM or nM range and is variable. Substrate-based inhibitors that have been further optimized often demonstrate IC50 values in the nM range. For instance, the PKCα inhibitor optimized by Lee et al. has an IC50 value of 1.9 nM, and the c-Src inhibitor reported by Hah et al. has an IC50 value of 40 nM.130,135 The TNIK inhibitor TP15 has an IC50 value of 14 nM.145 These peptides have also been reported to have good target selectivity. However, the less selective HIV-1 Tat peptide inhibitor had a higher IC50 value of 1.2 μM for PKA.143 In general, the more selective inhibitors have greater potency.
The effectiveness of peptide inhibitors against resistant mutants of kinases has not been extensively reported. In one report by Huang et al., the inhibitor MTAbl13 showed activity against the T315I drug-resistant mutant of Abl kinase.131 Further studies are necessary to evaluate the effectiveness of substrate-site peptide inhibitors against the emergence of resistance.
Alternative approaches to substrate-based inhibition
Bivalent inhibitors that interact with both the substrate and ATP binding sites of kinases have been developed to increase the specificity of ATP competitive inhibitors.17,153 For instance, Brandvold et al. conjugated the consensus substrate sequence of c-Src TK to an ATP-site inhibitor (compound 3), resulting in near-perfect selectivity against a panel of 213 kinases and an IC50 value of <30 nM.154 Poot et al. conjugated a high-affinity pseudosubstrate against PKC STK to an ATP inhibitor, resulting in bivalent inhibitors 2–4 times stronger than ATP-competitive inhibitors.155 Sõrmus et al. used the crystal structure of PKA bound to a peptide inhibitor to identify a substrate-site inhibitor fragment, which was then conjugated to an ATP-site inhibitor.156 Schnitzler et al. used structural information of heparin-CK2α complex to develop a bivalent inhibitor.157Although the major goal of bivalent inhibitors is to increase target specificity, detailed kinase selectivity panel assays have not been sufficiently utilized for most of the reported inhibitors. In addition, comparative analyses of their potency with clinically used kinase inhibitors are limited.153 The potential of the bivalent inhibitors against the emergence of single-point mutations and subsequent resistance is also largely understudied. The c-Src inhibitor compound 3, was tested against T338I mutant variant. The binding affinity, although found to be better than dasatinib, was 3500-fold less compared to the non-mutant c-Src.154 Shokat et al. ligated the first-generation mTOR inhibitor rapamycin with the second-generation MLN0128 to develop bivalent inhibitors.158 However, the newer generations of mTOR inhibitors are also susceptible to resistance-causing mutations.159
Future prospects
Inhibition of kinases through non-ATP mechanisms to achieve greater selectivity and resistance-resilience is defining a new era of kinase drug development for the treatment of cancer. Accordingly, the targeting of the substrate-binding site has attracted great interest. While it is tempting to speculate that the approval of tirbanibulin, a dual functional inhibitor, is a sign of more substrate-site inhibitors to come, some uncertainty on the significance of its substrate-competitive ability perhaps points to broader questions in the field. For example, yet to be established is a definitive understanding of the mechanisms of reported substrate-site inhibitors, with few studies showing comprehensive evidence spanning from competitive assays to structural data and functional validation. The small-molecule PDK1 inhibitor PS210118 and the peptide-based TNIK inhibitor TP15145 are among the best validated in vitro and have been shown to bind at or around the substrate-binding site.
With the number of studies on substrate-site inhibitors being far fewer than the vast efforts on targeting the ATP-binding site, it is premature to draw definitive comparisons between the targeting of the two sites and clinical outcomes. Nevertheless, some promising trends are emerging, as highlighted below.
The potential for higher selectivity by targeting the substrate-binding site is underpinned by the structural understanding that, while the kinase domain is conserved, the substrate-site has evolved to carry out specific functions for each kinase, unlike the ATP-binding site whose function is common to all kinases. However, although kinases recognize specific substrate motifs, it is important to note that there is some degree of crosstalk between the various kinases and the array of substrates, and therefore the general question arises as to the level of selectivity achievable or required. Encouragingly, substrate-site inhibitors have demonstrated high selectivity. For example, PS210 selectively inhibits PDK1 from within a panel of 121 kinases118 and TP15 shows good selectivity for TNIK.145
From these two examples, it can be seen that both small molecules and peptides can achieve high selectivity at the substrate site despite it being a shallow structure. Nevertheless, peptides are thought to be better equipped than small molecules for targeting such shallow surfaces. It is worth noting that selectivity is not the only important consideration for the development of substrate inhibitors. If we compare to the situation with ATP-competitive inhibitors, where there are examples of high selectivity but also many examples of poor selectivity, then one needs to consider if there are additional benefits in pursuing the substrate-binding site. One such benefit would be in the potential for reduced susceptibility to the development of resistance.
Inhibition at the substrate-binding sites presents an oncogenic kinase with a conundrum – acquire mutations to reduce inhibitor affinity but at the cost of endogenous substrate binding. More evidence is required to see if this consideration leads to a lower incidence of resistance for substrate-based compared to ATP-competitive inhibitors. There is also the possibility that other resistance mechanisms might emerge for substrate-based inhibitors. Promising observations so far are that substrate-site inhibitors are, as expected, effective against kinases resistant to ATP-competitive inhibitors. The small molecules ON012380123 and ON044580105 and the peptide MTAbl13131 all inhibit the notorious T315 mutant of BCR-Abl. Because peptides likely engage more interactions with the target kinase, it might be harder to acquire mutations that completely block peptide binding as opposed to small molecule binding, but this has yet to be demonstrated. One envisaged use of substrate-site inhibitors is co-administration with an ATP-competitive inhibitor to help reduce the acquisition of mutations.
There are differing drug design and discovery considerations relating to small molecules and peptides, important to realising the promise of substrate-site inhibition. In terms of lead discovery, peptides have an advantage over small molecules in that the natural substrates provide a guide to suitable sequences as starting points for drug design. Moreover, in the future, there is likely to be a greater emphasis on high throughput approaches for lead discovery that are tailored to peptides. For example, TP15, the TNIK inhibitor was discovered by screening a bioengineered peptide library using mRNA display.145 Such high-throughput techniques offer an efficient alternative over the manual sequence-based identification of substrate-site inhibitors. However, peptide-based leads typically need to be optimised. In the past, most efforts in this field utilized conventional chemical modification of substrate sequences for peptide inhibitor development. It is worth noting that mRNA display methods now offer the possibility of including non-canonical amino acids, not only for increasing the chemical diversity available in the screening libraries, but also for incorporating residues that might have more favourable biophysical properties than the 20 standard proteogenic amino acids.
While there are benefits to developing peptides as substrate-site inhibitors because they are naturally suited to binding the substrate-binding site, there are also challenges relating to their pharmacokinetic properties.160 Some aspects of these challenges can be met via strategic chemical modifications. For example, linear peptides are susceptible to proteolytic attack, but cyclization and other forms of chemical constraint have resulted in substrate-site inhibitors with improved stability. When constrained in the active conformation, stabilised peptides can have higher activity than their linear forms. This was observed in the study on synthetic c-Src substrate-based peptide inhibitors.133 Another major challenge is cellular delivery because the cell membrane acts as a molecular sieve preventing the passive entry of large molecular compounds. Other uptake pathways into cells have been characterized but often internalized molecules are trapped in endosomal compartments. Emerging technologies for quantifying cytosolic uptake promise to set the foundation for discovery of new molecular entities that not only cross cell membranes at high efficiency but also reach the cytosol at concentrations required for therapeutic efficacy.161
Conclusions
Substrate-site inhibitors might offer a solution to tackle the problems of selectivity and drug resistance and therefore could become some of the most important drugs in the next decade. Future work directed at the discovery of substrate-site inhibitors should consider thorough validation to clearly define their mechanisms of action at the molecular, cellular, and biological levels. This would allow for a better understanding of the potential of substrate-site inhibitors as therapeutics, building upon the promising and exciting data on their selectivity and activity already reported.Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Author contributions
Conceptualization: C. K. W. and D. J. C.; methodology: B. B., Y. H. H., and C. K. W.; software: B. B.; validation: B. B.; formal analysis: B. B., Y. H. H., and C. K. W.; investigation: B. B.; resources: C. K. W. and D. J. C.; data curation: B. B.; writing—original draft preparation: B. B.; writing—review and editing: Y. H. H., C. K. W. and D. J. C.; visualization: B. B., Y. H. H., and C. K. W.; supervision: Y. H. H., C. K. W. and D. J. C.; project administration: Y. H. H., C. K. W. and D. J. C.; funding acquisition: C. K. W. and D. J. C. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the work of many colleagues cited in the references for their valuable contributions to the field of protein kinases and kinase inhibitors. The work in our laboratory is supported by the Australian Research Council (ARC) Centre of Excellence for Innovations in Peptide and Protein Science (CE200100012). C.K.W. is an ARC Future Fellow (FT220100583). D.J.C. is supported by an Investigator Grant (GNT2009564) from the National Health and Medical Research Council (NHMRC) Australia. B.B. would like to acknowledge the contribution of the Graduate School of The University of Queensland, Brisbane 4072, Australia, and the Jashore University of Science and Technology, Jashore 7408, Bangladesh for the provision of this opportunity to study kinases as an HDR student.References
- K. S. Bhullar, N. O. Lagarón, E. M. McGowan, I. Parmar, A. Jha, B. P. Hubbard and H. P. V. Rupasinghe, Mol. Cancer, 2018, 17, 48 CrossRef PubMed.
- L. Castelo-Soccio, H. Kim, M. Gadina, P. L. Schwartzberg, A. Laurence and J. J. O’Shea, Nat. Rev. Immunol., 2023, 23, 787–806 CrossRef CAS PubMed.
- Y. L. Deribe, T. Pawson and I. Dikic, Nat. Struct. Mol. Biol., 2010, 17, 666–672 CrossRef CAS PubMed.
- E. D. Fleuren, L. Zhang, J. Wu and R. J. Daly, Nat. Rev. Cancer, 2016, 16, 83–98 CrossRef CAS PubMed.
- P. A. Futreal, L. Coin, M. Marshall, T. Down, T. Hubbard, R. Wooster, N. Rahman and M. R. Stratton, Nat. Rev. Cancer, 2004, 4, 177–183 CrossRef CAS PubMed.
- M. M. Attwood, D. Fabbro, A. V. Sokolov, S. Knapp and H. B. Schiöth, Nat. Rev. Drug Discovery, 2021, 20, 839–861 CrossRef CAS PubMed.
- P. Cohen, Nat. Rev. Drug Discovery, 2002, 1, 309–315 CrossRef CAS PubMed.
- P. Cohen, D. Cross and P. A. Jänne, Nat. Rev. Drug Discovery, 2021, 20, 551–569 CrossRef CAS PubMed.
- R. Roskoski Jr., Pharmacol. Res., 2024, 200, 107059 CrossRef PubMed.
- F. M. Ferguson and N. S. Gray, Nat. Rev. Drug Discovery, 2018, 17, 353–377 CrossRef CAS.
- M. Zhang, Y. Liu, H. Jang and R. Nussinov, J. Chem. Theory Comput., 2023, 19, 1615–1628 CrossRef CAS.
- S. Klaeger, S. Heinzlmeir, M. Wilhelm, H. Polzer, B. Vick, P.-A. Koenig, M. Reinecke, B. Ruprecht, S. Petzoldt and C. Meng, Science, 2017, 358, eaan4368 CrossRef PubMed.
- R. Martinez, A. Defnet and P. Shapiro, Avoiding or co-opting ATP inhibition: overview of type III, IV, V, and VI kinase inhibitors, in Next Generation Kinase Inhibitors: Moving Beyond the ATP Binding/Catalytic Sites, Springer, 2020, pp. 29–59 Search PubMed.
- M. E. Breen and M. B. Soellner, ACS Chem. Biol., 2015, 10, 175–189 CrossRef CAS PubMed.
- C. Arter, L. Trask, S. Ward, S. Yeoh and R. Bayliss, J. Biol. Chem., 2022, 298, 102247 CrossRef CAS PubMed.
- L. E. Hanold, M. D. Fulton and E. J. Kennedy, Pharmacol. Ther., 2017, 173, 159–170 CrossRef CAS PubMed.
- K.-C. Han, S. Yeon Kim and E. Gyeong Yang, Curr. Pharm. Des., 2012, 18, 2875–2882 CrossRef CAS PubMed.
- J. L. Johnson, T. M. Yaron, E. M. Huntsman, A. Kerelsky, J. Song, A. Regev, T.-Y. Lin, K. Liberatore, D. M. Cizin and B. M. Cohen, Nature, 2023, 613, 759–766 CrossRef CAS PubMed.
- K. C. Duong-Ly and J. R. Peterson, The human kinome and kinase inhibition, Curr. Protoc. Pharmacol., 2013, 60, 2.9.1–2.9.14 Search PubMed.
- D. Bradley, C. Viéitez, V. Rajeeve, J. Selkrig, P. R. Cutillas and P. Beltrao, Cell Rep., 2021, 34, 108602 CrossRef CAS.
- G. Manning, D. B. Whyte, R. Martinez, T. Hunter and S. Sudarsanam, Science, 2002, 298, 1912–1934 CrossRef CAS.
- S. S. Taylor, P. Zhang, J. M. Steichen, M. M. Keshwani and A. P. Kornev, Biochim. Biophys. Acta, Proteins Proteomics, 2013, 1834, 1271–1278 CrossRef CAS.
- B. Wang, H. Wu, C. Hu, H. Wang, J. Liu, W. Wang and Q. Liu, Signal Transduction Targeted Ther., 2021, 6, 423 CrossRef CAS.
- J. A. Ubersax and J. E. Ferrell Jr., Nat. Rev. Mol. Cell Biol., 2007, 8, 530–541 CrossRef CAS PubMed.
- P. Lahiry, A. Torkamani, N. J. Schork and R. A. Hegele, Nat. Rev. Genet., 2010, 11, 60–74 CrossRef CAS.
- S. Soverini, M. Mancini, L. Bavaro, M. Cavo and G. Martinelli, Mol. Cancer, 2018, 17, 1–15 CrossRef PubMed.
- J. D. Vasta, C. R. Corona, J. Wilkinson, C. A. Zimprich, J. R. Hartnett, M. R. Ingold, K. Zimmerman, T. Machleidt, T. A. Kirkland and K. G. Huwiler, Cell Chem. Biol., 2018, 25, 206–214 CrossRef CAS.
- P. Filippakopoulos, S. Müller and S. Knapp, Curr. Opin. Struct. Biol., 2009, 19, 643–649 CrossRef CAS.
- S. Panjarian, R. E. Iacob, S. Chen, J. R. Engen and T. E. Smithgall, J. Biol. Chem., 2013, 288, 5443–5450 CrossRef CAS.
- C. J. Miller and B. E. Turk, Trends Biochem. Sci., 2018, 43, 380–394 CrossRef CAS PubMed.
- S. S. Taylor, M. M. Keshwani, J. M. Steichen and A. P. Kornev, Philos. Trans. R. Soc., B, 2012, 367, 2517–2528 CrossRef CAS.
- A. P. Kornev and S. S. Taylor, Biochim. Biophys. Acta, Proteins Proteomics, 2010, 1804, 440–444 CrossRef CAS.
- V. Modi and R. L. Dunbrack Jr., Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 6818–6827 CrossRef CAS.
- A. P. Kornev, N. M. Haste, S. S. Taylor and L. F. Ten Eyck, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 17783–17788 CrossRef CAS PubMed.
- D. Huang, T. Zhou, K. Lafleur, C. Nevado and A. Caflisch, Bioinformatics, 2010, 26, 198–204 CrossRef CAS PubMed.
- S. S. Taylor and A. P. Kornev, Trends Biochem. Sci., 2011, 36, 65–77 CrossRef CAS PubMed.
- C. R. Guimarães, B. K. Rai, M. J. Munchhof, S. Liu, J. Wang, S. K. Bhattacharya and L. Buckbinder, J. Chem. Inf. Model., 2011, 51, 1199–1204 CrossRef PubMed.
- R. Y. Patel and R. J. Doerksen, J. Proteome Res., 2010, 9, 4433–4442 CrossRef CAS PubMed.
- J. A. Endicott, M. E. Noble and L. N. Johnson, Annu. Rev. Biochem., 2012, 81, 587–613 CrossRef CAS PubMed.
- A. Vulpetti and R. Bosotti, Il Farmaco, 2004, 59, 759–765 CrossRef CAS PubMed.
- Y. Zhu and X. Hu, Molecules, 2022, 27, 7124 CrossRef CAS PubMed.
- R. Vijayan, P. He, V. Modi, K. C. Duong-Ly, H. Ma, J. R. Peterson, R. L. Dunbrack Jr. and R. M. Levy, J. Med. Chem., 2015, 58, 466–479 CrossRef CAS PubMed.
- Y. Yang, S. Li, Y. Wang, Y. Zhao and Q. Li, Signal Transduction Targeted Ther., 2022, 7, 329 CrossRef CAS PubMed.
- L. Zhang, J.-C. Wang, L. Hou, P.-R. Cao, L. Wu, Q.-S. Zhang, H.-Y. Yang, Y. Zang, J.-P. Ding and J. Li, Sci. Rep., 2015, 5, 10115 CrossRef CAS PubMed.
- G. Zhu, K. Fujii, Y. Liu, V. Codrea, J. Herrero and S. Shaw, J. Biol. Chem., 2005, 280, 36372–36379 CrossRef CAS.
- A. Li, R. Voleti, M. Lee, D. Gagoski and N. H. Shah, Elife, 2023, 12, e82345 CrossRef CAS PubMed.
- B. E. Turk, Curr. Opin. Chem. Biol., 2008, 12, 4–10 CrossRef CAS PubMed.
- A. N. Bullock, J. Debreczeni, A. L. Amos, S. Knapp and B. E. Turk, J. Biol. Chem., 2005, 280, 41675–41682 CrossRef CAS PubMed.
- O. Wagih, J. Reimand and G. D. Bader, Nat. Methods, 2015, 12, 531–533 CrossRef CAS PubMed.
- C. Chen, B. H. Ha, A. F. Thévenin, H. J. Lou, R. Zhang, K. Y. Yip, J. R. Peterson, M. Gerstein, P. M. Kim and P. Filippakopoulos, Mol. Cell, 2014, 53, 140–147 CrossRef CAS PubMed.
- C. Chen, W. Nimlamool, C. J. Miller, H. J. Lou and B. E. Turk, ACS Chem. Biol., 2017, 12, 1194–1198 CrossRef CAS PubMed.
- S. Eid, S. Turk, A. Volkamer, F. Rippmann and S. Fulle, BMC Bioinf., 2017, 18, 1–6 CrossRef PubMed.
- S. Sehgal, Transplant. Proc., 2003, 35, S7–S14 CrossRef.
- N. Iqbal and N. Iqbal, Chemother. Res. Pract., 2014, 2014, 357027 Search PubMed.
- R. Roskoski Jr., Pharmacol. Res., 2016, 103, 26–48 CrossRef PubMed.
- C. M. Gower, M. E. Chang and D. J. Maly, Crit. Rev. Biochem. Mol. Biol., 2014, 49, 102–115 CrossRef CAS.
- A. Abdeldayem, Y. S. Raouf, S. N. Constantinescu, R. Moriggl and P. T. Gunning, Chem. Soc. Rev., 2020, 49, 2617–2687 RSC.
- B. Acharya, D. Saha, D. Armstrong, M. Hanafi, A. Herrera-Rueda, N. R. Lakkaniga and B. Frett, RSC Med. Chem., 2024, 15, 399–415 RSC.
- N. Uhlenbrock, S. Smith, J. Weisner, I. Landel, M. Lindemann, T. A. Le, J. Hardick, R. Gontla, R. Scheinpflug and P. Czodrowski, Chem. Sci., 2019, 10, 3573–3585 RSC.
- D. Réa and T. P. Hughes, Crit. Rev. Oncol. Hematol., 2022, 171, 103580 CrossRef PubMed.
- M. P. Smolinski, Y. Bu, J. Clements, I. H. Gelman, T. Hegab, D. L. Cutler, J. W. Fang, G. Fetterly, R. Kwan and A. Barnett, J. Med. Chem., 2018, 61, 4704–4719 CrossRef CAS PubMed.
- Z. A. Knight and K. M. Shokat, Chem. Biol., 2005, 12, 621–637 CrossRef CAS PubMed.
- C. C. Ayala-Aguilera, T. Valero, Á. Lorente-Macías, D. J. Baillache, S. Croke and A. Unciti-Broceta, J. Med. Chem., 2021, 65, 1047–1131 CrossRef PubMed.
- P. W. Manley, F. Blasco, J. Mestan and R. Aichholz, Bioorg. Med. Chem., 2013, 21, 3231–3239 CrossRef CAS PubMed.
- J. Schoepfer, W. Jahnke, G. Berellini, S. Buonamici, S. Cotesta, S. W. Cowan-Jacob, S. Dodd, P. Drueckes, D. Fabbro, T. Gabriel, J.-M. Groell, R. M. Grotzfeld, A. Q. Hassan, C. Henry, V. Iyer, D. Jones, F. Lombardo, A. Loo, P. W. Manley, X. Pellé, G. Rummel, B. Salem, M. Warmuth, A. A. Wylie, T. Zoller, A. L. Marzinzik and P. Furet, J. Med. Chem., 2018, 61, 8120–8135 CrossRef CAS PubMed.
- C. Robert, C. Mateus, A. Spatz, J. Wechsler and B. Escudier, J. Am. Acad. Dermatol., 2009, 60, 299–305 CrossRef PubMed.
- S. Ferreira, E. Guttman-Yassky and T. Torres, Am J Clin Dermatol., 2020, 21, 783–798 CrossRef PubMed.
- A. Mullally, J. Hood, C. Harrison and R. Mesa, Blood Adv., 2020, 4, 1792–1800 CrossRef CAS PubMed.
- M. I. Davis, J. P. Hunt, S. Herrgard, P. Ciceri, L. M. Wodicka, G. Pallares, M. Hocker, D. K. Treiber and P. P. Zarrinkar, Nat. Biotechnol., 2011, 29, 1046–1051 CrossRef CAS PubMed.
- S. Müller, A. Chaikuad, N. S. Gray and S. Knapp, Nat. Chem. Biol., 2015, 11, 818–821 CrossRef PubMed.
- Y. Cheng and H. Tian, Molecules, 2017, 22, 1551 CrossRef PubMed.
- Y. Yang, S. Li, Y. Wang, Y. Zhao and Q. Li, Signal Transduction Targeted Ther., 2022, 7, 329 CrossRef CAS PubMed.
- N. A. Cohen, T. S. Kim and R. P. DeMatteo, Ann. Surg., 2017, 265, 311–319 CrossRef PubMed.
- S. A. Rosenzweig, Adv. Cancer Res., 2018, 138, 71–98 CrossRef CAS PubMed.
- C. M. Lovly and A. T. Shaw, Clin. Cancer Res., 2014, 20, 2249–2256 CrossRef CAS PubMed.
- J. R. Sierra, V. Cepero and S. Giordano, Mol. Cancer, 2010, 9, 1–13 CrossRef PubMed.
- Q. Jiao, L. Bi, Y. Ren, S. Song, Q. Wang and Y.-s. Wang, Mol. Cancer, 2018, 17, 1–12 CrossRef PubMed.
- R. S. Herbst, M. Fukuoka and J. Baselga, Nat. Rev. Cancer, 2004, 4, 956–965 CrossRef CAS PubMed.
- N. S. Persky, D. Hernandez, M. Do Carmo, L. Brenan, O. Cohen, S. Kitajima, U. Nayar, A. Walker, S. Pantel, Y. Lee, J. Cordova, M. Sathappa, C. Zhu, T. K. Hayes, P. Ram, P. Pancholi, T. S. Mikkelsen, D. A. Barbie, X. Yang, R. Haq, F. Piccioni, D. E. Root and C. M. Johannessen, Nat. Struct. Mol. Biol., 2020, 27, 92–104 CrossRef CAS PubMed.
- T. P. Braun, C. A. Eide and B. J. Druker, Cancer Cell, 2020, 37, 530–542 CrossRef CAS PubMed.
- E. Jabbour, H. Kantarjian, D. Jones, M. Talpaz, N. Bekele, S. O'brien, X. Zhou, R. Luthra, G. Garcia-Manero and F. Giles, Leukemia, 2006, 20, 1767–1773 CrossRef CAS PubMed.
- C. A. Eide, M. S. Zabriskie, S. L. S. Stevens, O. Antelope, N. A. Vellore, H. Than, A. R. Schultz, P. Clair, A. D. Bowler and A. D. Pomicter, Cancer Cell, 2019, 36, 431–443 CrossRef CAS PubMed.
- C. Ricordel, L. Friboulet, F. Facchinetti and J.-C. Soria, Ann. Oncol., 2018, 29, i28–i37 CrossRef CAS PubMed.
- W. Zhao, A. Song, Y. Xu, Q. Wu, C. Liu, J. C. Yin, Q. Ou, X. Wu, Y. Shao and X. Zhao, BMC Med., 2023, 21, 73 CrossRef CAS PubMed.
- D. L. Wheeler, E. F. Dunn and P. M. Harari, Nat. Rev. Clin. Oncol., 2010, 7, 493–507 CrossRef CAS PubMed.
- V. Nelson, J. Ziehr, M. Agulnik and M. Johnson, OncoTargets Ther., 2013, 135–143 CAS.
- K. Tanaka, K. Nosaki, K. Otsubo, K. Azuma, S. Sakata, H. Ouchi, R. Morinaga, H. Wataya, A. Fujii and N. Nakagaki, Oncotarget, 2017, 8, 68123 CrossRef PubMed.
- H. L. Tumbrink, A. Heimsoeth and M. L. Sos, Oncogene, 2021, 40, 1–11 CrossRef CAS PubMed.
- A. Leonetti, S. Sharma, R. Minari, P. Perego, E. Giovannetti and M. Tiseo, Br. J. Cancer, 2019, 121, 725–737 CrossRef PubMed.
- J. Lategahn, M. Keul, P. Kloevekorn, H. L. Tumbrink, J. Niggenaber, M. P. Mueller, L. Hodson, M. Flasshoff, J. Hardick and T. Grabe, Chem. Sci., 2019, 10, 10789–10801 RSC.
- Y. Kobayashi, T. Fujino, M. Nishino, T. Koga, M. Chiba, Y. Sesumi, S. Ohara, M. Shimoji, K. Tomizawa and T. Takemoto, J. Thorac. Oncol., 2018, 13, 727–731 CrossRef PubMed.
- J. Veeraraghavan, S. Bose, R. Mistry, P. Selenica, S. Nanda, L. Qin, A. Gazzo, Y. Zhu, M. A. Mancini and F. Stossi, Cancer Res., 2022, 82, PD8-06 CrossRef.
- F. Gonzalvez, S. Vincent, T. E. Baker, A. E. Gould, S. Li, S. D. Wardwell, S. Nadworny, Y. Ning, S. Zhang and W.-S. Huang, Cancer Discovery, 2021, 11, 1672–1687 CrossRef CAS PubMed.
- Y. Gao, A. Maria, N. Na, A. da Cruz Paula, A. N. Gorelick, J. F. Hechtman, J. Carson, R. A. Lefkowitz, B. Weigelt and B. S. Taylor, Cancer Discovery, 2019, 9, 1182–1191 CrossRef CAS PubMed.
- F. Facchinetti, L. Lacroix, L. Mezquita, J.-Y. Scoazec, Y. Loriot, L. Tselikas, A. Gazzah, E. Rouleau, J. Adam and S. Michiels, Eur. J. Cancer, 2020, 132, 211–223 CrossRef CAS PubMed.
- H. Husain, M. Scur, A. Murtuza, N. Bui, B. Woodward and R. Kurzrock, Mol. Cancer Ther., 2017, 16, 265–272 CrossRef CAS PubMed.
- S. Boumahdi and F. J. de Sauvage, Nat. Rev. Drug Discovery, 2020, 19, 39–56 CrossRef CAS PubMed.
- T. Kim, M. S. Tyndel, Z. Zhang, J. Ahn, S. Choi, M. Szardenings, J. H. Lipton, H.-J. Kim and D. K. D. Hwan, Leuk. Res., 2017, 59, 142–148 CrossRef CAS PubMed.
- E. Traer, R. MacKenzie, J. Snead, A. Agarwal, A. M. Eiring, T. O'Hare, B. J. Druker and M. W. Deininger, Leukemia, 2012, 26, 1140–1143 CrossRef CAS PubMed.
- M. C. Papadimitriou, A. Pazaiti, K. Iliakopoulos, M. Markouli, V. Michalaki and C. A. Papadimitriou, Biochim. Biophys. Acta, Mol. Cell Res., 2022, 1869, 119346 CrossRef CAS PubMed.
- P. Gallipoli, A. Cook, S. Rhodes, L. Hopcroft, H. Wheadon, A. D. Whetton, H. G. Jørgensen, R. Bhatia and T. L. Holyoake, Blood, 2014, 124, 1492–1501 CrossRef CAS PubMed.
- J. H. Park, Y. J. Choi, S. Y. Kim, J.-E. Lee, K. J. Sung, S. Park, W. S. Kim, J. S. Song, C.-M. Choi and Y. H. Sung, Oncotarget, 2016, 7, 22005 CrossRef PubMed.
- J. Huang, L. Zheng, Z. Sun and J. Li, Int. J. Mol. Med., 2022, 50, 1–13 CAS.
- K. Gumireddy, S. J. Baker, S. C. Cosenza, P. John, A. D. Kang, K. A. Robell, M. R. Reddy and E. P. Reddy, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 1992–1997 CrossRef CAS PubMed.
- S. S. Jatiani, S. C. Cosenza, M. R. Reddy, J. H. Ha, S. J. Baker, A. K. Samanta, M. J. Olnes, L. Pfannes, E. M. Sloand, R. B. Arlinghaus and E. P. Reddy, Genes Cancer, 2010, 1, 331–345 CrossRef CAS PubMed.
- M. Imoto, K. Umezawa, K. Isshiki, S. Kunimoto, T. Sawa, T. Takeuchi and H. Umezawa, J. Antibiot., 1987, 40, 1471–1473 CrossRef CAS PubMed.
- H. Umezawa, M. Imoto, T. Sawa, K. Isshiki, N. Matsuda, T. Uchida, H. Iinuma, M. Hamada and T. Takeuchi, J. Antibiot., 1986, 39, 170–173 CrossRef CAS PubMed.
- T. Shiraishi, M. Koji Owada, M. Tatsuka, T. Yamashita, K. Watanabe and T. Kakunaga, Cancer Res., 1989, 49, 2374–2378 CAS.
- G. Blum, A. Gazit and A. Levitzki, Biochemistry, 2000, 39, 15705–15712 CrossRef CAS PubMed.
- R. L. Geahlen and J. L. McLaughlin, Biochem. Biophys. Res. Commun., 1989, 165, 241–245 CrossRef CAS PubMed.
- M. Cushman, D. Nagarathnam, D. Gopal and R. L. Geahlen, Bioorg. Med. Chem. Lett., 1991, 1, 215–218 CrossRef CAS.
- M. E. Breen, M. E. Steffey, E. J. Lachacz, F. E. Kwarcinski, C. C. Fox and M. B. Soellner, Angew. Chem., Int. Ed., 2014, 53, 7010–7013 CrossRef CAS PubMed.
- T. H. Marsilje, K. L. Milkiewicz and D. G. Hangauer, Bioorg. Med. Chem., 2000, 10, 477–481 CrossRef CAS PubMed.
- C. N. Hancock, A. Macias, E. K. Lee, S. Y. Yu, A. D. MacKerell and P. Shapiro, J. Med. Chem., 2005, 48, 4586–4595 CrossRef CAS PubMed.
- Z. Liang and Q. X. Li, ACS Chem. Neurosci., 2018, 9, 1166–1183 CrossRef CAS PubMed.
- J. L. Stebbins, S. K. De, T. Machleidt, B. Becattini, J. Vazquez, C. Kuntzen, L.-H. Chen, J. F. Cellitti, M. Riel-Mehan and A. Emdadi, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 16809–16813 CrossRef CAS PubMed.
- W. Davidson, L. Frego, G. W. Peet, R. R. Kroe, M. E. Labadia, S. M. Lukas, R. J. Snow, S. Jakes, C. A. Grygon and C. Pargellis, Biochemistry, 2004, 43, 11658–11671 CrossRef CAS PubMed.
- K. Busschots, L. A. Lopez-Garcia, C. Lammi, A. Stroba, S. Zeuzem, A. Piiper, P. M. Alzari, S. Neimanis, J. M. Arencibia and M. Engel, Chem. Biol., 2012, 19, 1152–1163 CrossRef CAS PubMed.
- J. Li, Z. Gao, D. Zhao, L. Zhang, X. Qiao, Y. Zhao, H. Ding, P. Zhang, J. Lu, J. Liu, H. Jiang, C. Luo and C. Chen, Cancer Res., 2017, 77, 6253–6266 CrossRef CAS PubMed.
- K. Gumireddy, M. R. Reddy, S. C. Cosenza, R. B. Nathan, S. J. Baker, N. Papathi, J. Jiang, J. Holland and E. P. Reddy, Cancer Cell, 2005, 7, 275–286 CrossRef CAS PubMed.
- M. R. Reddy, V. R. Pallela, S. C. Cosenza, M. R. Mallireddigari, R. Patti, M. Bonagura, M. Truongcao, B. Akula, S. S. Jatiani and E. P. Reddy, Bioorg. Med. Chem., 2010, 18, 2317–2326 CrossRef CAS PubMed.
- S. S. Jatiani, S. C. Cosenza, M. V. Reddy, J. H. Ha, S. J. Baker, A. K. Samanta, M. J. Olnes, L. Pfannes, E. M. Sloand, R. B. Arlinghaus and E. P. Reddy, Genes Cancer, 2010, 1, 331–345 CrossRef CAS PubMed.
- J. Wu, F. Meng, Y. Ying, Z. Peng, L. Daniels, W. Bornmann, A. Quintas-Cardama, D. Roulston, M. Talpaz and L. Peterson, Leukemia, 2010, 24, 869–872 CrossRef CAS PubMed.
- G. Blum, A. Gazit and A. Levitzki, J. Biol. Chem., 2003, 278, 40442–40454 CrossRef CAS PubMed.
- J. M. Holub, Novel peptide-based inhibitors of protein kinases, in Next Generation Kinase Inhibitors: Moving Beyond the ATP Binding/Catalytic Sites, Springer, 2020, pp. 169–206 Search PubMed.
- J. Alfaro-Lopez, W. Yuan, B. C. Phan, J. Kamath, Q. Lou, K. S. Lam and V. J. Hruby, J. Med. Chem., 1998, 41, 2252–2260 CrossRef CAS PubMed.
- T. Obata, M. B. Yaffe, G. G. Leparc, E. T. Piro, H. Maegawa, A. Kashiwagi, R. Kikkawa and L. C. Cantley, J. Biol. Chem., 2000, 275, 36108–36115 CrossRef CAS PubMed.
- Y. Luo, R. A. Smith, R. Guan, X. Liu, V. Klinghofer, J. Shen, C. Hutchins, P. Richardson, T. Holzman and S. H. Rosenberg, Biochemistry, 2004, 43, 1254–1263 CrossRef CAS PubMed.
- N. E. Ward and C. A. O'Brian, Biochemistry, 1993, 32, 11903–11909 CrossRef CAS PubMed.
- J. H. Lee, S. K. Nandy and D. S. Lawrence, J. Am. Chem. Soc., 2004, 126, 3394–3395 CrossRef CAS PubMed.
- Y.-H. Huang, S. T. Henriques, C. K. Wang, L. Thorstholm, N. L. Daly, Q. Kaas and D. J. Craik, Sci. Rep., 2015, 5, 1–15 Search PubMed.
- P. Litman, O. Ohne, S. Ben-Yaakov, L. Shemesh-Darvish, T. Yechezkel, Y. Salitra, S. Rubnov, I. Cohen, H. Senderowitz and D. Kidron, Biochemistry, 2007, 46, 4716–4724 CrossRef CAS PubMed.
- A. Kumar, G. Ye, Y. Wang, X. Lin, G. Sun and K. Parang, J. Med. Chem., 2006, 49, 3395–3401 CrossRef CAS PubMed.
- J. Kamath, R. Liu, A. Enstrom, Q. Lou and K. Lam, J. Pept. Res., 2003, 62, 260–268 CrossRef CAS PubMed.
- J.-M. Hah, V. Sharma, H. Li and D. S. Lawrence, J. Am. Chem. Soc., 2006, 128, 5996–5997 CrossRef CAS PubMed.
- F. Tavakoli and M. R. Ganjalikhany, PLoS One, 2019, 14, e0217031 CrossRef CAS PubMed.
- A. Licht-Murava, B. Plotkin, M. Eisenstein and H. Eldar-Finkelman, J. Mol. Biol., 2011, 408, 366–378 CrossRef CAS PubMed.
- B. Plotkin, O. Kaidanovich, I. Talior and H. Eldar-Finkelman, J. Pharmacol. Exp. Ther., 2003, 305, 974–980 CrossRef CAS PubMed.
- D. B. Glass, H.-C. Cheng, L. Mende-Mueller, J. Reed and D. Walsh, J. Biol. Chem., 1989, 264, 8802–8810 CrossRef CAS PubMed.
- G. D. Dalton and W. L. Dewey, Neuropeptides, 2006, 40, 23–34 CrossRef CAS PubMed.
- C. House and B. E. Kemp, Cell Signaling, 1990, 2, 187–190 CrossRef CAS PubMed.
- R. Dajani, E. Fraser, S. M. Roe, N. Young, V. Good, T. C. Dale and L. H. Pearl, Cell, 2001, 105, 721–732 CrossRef CAS PubMed.
- E. Ekokoski, O. Aitio, K. Törnquist, J. Yli-Kauhaluoma and R. K. Tuominen, Eur. J. Pharm. Sci., 2010, 40, 404–411 CrossRef CAS PubMed.
- A. N. Shirazi, R. K. Tiwari, A. Brown, D. Mandal, G. Sun and K. Parang, Bioorg. Med. Chem., 2013, 23, 3230–3234 CrossRef PubMed.
- A. A. Vinogradov, Y. Zhang, K. Hamada, J. S. Chang, C. Okada, H. Nishimura, N. Terasaka, Y. Goto, K. Ogata and T. Sengoku, J. Am. Chem. Soc., 2022, 144, 20332–20341 CrossRef CAS PubMed.
- H. Nagao, D. Kitagawa, F. Nakajima, M. Sawa and T. Kinoshita, Bioorg. Med. Chem., 2021, 43, 128056 CrossRef CAS PubMed.
- L. Niu, K.-C. Chang, S. Wilson, P. Tran, F. Zuo and D. C. Swinney, Biochemistry, 2007, 46, 4775–4784 CrossRef CAS PubMed.
- R. K. Barr, T. S. Kendrick and M. A. Bogoyevitch, J. Biol. Chem., 2002, 277, 10987–10997 CrossRef CAS PubMed.
- K. R. Ngoei, B. Catimel, N. Milech, P. M. Watt and M. A. Bogoyevitch, Int. J. Biochem. Cell Biol., 2013, 45, 1939–1950 CrossRef CAS PubMed.
- E. Park, N. Kim, S. B. Ficarro, Y. Zhang, B. I. Lee, A. Cho, K. Kim, A. K. Park, W.-Y. Park and B. Murray, Nat. Struct. Mol. Biol., 2015, 22, 703–711 CrossRef CAS PubMed.
- C. Liu, P. Ke, J. Zhang, X. Zhang and X. Chen, Front. Physiol., 2020, 11, 574030 CrossRef PubMed.
- D. A. Walsh, C. D. Ashby, C. Gonzalez, D. Calkins, E. H. Fischer and E. G. Krebs, J. Biol. Chem., 1971, 246, 1977–1985 CrossRef CAS PubMed.
- S. Lee, J. Kim, J. Jo, J. W. Chang, J. Sim and H. Yun, Eur. J. Med. Chem., 2021, 216, 113318 CrossRef CAS PubMed.
- K. R. Brandvold, S. M. Santos, M. E. Breen, E. J. Lachacz, M. E. Steffey and M. B. Soellner, ACS Chem. Biol., 2015, 10, 1387–1391 CrossRef CAS PubMed.
- A. J. Poot, J. van Ameijde, M. Slijper, A. van den Berg, R. Hilhorst, R. Ruijtenbeek, D. T. Rijkers and R. M. Liskamp, Chembiochem, 2009, 10, 2042–2051 CrossRef CAS PubMed.
- T. Sõrmus, D. Lavogina, A. Teearu, E. Enkvist, A. Uri and K. Viht, J. Med. Chem., 2022, 65, 10975–10991 CrossRef PubMed.
- A. Schnitzler and K. Niefind, Eur. J. Med. Chem., 2021, 214, 113223 CrossRef CAS PubMed.
- V. S. Rodrik-Outmezguine, M. Okaniwa, Z. Yao, C. J. Novotny, C. McWhirter, A. Banaji, H. Won, W. Wong, M. Berger and E. de Stanchina, Nature, 2016, 534, 272–276 CrossRef CAS PubMed.
- L. Formisano, F. Napolitano, R. Rosa, V. D'Amato, A. Servetto, R. Marciano, P. De Placido, C. Bianco and R. Bianco, Crit. Rev. Oncol. Hematol., 2020, 147, 102886 CrossRef PubMed.
- M. Muttenthaler, G. F. King, D. J. Adams and P. F. Alewood, Nat. Rev. Drug Discovery, 2021, 20, 309–325 CrossRef CAS PubMed.
- M. Sánchez-Navarro, Adv. Drug Delivery Rev., 2021, 171, 187–198 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2024 |