
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A perspective on the reactions of organic peroxy radicals with HO2
Niklas Illmann *
*
University of Wuppertal, Institute for Atmospheric and Environmental Research, Gaußstraße 20, 42119 Wuppertal, Germany. E-mail: illmann@uni-wuppertal.de
First published on 25th June 2025
Abstract
The chemistry of organic peroxy radicals (RO2) is crucial for ozone and secondary organic aerosol formation in the troposphere. The level of nitrogen monoxide (NO) exerts a major control on further reactions of peroxy radicals. The research on these reactions in the absence of NO has been receiving increasing attention recently. The current studies under these conditions, typically associated with pristine environments, are focused on understanding the formation of highly oxygenated organic molecules (HOMs) via autoxidation and generation of accretion products, which supposedly result from peroxy radical permutation reactions (RO2 + RO2). Apart from the potential OH production from some oxygenated peroxy radicals, there is less research activity on the reactions of peroxy radicals with HO2. This article reviews the existing literature data available on RO2 + HO2 reactions and highlights the gaps where future research is required. To date, limited information has been provided on the reactions of HO2 with functionalized RO2, particularly for β-hydroxyalkyl peroxy radicals, carbonyl-substituted peroxy radicals other than acyl peroxy, and peroxy radicals containing at least two functionalities. In addition, the temperature dependence of product branching ratios is not well established. Future studies targeting the influence of RO2 + HO2 on the tropospheric HOx (![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) OH + HO2) budget should ideally enlarge the dataset of OH yields from various peroxy radical structures. This also highlights the need to broaden the investigations on the formed hydroperoxides, whose gas-phase chemistry is not well known.
OH + HO2) budget should ideally enlarge the dataset of OH yields from various peroxy radical structures. This also highlights the need to broaden the investigations on the formed hydroperoxides, whose gas-phase chemistry is not well known.
Environmental significanceOrganic peroxy radicals (RO2) are key species involved in chain reactions that yield tropospheric ozone and secondary organic aerosols. Emission control strategies, upon international policy agreements, lead to a continuous reduction in nitrogen oxides. Chemical conditions commonly associated with pristine regions are thus expected to become more frequent. Peroxy radical reactions favoured under these conditions, such as unimolecular isomerization, permutation reactions or reactions of RO2 with HO2, are complex, and experiments targeting these chemical regimes are challenging. The present work offers an overview of the current state of knowledge and uncovers gaps that need to be addressed to draw a comprehensive picture of the importance of peroxy radical reactions with HO2 in atmospheric chemistry. |
Introduction
Organic peroxy radicals (RO2) are essential intermediates in the atmospheric oxidation of volatile organic compounds (VOCs). These are emitted into the troposphere predominantly from biogenic1 but also from anthropogenic sources. Besides its central role in the formation of tropospheric ozone and secondary organic aerosol (SOA), RO2 chemistry controls the recycling of reactive radicals such as OH (e.g. ref. 2 and 3). Once formed through VOC oxidation, e.g. in the OH-initiated oxidation either via abstraction| RH + OH → R + H2O | (R1) |
| R + O2 + M → RO2 + M | (R2) |
|
R2CCR2 + OH + M → R2C(OH)CR2 + M
| (R3) |
| R2C(OH)CR2 + O2 + M → R2C(OH)C(O2)R2 + M, | (R4) |
NO + NO2) plays a central role in efficient radical-recycling, including the regeneration of OH and causing the formation of tropospheric ozone (Fig. 1). The reaction with NO typically represents the major RO2 loss in air masses influenced by anthropogenic activities.
 | ||
| Fig. 1 Simplified, generic scheme for the major paths of RO2 chemistry under NOx-dominated conditions (blue) and NOx-poor conditions, where other pathways become competitive (red). | ||
The knowledge of peroxy radical chemistry, which is incorporated into chemistry-transport models, is crucial for both forecasting local air pollution episodes on regional scale and predicting global earth-climate interactions. Although our understanding of RO2 chemistry is continuously evolving and has led to improvements in chemistry models, there is always a need to critically review the available chemical mechanisms and experimental set-ups. One of the famous turn-around examples is the unimolecular isomerization of peroxy radicals, which is found to be competitive with bimolecular processes in the troposphere,4,5 but, to be observed, requires simulating atmospheric conditions (e. g. concentrations) in the experiment as closely as possible.
Apart from elucidating the HOx (OH + HO2) budget in forested regions (e.g. ref. 4 and 6), research on the atmospheric oxidation of VOC during the last 15 years has considerably focused on understanding the mechanisms yielding SOA. The detection of highly oxygenated organic molecules (HOMs) formed through a chain of unimolecular isomerization steps called autoxidation initiated the allocation of tremendous research effort in a rapidly growing scientific community (see Bianchi et al.7 and references therein). However, the formation of low-volatility vapours, which act as aerosol precursors, is not limited to unimolecular processes. Several observations suggest the relevance of accretion products (organic peroxides, ROOR) formation in the permutation reactions (self- and cross-reactions) of (HOM) peroxy radicals, which terminate autoxidation (e.g. ref. 8–10).
In contrast to the increasing number of reports on RO2 isomerization and permutation reactions, there are very few recent studies on the reactions of peroxy radicals with HO2 except for some short, renewed interest following the detection of discrepancies in the HOx budget in forested regions dominated by isoprene emissions (e.g. ref. 6 and 11). The present perspective article reviews the available information on this particular class of organic peroxy radical reactions. In addition, it aims to discuss the challenges that occur when RO2 + HO2 are being investigated experimentally and, finally, gaps that require further research are identified.
Experimental and analytical approaches
Retrieving information on RO2 + HO2 reactions requires experimental approaches and analytical instrumentation that allow the time-resolved detection of either radicals or closed-shell species or both. Although the sections below are spiked with both experimental and analytical information, a brief overview is provided in this section. However, a detailed evaluation and a complete representation of the available methods are beyond the scope of the present work. In addition, there exist a number of developments in radical detection that have not yet been applied to investigate RO2 + HO2 chemistry.Traditionally, product yields and branching ratios have been determined in static systems, such as simulation chambers, while flow tube applications are commonly used for absolute rate coefficient determination. The first investigations on RO2 + HO2 reactions date back to the work by Cox and Tyndall,12,13 who investigated the kinetics of CH3O2 + HO2 using molecular modulation spectrometry. In these studies, methyl peroxy radicals were generated by the photolysis of CH4/Cl2 mixtures in the presence of oxygen and a relative increase in HO2 was achieved by the addition of H2. This pioneering experimental approach has been adapted ever since in many of the later studies targeting RO2 + HO2 reactions, namely the generation of a specific peroxy radical by the reaction of an organic precursor with Cl atoms and the increase in HO2 relative to RO2 by the addition of an HO2 precursor, such as H2, methanol, and formaldehyde. In simulation chamber studies, this approach has been applied to determine product branching ratios by systematically varying the HO2/RO2 ratio. This ranges from a system usually dominated by RO2 permutation reactions, at zero HO2 precursor, to a system that favours RO2 + HO2 reactions when the concentrations of the HO2 precursor are sufficiently high. In the ideal case, the product branching ratios for RO2 + HO2 reactions are then obtained from the observed product yields.
In chamber studies, branching ratios were derived from stable end-product analysis of continuously irradiated reaction mixtures mostly by means of Fourier transform infrared spectroscopy (FTIR).14–33 In some instances, organic hydroperoxides were measured by the selective reaction of hydroperoxides with peroxidase.27,32 The organic hydroperoxides are first trapped in the liquid phase inside a stripping coil attached to the simulation chamber and collected for high performance liquid chromatography (HPLC) analysis. After separation on the column, a reagent consisting of horseradish peroxidase (HRP) and p-hydroxyphenylacetic acid is added. The enzyme catalyses a reaction between the hydroperoxide and p-hydroxyphenylacetic acid, yielding a dimer whose fluorescence is measured following excitation.34 It is worth noting that the HRP-fluorescence method coupled to HPLC has been applied more regularly to investigations of ozonolysis systems, particularly for detecting H2O2 and hydroxyalkyl hydroperoxides (e.g. Hasson et al.35 and references therein). The Caltech group has developed a chemical ionisation mass spectrometry (CIMS) instrument based on clustering with CF3O− for detecting hydroperoxides.36 The technique was successfully tested against the HPLC-fluorescence method in the field and subsequently used in numerous investigations, including RO2 + HO2 chemistry.36–39 Apart from stable end-products, Winiberg et al. were the first to directly quantify OH formation from RO2 + HO2 chemistry in a chamber study through detection with the Fluorescence Assay by Gas Expansion (FAGE) method based on laser-induced fluorescence (LIF).33
In flow tube applications, peroxy radical generation was commonly initiated by flash18,19,40–52 or laser photolysis24,53–61 of VOC/Cl2/HO2-precursor or VOC/H2O2 mixtures or, less often, by molecular modulation methods12,13,62,63 or discharge flow.64,65 Time-resolved ultraviolet (UV) absorption spectroscopy has often been used to monitor peroxy radical species. Rate coefficients are finally obtained by fitting kinetic parameters to the decay rates recorded for the respective species. The UV spectra of small peroxy radicals have been critically reviewed in the literature.66 A larger set of absorption cross sections can be found in the Mainz database.67
Information on branching ratios was obtained from flow tube experiments in some cases for OH- and O3-forming pathways by means of either UV (O3),42,58,60 transient absorption (O3),59 or IR-wavelength-modulated spectroscopy (OH),60,61 CIMS (OH, O3),64,65 and LIF (OH).56,59 In contrast, there are a few examples of absolute rate coefficient determinations by simulation chamber experiments. Moortgat et al.68 obtained the rate coefficient for CH3O2 + HO2 by the modulated photolysis of acetaldehyde/air mixtures in a 44 L quartz cell. Winiberg et al.33 obtained the absolute rate coefficient for CH3C(O)O2 + HO2 by global modelling of the reaction system. Rate coefficients for all pathways of the CH3C(O)O2 + HO2 reaction were optimized individually to match the experimental data over a range of conditions. More recently, Østerstrøm et al.69 redetermined the rate coefficient for the reaction of methyl peroxy with HO2. Both CH3O2 and HO2 were detected by FAGE-LIF, and rate coefficients were obtained by applying a numerical model used to fit the temporal decay of both radical species. In contrast to common flash photolysis applications, the authors used a simulation chamber (HIRAC, University of Leeds) to build up steady-state radical concentrations by irradiating CH4/CH3OH/Cl2/air mixtures for about 5 min before monitoring the temporal decay of CH3O2 and HO2 after switching off the lamps. This approach has led to substantially lower peroxy radical concentrations than those typically found for kinetic studies in flow tube systems.
Although most kinetic studies relied on UV absorption, a number of methods for peroxy radical detection, including matrix isolation and electron spin resonance spectroscopy (MIESR),70,71 laser-induced fluorescence,72–78 peroxy radical chemical amplification (PERCA),79–91 CIMS92–116 and spectroscopic approaches such as cavity ring-down (CRD)117–125 or wavelength modulation techniques,57,60,61,126–129 were deployed in published works, increasing in the last two decades. These methods differ primarily in the level of chemical speciation of the detected radical species.
The MIESR method is an offline technique that relies on cryofocusing of radicals at 77 K prior to quantifying using electron spin resonance spectroscopy.70,71 MIESR can differentiate HO2 and CH3C(O)O2 from other RO2. The method was developed and predominantly used by the research groups at Forschungszentrum Jülich.
The original FAGE-LIF method is selective for OH but also represents an established method for HO2 quantification. In essence, HO2 is detected as OH after titrating the HO2 with NO. The approach has been expanded for the detection of RO2 in a second channel (ROxLIF) by applying a two-stage process that consists of chemical conversion of ROx (RO2 + RO + HO2 + OH) into OH.76 Discrimination between HO2 and RO2 is achieved in this set-up by modulating the chemical conversion.76 The fundamentals of the FAGE technique and possible interferences have been described in detail in several reviews.130–132 More recently, Onel et al.78 presented an adaptation of FAGE for the sensitive and selective quantification of CH3O2 by detecting the off-resonant LIF of CH3O formed upon the addition of NO.
Chemical amplification techniques rely on efficient radical recycling in the presence of NO. In the original PERCA approach, pioneered by Cantrell and Stedman,79,80 amplified NO2 formed upon the addition of NO and CO is used for peroxy radical quantification. Several techniques have been applied to quantify the resultant amplified NO2, including luminol-chemiluminescence,79–83,85,87 laser-induced fluorescence,86 cavity-enhanced absorption spectroscopy (CEAS)90 and cavity ring-down spectroscopy (CRDS).88,89 PERCA instruments consequently yield a sum parameter ROx. Wood et al.91 presented a chemical amplification technique (ECHAMP) based on ethane instead of carbon monoxide. In the approach presented by Reiner et al.,84 carbon monoxide is replaced by SO2 and the resultant H2SO4 is detected by CIMS. A separation between HO2 and RO2 is achieved by modulating the NO/O2 ratio.133
CIMS is applied to detect peroxy radicals using various reagent ions, such as H3O+(H2O)n,97,99,101–103,106–108,113,115 NH4+,114 O2+,92 NO3−,109,110 I−,94,111 Br−,112,116 O2−95,96 or SF6−.64,65,93,94,98,104,106,107 These ionisation schemes differ primarily in the resulting ions and their ability to detect specific classes of peroxy radicals. For instance, although H3O+(H2O)n, NH4+ and NO3− result in cluster ions, RO2 were detected as parent ions in O2− and SF6− mode.93–96,98,100 Exceptions to the SF6− mode are HO2 and CH3O2, which are detected as SF4O2− and FO2−, respectively.64,65,106,107
A number of studies have provided evidence that H3O+(H2O)n can detect different alkyl peroxy97,99,101–103,106–108,113,115 and bicyclic peroxy radicals.106–108 Both I− and SF6− were found to be suitable for measuring acyl peroxy radicals, particularly acetyl peroxy.93,94,98 HO2 detection has been successfully proven by clustering with I− (ref. 111) or Br−,112,116 while the detection of highly oxidized RO2 has been reported by clustering with NO3−.109,110 CRD approaches have been developed in the near-infrared and used for the detection of CH3O2,117,121,124,125 C2H5O2,117,123 hydroxyalkyl peroxy,119 acetyl peroxy,124,125 acetonyl peroxy125 and particularly HO2.118,120,122,123,125
The applicability of these techniques to pulsed photolysis/flow tube applications and chamber experiments depends on the respective detection limits, time resolution and the chemical speciation of radical species. For example, flow tube experiments require a high time resolution to enable monitoring of the temporal decay of radicals but result in radical concentrations that are typically orders of magnitude larger than those in simulation chambers.
An overview of RO2 + HO2
Based on the body of kinetic data published since those of Cox and Tyndall12,13 (Table 1), it is established that rate coefficients for the title reactions should lie in the range of (0.5–2) × 10−11 cm3 molecule−1 s−1 at room temperature. They tend to be larger in the case of β-hydroxy-substituted peroxy radicals, such as those resulting from the addition of OH to an unsaturated organic compound, while substitution with halogen atoms appears to slow down the reactivity towards HO2 for specific substitution patterns (Table 1). Overall, RO2 + HO2 reactions are thus rather rapid. The variation in the rate coefficient values spans a factor of roughly only 4–5 and is obviously significantly less pronounced than that of RO2 permutation reactions, whose rate coefficients span several orders of magnitude (e.g. ref. 135 and 136). The reader should note that in all instances, the values recommended by the International Union of Pure and Applied Chemistry (IUPAC) for room temperature rate coefficients are uncertain by a factor of 1.6–2.134 These recommended uncertainty ranges reflect either a limited number of determinations or significant variation in the available data. In the case of methyl and acetonyl peroxy, the IUPAC recommendation is identical to the values of the Jet Propulsion Laboratory (JPL).137 In contrast, although within the large error limits, the IUPAC and JPL recommendations differ by about 30% for the room temperature rate coefficient for acetyl peroxy + HO2. Nevertheless, it is well established that room temperature rate coefficients appear to increase with size in the case of the reaction of alkyl peroxy radicals with HO2. For example, the rate coefficients for both neopentyl and cyclopentyl peroxy (C5) are almost identical while being a factor of about three larger than that for methyl peroxy (Table 1).| Peroxy radical | k298 K × 1012 (cm3 s−1) | A × 1013 (cm3 s−1) | Ea/R (K) | Ref. |
|---|---|---|---|---|
| a Represents the mixture of peroxy radicals generated from decane + OH.b Represents the mixture of peroxy radicals generated from tetradecane + OH.c Peroxy radicals generated from the reaction of the parent VOC with OH. The β-hydroxyalkyl peroxy radical isomers are the predominant RO2 in the system.d Represents the mixture of HOC5H8O2 isomers formed from isoprene + OH. | ||||
| Alkyl peroxy | ||||
| CH3O2 | 5.2 | 3.8 | −780 | 134 |
| CH3CH2O2 | 6.9 | 6.4 | −710 | 134 |
| neo-C5H11O2 | 15 | 1.4 | −1380 | 18 and 55 |
| cyclo-C5H9O2 | 18 | 2.1 | −1320 | 19 |
| cyclo-C6H11O2 | 17 | 2.6 | −1250 | 19 |
| C10H21O2a | 20 | 55 | ||
| C14H29O2b | 21 | 55 | ||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
||||
| Haloalkyl peroxy | ||||
| CH2ClO2 | 5.0 | 3.2 | −820 | 48 |
| CHCl2O2 | 5.9 | 5.6 | −700 | 22 |
| CCl3O2 | 5.1 | 4.8 | −706 | 22 |
| CF2ClO2 | 3.4 | 50 | ||
| CF2ClCH2O2 | 6.8 | 50 | ||
| CFCl2CH2O2 | 9.2 | 50 | ||
| CF3CCl2O2 | 1.9 | 49 | ||
| CF3CHFO2 | 4.3 | 2.0 | −910 | 134 |
| CF3CF2O2 | 1.2 | 50 | ||
| CF2ClCF2O2 | 1.2 | 50 | ||
|
||||
| β-Aryl peroxy | ||||
| C6H5CH2O2 | 12 | 1.5 | −1310 | 134 |
|
||||
| Alkoxyalkyl peroxy | ||||
| CH3OCH2O2 | 12 | 134 | ||
|
||||
| Acyl peroxy | ||||
| CH3C(O)O2 | 20 | 1.73 | −730 | 134 |
| C6H5C(O)O2 | 38 | 110 | −364 | 58 |
|
||||
| Carbonyl-containing peroxy | ||||
| CH3C(O)CH2O2 | 9.0 | 134 | ||
|
||||
| α-Hydroxyalkyl peroxy | ||||
| HOCH2O2 | 12 | 0.056 | −2300 | 134 |
|
||||
| β-Hydroxyalkyl peroxy | ||||
| HOCH2CH2O2 | 13 | 134 | ||
| HO–(1,2-C2H2Cl2)–O2 | 6 | 55 | ||
| (CH3)2C(OH)CH2O2 | 14 | 0.56 | −1650 | 134 |
| CH3CH(OH)CH(O2)CH3 | 15 | 54 | ||
| (CH3)2C(OH)C(O2)(CH3)2 | 15 | 55 | ||
| HO–(cyclohexene)–O2c | 22 | 55 | ||
| HO–(α-pinene)–O2c | 21 | 55 | ||
| HO–(γ-terpinene)–O2c | 20 | 55 | ||
| HO–(D-limonene)–O2c | 21 | 55 | ||
|
||||
| Hydroxyalkenyl peroxy | ||||
| HO–(isoprene)–O2d | 17 | 55 | ||
This observation led to the construction of expressions relating the rate coefficient k to the number of carbon atoms.55,138,139 For example, Boyd et al.55 presented a relationship of the form k = 2.2 × 10−11 × [1 − exp(−0.26 × n)] for the room temperature rate coefficients, where n is the number of carbon atoms. Subsequently, this equation was updated by Calvert et al.140 The more recent estimation method by Jenkin et al.136 introduced a modified expression by relating the rate coefficient to a newly developed parameter nCON, which is the number of carbon, oxygen and nitrogen atoms of the organic group R without counting the peroxy radical oxygen atoms. This approach resulted in two parameterisations that differentiate between acyl peroxy and non-acyl peroxy radicals, considering the fact that the rate coefficients of acyl peroxy radicals appear to be larger than their alkyl analogues. For completeness, it is worth mentioning that two other correlation-type structure–activity relationships have been reported for the prediction of RO2 + HO2 rate coefficients. The concept by King et al.141 relies on perturbation frontier molecular orbital (PFMO) theory, and RO2 + HO2 rate coefficients are correlated with the single occupied molecular orbital (SOMO) energy of the peroxy radical. Johnson et al.142 proposed a correlation between the logarithm of k and the (calculated) ionisation potential of a structural analogue R–CHCH2, where R corresponds to the same organic substituent present in the peroxy radical.
As discussed in several studies, RO2 + HO2 reactions exhibit a negative temperature-dependence (with rather large negative activation energies, Table 1), suggesting that these reactions proceed via an intermediate complex. In contrast to RO2 + NO reactions, with NO being an electrophile, the HO2 radical acts rather as a nucleophile in RO2 + HO2 reactions according to frontier molecular orbital theory.141 In a simplified manner, these reactions are often described as occurring through a single channel yielding quantitatively a hydroperoxide although this turned out to be merely true for alkyl peroxy radicals (e.g. ref. 3 and 135). Meanwhile, product studies have shown the occurrence of other reaction channels, where the accessibility of some of the pathways depends on the peroxy radical structure.
| RO2 + HO2 → ROOH + O2 | (R5a) |
| RO2 + HO2 → ROH + O3 | (R5b) |
| RO2 + HO2 → RO + OH + O2 | (R5c) |
|
RO2 + HO2 → R–HO + OH + HO2
| (R5d) |
|
RO2 + HO2 → R–HO + H2O + O2
| (R5e) |
Interestingly, the first product study focusing on RO2 + HO2 investigated the acetyl peroxy radical and not the simpler CH3O2, as might be intuitively expected.14 In the early 1980s, information on CH3O2 + HO2 was obtained as an offshoot of investigations targeting the CH3O2 self-reaction.143,144 In fact, Calvert and co-workers had already observed methyl hydroperoxide formation from CH3O2 radicals generated by the photolysis of azomethane in the early 1960s.145,146 However, at that time, the occurrence and participation of HO2 in the reaction system were not yet known, and methyl hydroperoxide was postulated to occur through either the CH3O2 self-reaction, the reaction of CH3O2 with CH3O, or H atom abstraction from an organic species by excited CH3O2.145,146
In 1985, Niki et al. investigated the oxidation of acetaldehyde by irradiating CH3CHO/Cl2/air mixtures at atmospheric pressure and room temperature.14 In these experiments, the HO2 radical concentrations were increased by the addition of formaldehyde, and the reaction mixtures were monitored by Fourier-transform infrared (FTIR) spectroscopy. Based on the product analysis, the authors showed that the reaction of acetyl peroxy with HO2 must proceed via at least two channels yielding either peracetic acid (≈75%) or O3 (≈25%).
| CH3C(O)O2 + HO2 → CH3C(O)OOH + O2 | (R6a) |
| CH3C(O)O2 + HO2 → CH3C(O)OH + O3 | (R6b) |
As pointed out by Calvert et al.,3 channel (R6b) is intriguing in the sense that it represents one of the few known chemical reactions yielding ozone directly, even though its contribution to tropospheric ozone is, however, rather small. In essence, this early investigation has already drawn a more complex picture of RO2 + HO2 reactions.
Subsequently, these results were confirmed by Moortgat et al.42 who generated the target radicals by the flash photolysis of Cl2/CH3CHO/CH3OH/N2/O2 mixtures and detected the species by time-resolved UV spectroscopy. Based on this analysis, they obtained the rate coefficient for the target reaction (over the temperature range 253–368 K) and the branching ratio for channel (R6b). They suggested that an H-bond in the tetroxide intermediate (ROOOOH), possible only due to the carbonyl group, allows for decomposition into CH3C(O)OH + O3.42
The occurrence of a third reaction channel, producing OH, was first reported for a perfluorinated acyl peroxy radical.26 The branching ratio obtained in this study has been corrected in follow-up work.28 This observation, together with a revised infrared cross section for peracetic acid147 and thermochemical considerations, has motivated Hasson et al.27 to undertake a reinvestigation of the CH3C(O)O2 + HO2 system. Based essentially on two observations, they did prove the existence of channel (R6c):
| CH3C(O)O2 + HO2 → CH3 + CO2 + OH + O2. | (R6c) |
First, using the updated infrared cross section reduced the peracetic yield and led to a carbon balance well below 100% when considering only the well-established reaction products (peracetic acid + acetic acid). Second, the change observed for both the methyl hydroperoxide and the CO2 yield while transitioning from an RO2-dominated to an HO2-dominated system was inconsistent with a reaction scheme that considered only channels (R6a) and (R6b) for CH3C(O)O2 + HO2.
In these first investigations, the OH-forming channel was identified by closed-shell co-products. Meanwhile, OH formation is quantified by both direct and indirect measurements and, as presented in the section below, is also reported for other RO2 + HO2 reactions. One of the established methods to scavenge OH in systems where organic precursor/Cl2 mixtures are irradiated is the addition of excess benzene. The reaction of benzene with Cl atoms is extremely slow (kCl < 2 × 10−16 cm3 molecule−1 s−1),20 while benzene is reasonably reactive towards OH radicals (kOH ≈ 1 × 10−12 cm3 molecule−1 s−1)148 and consequently allows scavenging of OH without representing a significant sink for Cl atoms. Jenkin et al.29 adapted this technique for investigations on CH3C(O)O2 + HO2 and used phenol formation (from benzene + OH) to diagnose OH. It was previously found that, under these conditions, the phenol yield is about 53% ± 7%.149 The authors performed a detailed analysis showing that benzene chemistry does not affect the removal of acetyl peroxy radicals under HO2-dominated conditions. In later studies, direct measurements of OH were performed by laser-induced fluorescence or mid-infrared wavelength modulation spectroscopy, resulting in branching ratios consistent with the former indirect measurements within the assigned uncertainties.33,56,59,60
The apparent differences between the reactions of alkyl peroxy radicals and functionalized peroxy radicals with HO2 were addressed in several theoretical studies.52,150–154 Although it was initially believed that RO2 + HO2 reactions proceed via a tetroxide intermediate (ROOOOH) similar to RO2 permutation reactions, it was found that hydroperoxide formation occurs via a hydrogen-bonded pre-reactive complex (ROO··HOO) on a triplet potential energy surface (PES).150,151,153 Other reaction channels, such as the formation of OH or O3, proceed through the tetroxide intermediate on a singlet PES (Fig. 2).52,150,152 As pointed out by Vereecken and Francisco,155 the hydrogen bonding results in a submerged barrier for H migration (on the triplet surface); consequently, the accessibility of pathways other than hydroperoxide formation depends on the barriers for the decomposition of the tetroxide intermediate (formed on the singlet surface). In the case of simple alkyl peroxy radicals, these barriers cause all pathways proceeding via the ROOOOH intermediate to appear uncompetitive. However, for oxygenated peroxy radicals, such as those containing a carbonyl group, several theoretical calculations suggest lower energy barriers owing to strong hydrogen bonding in the tetroxide intermediate, hence facilitating decomposition towards other reaction products.52,150,154
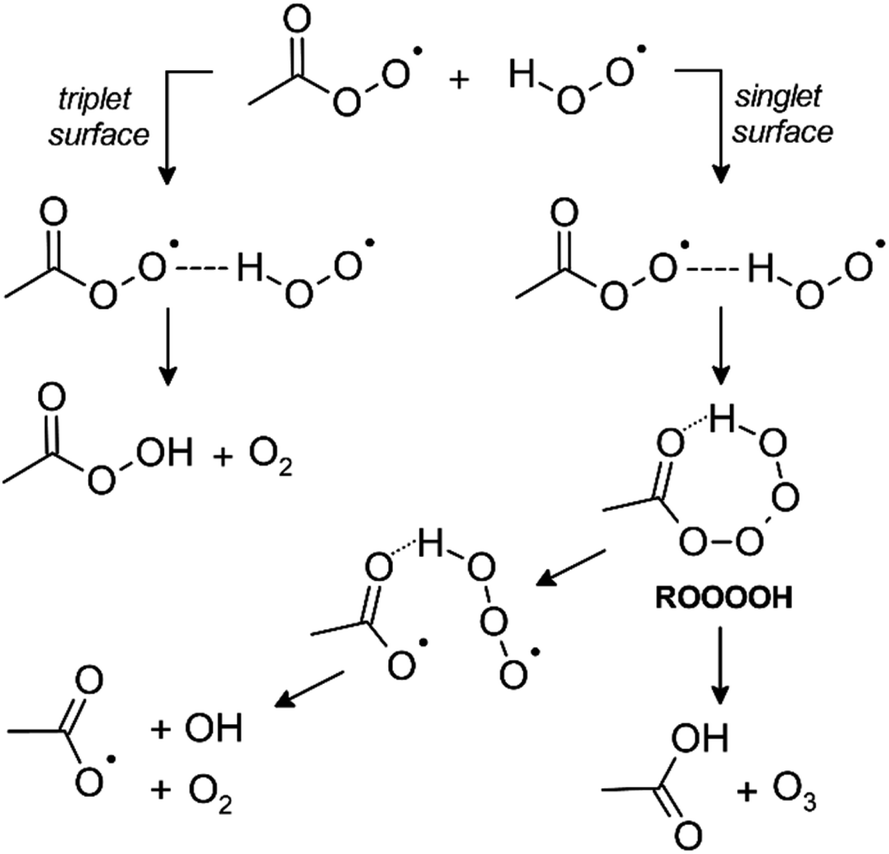 | ||
| Fig. 2 Pathways of RO2 + HO2 reactions illustrated by CH3C(O)O2 + HO2. The pathway yielding the hydroperoxide (peracid) proceeds over a triplet potential energy surface, while tetroxide formation proceeds over a singlet surface. The acetoxy radical formed in the OH channel immediately decomposes and yields CO2 + CH3O2 under atmospheric conditions. | ||
Interestingly, the number of alkyl peroxy radicals investigated in experimental product studies is rather small; so far, such studies have mainly addressed methyl15,17,156 and ethyl peroxy,16,25,27,65 followed by single investigations on neopentyl,18 cyclopentyl19 and cyclohexyl19 peroxy, and at least for the methyl peroxy radical, the occurrence of a second channel, yielding a carbonyl + H2O (R5e), has been postulated. Jenkin et al.62 reported significant HDO formation from the photolysis of Cl2/CD4/CH3OH/O2 mixtures analysed by UV and infrared diode laser spectroscopy. Although the authors presented a detailed error analysis and identified potentially interfering processes yielding HDO, they were able to explain their results only by HDO formation (about 40%) from CD3O2 + HO2. Subsequently, Wallington and Japar15 investigated CH3O2 + HO2 by FTIR spectroscopy and concluded, based on the quantification of methyl hydroperoxide, that this reaction produces only a hydroperoxide. In their preceding work, the authors presented similar results on ethyl peroxy.16 In contrast, Elrod et al.156 reported an 11% contribution of the carbonyl + H2O channel from CH3O2 + HO2 at 298 K, increasing to about 31% at 218 K.
Theoretical work on CH3O2 + HO2 has been performed by applying different computational methods.151,153,157,158 Although most of these studies found methyl hydroperoxide formation to be the only reaction channel,151,157 Drougas158 concluded that formaldehyde formation is also accessible, particularly at lower temperatures. At present, IUPAC recommends a branching ratio for HCHO formation of 0.1 ± 0.1 at 298 K, while JPL continues to recommend a 100% yield for methyl hydroperoxide.134,137 Owing to the ubiquity of methyl peroxy radicals throughout the troposphere, further studies on this subject, particularly on the temperature-dependence, would be very valuable.
Similarly, carbonyl formation of 71–100% has been reported for the reactions of halogenated alkyl peroxy radicals, such as CH3−nClnO2 (n = 1–3) or CH2FO2 with HO2.21–23 The co-product of this pathway was originally proposed to be H2O. Meanwhile, theoretical work has shown that the channel proceeds over a singlet PES via a five-membered ring intermediate with the halogen atom exhibiting a stabilizing effect that lowers energy barriers.151,152 The expected co-products are then OH + HO2. These calculations reproduce the experimental data for CH2FO2 and CH2ClO2 radicals. Interestingly, no hydroperoxide was detected in the experiments on CHCl2O2 and CCl3O2 + HO2.22 However, as pointed out by Hou et al.,152 these observations might rely on chemical activation in the nascent hydroperoxide product. Subsequent decomposition of the hydroperoxide yields carbonyl products, which will add to the carbonyl yield of the carbonyl + OH + HO2 channel (R5d).152 Clearly, halogenated alkyl peroxy radicals behave differently from alkyl peroxy in their reaction with hydroperoxy radicals.
How relevant are RO2 + HO2 reactions to tropospheric chemistry?
Based on the reports of various field campaigns, it has been established that tropospheric HO2 is of the order of 108 cm−3 with peak daytime concentrations of up to 1 × 109 cm−3 (e.g. Stone et al.132 and references therein). Since the rate coefficients for the reactions of organic peroxy radicals with HO2 are close to those with NO (≈9 × 10−12 cm3 molecule−1 s−1 at 298 K), RO2 + HO2 becomes thus competitive to RO2 + NO once NO concentrations are close to HO2. Before it was discovered that unimolecular isomerization of peroxy radicals is competitive under atmospheric conditions, RO2 + HO2 reactions were generally considered to be one of the major loss processes in so-called “low NO” environments. One should note that, although the term “low NO” is insufficient to define a chemical regime, it is used here for simplicity and represents the conditions under which the reaction of peroxy radicals with NO no longer represents the major peroxy radical loss process.H migration was found to be particularly rapid, e.g. for allylic peroxy radicals or migration of aldehydic H atoms.5 For example, unimolecular isomerisation was shown to account for about 50% of the loss of isoprene peroxy radicals at NO levels below 200 pptv.159 Overall, rate coefficients for H migration depend heavily on the peroxy radical structure and thus span several orders of magnitude in contrast to rate coefficients for reactions of organic peroxy radicals with HO2.5 As a rule of thumb, RO2 + HO2 appears competitive to unimolecular isomerisation at room temperature once H migration is <0.01 s−1.
Low concentrations of NO, which are essential for the occurrence of RO2 + HO2 reactions, are commonly found in air masses that are not impacted by anthropogenic activities; for example, those found over tropical and boreal forests. However, this traditional perspective appears to change, e.g. as a consequence of changing emission profiles, and examples of “low NO” chemistry are also reported for urban areas. Praske et al. concluded that autoxidation becomes increasingly competitive to RO2 + NO chemistry in urban and suburban areas of the United States owing to the overall reduction in NOx emissions.160 Although the authors have shown this effect mainly for peroxy radicals exhibiting large H-shift rate coefficients, the competitiveness of RO2 + HO2 reactions is also supposed to increase if NOx emissions are further declining. Newland et al. provided evidence for traditional “low NO” chemistry in the polluted air masses of central Beijing in the afternoon when high levels of photochemically produced O3 limit the availability of NO.161 Products of peroxy radical reactions with HO2, namely hydrogen peroxide (HO2 + HO2) and peracetic acid (acetyl peroxy + HO2), have also been identified in biomass burning plumes. Yokelson et al. observed initial fire emissions of both these peroxide species from biomass burning in the Yucatán; however, the authors highlighted that fast initial photochemistry may account partly for the significant amounts present in the young smoke.162 Indeed, according to current emission inventories, peracetic acid is not emitted directly from biomass burning,163 yet elevated levels of both hydrogen peroxide and peracetic acid are observed when air masses are influenced by biomass burning plumes.164 In essence, the occurrence of chemical conditions allowing for RO2 + HO2 appears to be less limited to typical pristine conditions and becomes more frequently encountered.
Role of RO2 + HO2 in the tropospheric HOx budget
The reactions of peroxy radicals with HO2 are primarily considered a sink for HOx owing to the formation of an organic hydroperoxide, which (supposedly) terminates radical-chain reactions. This perspective appears simplified since OH formation was discovered for some RO2 + HO2 reactions. After model-to-measurement discrepancies indicated that significant unidentified HOx recycling must occur in regions dominated by isoprene emissions (e.g. ref. 6, 11 and 165–167), it has been initially hypothesized that this might be the effect of peroxy radical reactions with HO2.6,11 However, following tremendous research activities during the last 15 years, there seems now to be a consensus that the efficient OH recycling emerges primarily from unimolecular isomerization of isoprene peroxy radicals (e.g. ref. 4, 159 and 168–170). Simultaneously, these radicals were shown to yield predominantly isoprene hydroperoxides (ISOPOOHs) when reacting with HO2.171 The reader should note that this is different for peroxy radicals resulting from the major isoprene oxidation products, methacrolein and methyl vinyl ketone. Both methacrolein and methyl vinyl ketone peroxy radicals synthesized by the photolysis of VOC/alcohol/Cl2/O2/N2 mixtures were shown to generate OH with yields of about 80% ± 20%.32 Praske et al.39 confirmed significant OH formation from methyl vinyl ketone + OH under HO2-dominated conditions, which consequently adds to the OH recycling by unimolecular isomerization.Table 2 summarizes OH yields reported for reactions of HO2 with different organic peroxy radicals. These were mostly determined for carbonyl-substituted, particularly acyl peroxy radicals. However, in addition to these data, some outstanding observations should be considered. Significant OH formation was reported to occur from the reaction of HO2 with the β-hydroxy peroxy radicals of α-pinene (Fig. 3), following the observation of substantial pinonaldehyde formation under HO2-dominated conditions, which is in contrast to the very low OH yield for simple β-hydroxy peroxy radicals, such as HOCH2CH2O2.37,56 One should note that, although a computational study showed that OH production appears thermodynamically accessible, at least for some of the α-pinene-derived peroxy radical isomers, a more recent experimental study performed under “low NO” conditions determined a substantially lower pinonaldehyde yield.172,173 This stresses the need to extend the studies on OH recycling to other monoterpenes. Rollins et al. argued that the carbonyl/hydroperoxide ratio observed in their experiments suggests pathways other than hydroperoxide formation for the reaction of HO2 with peroxy radicals formed in the isoprene + NO3 system; subsequently, OH formation was reported for different nitrooxy alkylperoxy radicals derived from isoprene.38,174 In addition, Birdsall and co-workers found OH regeneration in the oxidation of aromatics under HO2-dominated conditions and proposed the reaction of bicyclic peroxy radicals with HO2 to produce OH.106,107
| Peroxy radical | OH yield (%) | Reference |
|---|---|---|
| a Benzoyl peroxy.b Surrogate for OH–(methacrolein)–O2.c Surrogate for OH–(methyl vinyl ketone)–O2. | ||
| CH3C(O)O2 | 40 ± 16 | 27 |
| <10 | 52 | |
| 43 ± 10 | 29 | |
| 50 ± 20 | 56 | |
| 61 ± 9 | 59 | |
| 51 ± 12 | 33 | |
| 48 ± 9 | 60 | |
| CH3CH2C(O)O2 | 40 ± 10 | 32 |
| CH3CH2CH2C(O)O2 | 47 ± 15 | 32 |
| CF3C(O)O2 | 52 ± 5 | 28 |
| CF3CF2C(O)O2 | 50 ± 8 | 28 |
| CF3CF2CF2C(O)O2 | 47 ± 11 | 28 |
| CF3CF2CF2CF2C(O)O2 | 27 ± 18 | 28 |
| C6H5C(O)O2a | ≈20 | 56 |
| 20 ± 5 | 58 | |
| CH3C(O)CH2O2 | 67 ± 20 | 27 |
| ≈15 | 56 | |
| 15 ± 8 | 30 | |
| 25 ± 13 | 32 | |
| 30 ± 4 | 61 | |
| CH3C(O)CH(O2)CH3 | ≈20 | 56 |
| 58 ± 10 | 32 | |
| CH2ClC(CH3)(O2)CHOb | 80 ± 20 | 32 |
| CH2ClCH(O2)C(O)CH3c | 80 ± 20 | 32 |
| CH2OHCH(O2)C(O)CH3 | 48 | 39 |
| HOCH2O2 | 20 ± 5 | 29 |
| HOCH2CH2O2 | <4 | 56 |
| CH3OCH2O2 | 19 ± 8 | 31 |
| O2NO–(isoprene)–O2 | 53 | 38 |
 | ||
| Fig. 3 OH production via the reaction of α-pinene-derived peroxy radicals with HO2, as suggested by Eddingsaas et al.37 | ||
It appears that two pathways define the role of the reactions of organic peroxy radicals with HO2 in the HOx budget. One is obviously OH production in the radical-propagating channel (R5c), which seems accessible for various oxygenated peroxy radicals and hence reduces the strength of acting as a HOx sink. However, it is worth mentioning that, apart from the reaction of acyl peroxy radicals with HO2, the further chemistry of the alkoxy co-product also recycles HO2 as long as alkoxy isomerization is unimportant. The second major control for the HOx budget is organic hydroperoxide formation, as mentioned earlier. Classifying the reactions of organic peroxy radicals with HO2 as a HOx sink relies purely on the fact that organic hydroperoxides supposedly act as temporary HOx reservoirs on a significant timescale before possibly regenerating OH by photolysis. In addition, when gas-phase photochemical and photophysical processes are slow, the removal of organic hydroperoxides is likely dominated by heterogeneous losses. For example, the atmospheric lifetime of the most abundant organic hydroperoxide, methyl hydroperoxide, is on the order of 1 day; although its solubility is comparatively low, it was found in water droplets.175–177 It was already demonstrated that dissolved organic hydroperoxides are still able to act as a source of HOx and initiate photochemistry in the liquid phase.178,179 The overall picture might change when considering more complex or multi-functional hydroperoxides. It is intriguing that, although formed through unimolecular processes, rapid photolysis of hydroperoxy aldehydes (HPALDs) is one of the key routes of OH recycling in isoprene chemistry.159,180,181 In addition, Praske et al.39 observed rapid photolysis of a multifunctional hydroperoxide formed in the reaction of HO2 with a peroxy radical resulting from OH addition to methyl vinyl ketone. These results indicate that HOx recycling from the reactions of organic peroxy radicals with HO2 may also depend strongly on the identity of the organic hydroperoxide, and further studies on a larger set of species can help refine the role of RO2 + HO2 reactions in the tropospheric HOx budget.
What are the challenges?
The (mainly historical) overview presented above, in particular on the reaction of the acetyl peroxy radical with HO2, highlights that the quantification of organic hydroperoxides is not only key for understanding RO2 + HO2 reactions but certainly represents one of the major experimental challenges. Quantifying organic hydroperoxides is demanding for three reasons: the behaviour of organic hydroperoxides under experimental conditions, the accessibility of standards, and the rather limited number of analytical methods for gaseous organic hydroperoxides.It is well known that hydroperoxides decompose readily; for example, they are thermally sensitive in general. However, two observations illustrate another feature besides thermal decomposition. Conversion processes of hydroperoxides into the corresponding carbonyl were reported to occur inside analytical instrumentation, such as GC systems or PTR-MS, leading to bias in both atmospheric and experimental observations if they remain unrecognized.182 Similar observations have been made in atmospheric simulation chamber experiments. Bernhammer et al.183 investigated the formation and loss processes of isoprene oxidation products under low NOx conditions. The authors showed that isoprene hydroperoxides (ISOPOOHs) are partly converted into methyl vinyl ketone and methacrolein on the stainless steel surface of the simulation chamber. Hence, a quantitative description of isoprene hydroperoxides was achieved indirectly by considering the larger-than-expected methyl vinyl ketone and methacrolein yields observed in the oxidation experiments.
Both observations rely on the same chemical principle: metal ions, such as copper, iron, and manganese, can initiate hydroperoxide decomposition by electron-transfer (e.g. Sanchez and Myers184 and references therein):
| ROOH + Mn+ → RO + OH− + M(n+1)+ | (R7) |
| ROOH + M(n+1)+ → RO2 + H+ + Mn+ | (R8) |
If both reactions are accessible, that is both valence states can react as is the case for most transition metals, this results in the catalytic destruction of the hydroperoxide, which affects the experimental results.
The accessibility and usage of hydroperoxide standards are twofold challenging. First, it is obvious that special care must be taken in the preparation and handling of pure hydroperoxide samples, owing to the explosive nature of organic hydroperoxides, at least for hydroperoxides with a high peroxide oxygen content, such as methyl and ethyl hydroperoxide. An option to overcome this issue is the handling of diluted solutions, which, to a certain extent, limits their application to analytical methods quantifying hydroperoxides in the liquid phase. Although various synthesis routes exist, e.g. methylation of H2O2 by dimethyl sulphate for the preparation of methyl hydroperoxide,185 the reaction of alkyl methanesulfonates (mesylates) with H2O2 for larger and branched alkyl hydroperoxides,186,187 and the reaction of tertiary alcohols with H2O2 for tertiary alkyl hydroperoxides, the preparation of higher functionalized hydroperoxides is demanding. However, it should be mentioned that research efforts in the last decade have made accessible, for example, the preparation of isoprene hydroperoxides.182,188,189
Additionally, achieving a quantitative conversion of a precursor compound into the desired hydroperoxide is not always possible. For example, peracids are easily synthesized by mixing concentrated H2O2 solutions with the corresponding acid often in the presence of an acidic catalyst (commonly sulphuric acid).190 In the case of peracetic acid, this results in equilibrium mixtures containing a peracid fraction on the order of 40%. The determination of infrared absorption cross sections based on the vaporization of these samples requires scrupulously accounting for other volatilized components, such as H2O, acetic acid and the acetic acid dimer, which hence represents a potential source of error. As pointed out by Orlando et al.,147 this might have been one of the reasons for the low absorption cross section reported for peracetic acid by Crawford et al.24 FTIR spectroscopy is one of the workhorses for analysing gaseous reaction systems in atmospheric simulation chamber experiments and has consequently been used regularly for the quantification of organic hydroperoxides. However, as discussed above, the reliability of the retrieved information depends on well-established absorption cross sections and hence on well-characterized procedures for vaporizing samples with known organic hydroperoxide content.
As presented in the overview section, the product study of Hasson et al.27 was the first to use the revised absorption cross section for peracetic acid. However, the authors validated the FTIR data against the HPLC-fluorescence method. In separate experiments, by preparing gas phase mixtures of peracetic acid in the chamber, Hasson et al.27 showed that FTIR and HPLC analyses agree within a factor of about 1.2. Based on the reported product yields in different experiments, both methods agreed on average within a factor of 1.2, 1.1 and 1.5 for methyl hydroperoxide, ethyl hydroperoxide and peracetic acid, respectively, in the oxidation experiments. There is no doubt about these experimental results, yet these data highlight that the quantification of organic hydroperoxides remains a challenging task. Hasson et al.35 pointed out that hydroperoxide yields obtained from different ozonolysis systems (where RO2 + HO2 reactions occur) in several studies vary considerably although both experimental methodologies and analytical instrumentation were similar. The authors presumed that this might be due to the complexity of applying the HPLC-fluorescence technique to simulation chamber experiments. In addition, the selectivity of horseradish peroxidase (HRP) limits the detectability of organic hydroperoxides. Although linear n-alkyl hydroperoxides up to C18 were found to react with HRP with similar efficiencies, secondary and tertiary hydroperoxides exhibit either significantly lower responses or are undetectable.191,192
Overall, there is a need for additional analytical methods preferably preserving the chemical integrity of the sample. Chemical ionisation mass spectrometry (CIMS), which is increasingly used in atmospheric science, is certainly one of these methods. Generally, the number of ionisation schemes used in CIMS instruments is increasing; particularly, reagent ions, such as NH4+ or I−, are promising. It is realistic to estimate that a large body of work will be necessary to reduce overall uncertainties and that the lack of hydroperoxide standards might remain a limiting factor.
Another major experimental challenge relies on the procedures used for peroxy radical generation and the subsequent radical chemistry. Most of the data on reaction products and branching ratios were obtained from atmospheric simulation chamber experiments by either Cl atom reactions of an organic precursor or VOC + OH oxidation experiments performed under HO2-dominated conditions. It is obvious that the generation of (almost exclusively) a single peroxy radical in the initial step is limited in these approaches to small organics such as methane (for methyl peroxy), ethane (for ethyl peroxy), acetaldehyde (for acetyl peroxy), acetone (for acetonyl peroxy) or unsubstituted cycloalkanes (for cycloalkyl peroxy). For larger molecules, the reaction with Cl, and even OH, concurrently generates several different peroxy radicals, initiating a series of RO2 permutation reactions. As illustrated by the reaction sequence of the acetyl peroxy radical (Fig. 4), even the generation of a single RO2 species results in a cascade of reactions producing additional RO2 species. Moreover, this is complicated by the fact that further oxidation of closed-shell first-generation products adds to the radical pool by generating peroxy radicals and possibly OH. Advances in the understanding of site-specific chemistry (e.g. ref. 193) and refined structure–activity relationships (e.g. ref. 194) will help at least to determine the relative ratio of peroxy radicals initially formed in oxidation systems more accurately; however, as pointed out by Ervens et al.,195 further experimental work on this subject is highly required. In addition, there is a growing number of reports on the generation of specific peroxy radicals from photolytic precursors, such as iodine-containing organic compounds.63,196–200
 | ||
| Fig. 4 Reaction sequence for CH3C(O)O2 in the absence of NO. The co-product O2 is omitted for readability reasons. Branching ratios (in bold) represent the current IUPAC recommendations.134 | ||
The addition of an HO2 precursor aims at shifting the reaction system towards conditions where the reaction of RO2 with HO2 is favoured, and in the limiting case, RO2 reacts almost exclusively with HO2. However, both the experimental set-up and the chemistry often impede the attainment of this rather ideal case. The amount of HO2 precursor is often limited by the deployed analytical instrumentation; for example, high concentrations of methanol complicate the analysis of FTIR spectra or might deplete the reagent ions in CIMS instruments. In addition, when increasing the HO2 concentration, the self-reaction and the reaction with aldehydes, particularly with formaldehyde, become significant loss processes for HO2.
The overall consequence is that the kinetic information on RO2 + HO2 reactions is mostly obtained from the modelling of a complex chemical reaction system. This not only requires accurate analytics data but also information on rate coefficients and branching ratios of all involved reactions. Missing or incomplete data add uncertainty to the branching ratio determination for the target reaction. For instance, acetic acid is formed from CH3C(O)O2 + HO2, as discussed above, but it also appears to arise from the cross-reaction of acetyl peroxy with methyl peroxy (Fig. 4). According to the currently recommended values, this channel accounts for about 10% of the CH3C(O)O2 + CH3O2 reaction.134 However, the relative error is 100%. Recently, Assali and Fittschen124 reported a rate coefficient for the cross-reaction, which is a factor of two larger than the current recommendations. Simultaneously, they obtained a lower contribution from the radical-propagating channel.
The above-discussed challenges, partly or entirely, also apply to the absolute rate coefficient determination work. Most of the kinetic data presented in Table 1 were obtained from flash photolysis or pulsed laser photolysis experiments combined with UV absorption spectroscopy in the 1990s and early 2000s. Accordingly, accurate absorption cross sections of the radical species present in the system and a successful separation of overlapping absorption bands are crucial for the determination of rate coefficients. Since rate coefficients are finally obtained by fitting kinetic parameters to the decay rates recorded for the respective species, the uncertainty of the rate coefficient depends on the accuracy of the available kinetic mechanism. For example, by trend smaller rate coefficients and stronger temperature dependence were reported for CH3C(O)O2 + HO2 before the radical-propagating channel was discovered and included in the analysis.24,33,42,52,56,59,60
Some of these issues were minimized by Boyd et al.55 by determining the rate coefficients under a large excess of HO2. The authors employed nearly pseudo-first order conditions using an excess of H2O2, hence suppressing RO2 self-reactions and subsequent chemistry. However, this approach is limited to reaction systems without significant radical recycling. Østerstrøm et al.69 reduced the uncertainties in the CH3O2 + HO2 rate coefficient significantly by detecting methyl peroxy and HO2 using the FAGE-LIF method and applying an updated kinetic model, which uses a revised rate coefficient for the CH3O2 self-reaction they have obtained in preceding work.201 The authors determined temperature-dependent rate coefficients of about 15% lower than current recommendations.
The progress made in the detection of peroxy radicals and possibly the rethinking of experimental designs will help reduce uncertainties. For example, Zuraski et al.61 investigated the kinetics and OH production from the acetonyl peroxy + HO2 reaction by monitoring the three radical species independently using a combination of IR and UV measurements. However, further advances in the detection of radicals, particularly with respect to chemical speciation, are still desirable.
Gaps in knowledge
Substantial progress has been made over the last approximately 25 years in understanding the reactions of organic peroxy radicals with HO2, particularly with respect to the occurrence of pathways other than organic hydroperoxide formation. However, there remain several areas that require further research as a consequence of either a lack of data or significant uncertainties and divergence in existing data:(1) Rate coefficients
Currently, there are few recommended values (assigned with large uncertainty ranges) for the rate coefficients of the title reactions, which are insufficient to cover the variety of peroxy radicals that are potentially produced in the troposphere.(2) OH formation
As evident from Table 2, significant scattering is found for the OH yield of some RO2 + HO2 reactions, and additional measurements are required to reduce overall uncertainties. Thus far, most data on acyl peroxy radicals have been reported. Systematic investigations of a broader range of peroxy radicals would help draw a more comprehensive picture of OH formation from RO2 + HO2 and, more specifically, assess the influence of functional groups and structures on OH yield.(3) β-Hydroxy peroxy radicals
The dataset on β-hydroxy peroxy radicals is scarce. Given the importance of this class of peroxy radicals in the atmosphere (they are formed following the addition of OH to nearly all unsaturated VOC) and the apparent discrepancies reported in the literature (e.g. ethene-derived vs. α-pinene-derived peroxy radicals), there is a need for systematic investigations on β-hydroxy peroxy radicals with respect to structural variation.(4) Multifunctional peroxy radicals
Investigations reported in the literature are limited to peroxy radicals containing very few different functional groups. Given the partial focus on acyl peroxy radicals, additional systematic studies on the different functional groups of oxygenated peroxy radicals would be valuable for refining our understanding of RO2 + HO2 reactions. In particular, it is entirely unknown at present how reactivity and product branching ratios are affected by a combination of different functionalities.(5) Temperature-dependence
Although the temperature-dependence was reported for the absolute rate coefficients of a range of peroxy radicals (Table 1), the temperature-dependence of product branching ratios, in particular for oxygenated peroxy radicals, is not well established. Dedicated temperature-dependent product studies were, to the best of the author's knowledge, reported only for ethyl peroxy25 and acetyl peroxy.60 There is a clear need for further research to assess the relative change in branching ratios with temperature.(6) Hydroperoxides
As discussed above, the atmospheric fate of an organic hydroperoxide is crucial for the HOx budget related to a specific RO2 + HO2 reaction. Accordingly, organic hydroperoxides may be regarded as an additional area of research, particularly in the case of multifunctional hydroperoxides, which are potentially more sensitive to photolysis.(7) Theoretical investigations
Few studies have applied computational methods to RO2 + HO2 reactions. As with experimental data, further theoretical studies, particularly on the accessibility of pathways other than hydroperoxide formation for different peroxy radical structures, are desirable.Conclusions
The present work highlights that despite the progress made in understanding RO2 + HO2 reactions, there are several areas requiring further research activities. Although H-shift isomerization is now accepted to represent a significant peroxy radical loss process for certain peroxy radical classes, it might be less competitive for others. Thus, under pristine-like conditions, the reaction with HO2 still represents one of the major loss processes for a large range of peroxy radicals formed in the atmosphere. This, together with the observed increased incidence of low NOx tropospheric conditions, indicates that the systematic scrutiny of the title reaction is essential to draw a comprehensive image of the peroxy radical chemistry.Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this perspective.Conflicts of interest
There are no conflicts to declare.Acknowledgements
NI gratefully acknowledges Iulia Patroescu-Klotz for proofreading this manuscript.References
- A. Guenther, C. N. Hewitt, D. Erickson, R. Fall, C. Geron, T. Graedel, P. Harley, L. Klinger, M. Lerdau, W. A. McKay, T. Pierce, B. Scholes, R. Steinbrecher, R. Tallamraju, J. Taylor and P. Zimmerman, A global model of natural volatile organic compound emissions, J. Geophys. Res., 1995, 100, 8873–8892 CrossRef CAS.
- R. Atkinson and J. Arey, Atmospheric Degradation of Volatile Organic Compounds, Chem. Rev., 2003, 103, 4605–4638 CrossRef CAS PubMed.
- J. G. Calvert, J. J. Orlando, W. R. Stockwell and T. J. Wallington, The Mechanisms of Reactions Influencing Atmospheric Ozone, Oxford University Press, Oxford, 2015 Search PubMed.
- H. Fuchs, A. Hofzumahaus, F. Rohrer, B. Bohn, T. Brauers, H.-P. Dorn, R. Häseler, F. Holland, M. Kaminski, X. Li, S. Nehr, R. Tillmann, R. Wegener and A. Wahner, Experimental evidence for efficient hydroxyl radical regeneration in isoprene oxidation, Nat. Geosci., 2013, 6, 1023–1026 CrossRef CAS.
- L. Vereecken and B. Nozière, H migration in peroxy radicals under atmospheric conditions, Atmos. Chem. Phys., 2020, 20, 7429–7458 CrossRef CAS.
- J. Lelieveld, T. M. Butler, J. N. Crowley, T. J. Dillon, H. Fischer, L. Ganzeveld, H. Harder, M. G. Lawrence, M. Martinez, D. Taraborrelli and J. Williams, Atmospheric oxidation capacity sustained by a tropical forest, Nature, 2008, 452, 737–740 CrossRef CAS PubMed.
- F. Bianchi, T. Kurtén, M. Riva, C. Mohr, M. P. Rissanen, P. Roldin, T. Berndt, J. D. Crounse, P. O. Wennberg, T. F. Mentel, J. Wildt, H. Junninen, T. Jokinen, M. Kulmala, D. R. Worsnop, J. A. Thornton, N. Donahue, H. G. Kjaergaard and M. Ehn, Highly Oxygenated Organic Molecules (HOM) from Gas-Phase Autoxidation Involving Peroxy Radicals: A Key Contributor to Atmospheric Aerosol, Chem. Rev., 2019, 119, 3472–3509 CrossRef CAS PubMed.
- M. Ehn, J. A. Thornton, E. Kleist, M. Sipilä, H. Junninen, I. Pullinen, M. Springer, F. Rubach, R. Tillmann, B. Lee, F. Lopez-Hilfiker, S. Andres, I.-H. Acir, M. Rissanen, T. Jokinen, S. Schobesberger, J. Kangasluoma, J. Kontkanen, T. Nieminen, T. Kurtén, L. B. Nielsen, S. Jørgensen, H. G. Kjaergaard, M. Canagaratna, M. Dal Maso, T. Berndt, T. Petäjä, A. Wahner, V.-M. Kerminen, M. Kulmala, D. R. Worsnop, J. Wildt and T. F. Mentel, A large source of low-volatility secondary organic aerosol, Nature, 2014, 506, 476–479 CrossRef CAS PubMed.
- T. F. Mentel, M. Springer, M. Ehn, E. Kleist, I. Pullinen, T. Kurtén, M. Rissanen, A. Wahner and J. Wildt, Formation of highly oxidized multifunctional compounds: autoxidation of peroxy radicals formed in the ozonolysis of alkenes – deduced from structure–product relationships, Atmos. Chem. Phys., 2015, 15, 6745–6765 CrossRef CAS.
- T. Berndt, W. Scholz, B. Mentler, L. Fischer, H. Herrmann, M. Kulmala and A. Hansel, Accretion Product Formation from Self- and Cross-Reactions of RO2 Radicals in the Atmosphere, Angew. Chem., Int. Ed., 2018, 57, 3820–3824 CrossRef CAS PubMed.
- J. A. Thornton, P. J. Wooldridge, R. C. Cohen, M. Martinez, H. Harder, W. H. Brune, E. J. Williams, J. M. Roberts, F. C. Fehsenfeld, S. R. Hall, R. E. Shetter, B. P. Wert and A. Fried, Ozone production rates as a function of NOx abundances and HOx productions rates in the Nashville urban plume, J. Geophys. Res., 2002, 107, 4146 Search PubMed.
- R. A. Cox and G. S. Tyndall, Rate constants for the reactions of CH3O2 in the gas phase, Chem. Phys. Lett., 1979, 65, 357–360 CrossRef CAS.
- R. A. Cox and G. S. Tyndall, Rate constants for the reactions of CH3O2 with HO2, NO and NO2 using molecular modulation spectrometry, J. Chem. Soc., Faraday Trans. 2, 1980, 76, 153–163 RSC.
- H. Niki, P. D. Maker, C. M. Savage and L. P. Breitenbach, FTIR Study of the Kinetics and Mechanism for Cl-Atom-Initiated Reactions of Acetaldehyde, J. Phys. Chem., 1985, 89, 588–591 CrossRef CAS.
- T. J. Wallington and S. M. Japar, Reaction of CH3O2+ HO2 in air at 295 K: a product study, Chem. Phys. Lett., 1990, 167, 513–518 CrossRef CAS.
- T. J. Wallington and S. M. Japar, FTIR product study of the reaction of C2H5O2 + HO2 in air at 295 K, Chem. Phys. Lett., 1990, 166, 495–499 CrossRef CAS.
- T. J. Wallington, Fourier-transform Infrared Product Study of the Reaction of CH3O2+ HO2 over the Pressure Range 15–700 Torr at 295 K, J. Chem. Soc., Faraday Trans., 1991, 87, 2379–2382 RSC.
- D. M. Rowley, R. Lesclaux, P. D. Lightfoot, K. Hughes, M. D. Hurley, S. Rudy and T. J. Wallington, A Kinetic and Mechanistic Study of the Reaction of Neopentylperoxy Radicals with HO2, J. Phys. Chem., 1992, 96, 7043–7048 CrossRef CAS.
- D. M. Rowley, R. Lesclaux, P. D. Lightfoot, B. Nozière, T. J. Wallington and M. D. Hurly, Kinetic and Mechanistic Studies of the Reactions of Cyclopentylperoxy and Cyclohexylperoxy Radicals with HO2, J. Phys. Chem., 1992, 96, 4889–4894 CrossRef CAS.
- B. Nozière, R. Lesclaux, M. D. Hurley, M. A. Dearth and T. J. Wallington, A Kinetic and Mechanistic Study of the Self-Reaction and Reaction with HO2 of the Benzylperoxy Radical, J. Phys. Chem., 1994, 98, 2864–2873 CrossRef.
- T. J. Wallington, M. D. Hurley, W. F. Schneider, J. Sehested and O. J. Nielsen, Mechanistic study of the gas-phase reaction of CH2FO2 radicals with HO2, Chem. Phys. Lett., 1994, 218, 34–42 CrossRef CAS.
- V. Catoire, R. Lesclaux, W. F. Schneider and T. J. Wallington, Kinetics and Mechanisms of the Self-Reactions of CCl3O2 and CHCl2O2 Radicals and Their Reactions with HO2, J. Phys. Chem., 1996, 100, 14356–14371 CrossRef CAS.
- T. J. Wallington, M. D. Hurley and W. F. Schneider, Atmospheric chemistry of CH3Cl: mechanistic study of the reaction of CH2ClO2 radicals with HO2, Chem. Phys. Lett., 1996, 251, 164–173 CrossRef CAS.
- M. A. Crawford, T. J. Wallington, J. J. Szente, M. M. Maricq and J. S Francisco, Kinetics and Mechanism of the Acetylperoxy + HO2 Reaction, J. Phys. Chem. A, 1999, 103, 365–378 CrossRef CAS.
- M. Spittler, I. Barnes, K. H. Becker and T. J. Wallington, Product study of the C2H5O2 + HO2 reaction in 760 Torr of air at 284–312 K, Chem. Phys. Lett., 2000, 321, 57–61 CrossRef CAS.
- M. P. S. Andersen, M. D. Hurley, T. J. Wallington, J. C. Ball, J. W. Martin, D. A. Ellis and S. A. Mabury, Atmospheric chemistry of C2F5CHO: mechanism of the C2F5C(O)O2 + HO2 reaction, Chem. Phys. Lett., 2003, 381, 14–21 CrossRef.
- A. S. Hasson, G. S. Tyndall and J. J. Orlando, A Product Yield Study of the Reaction of HO2 Radicals with Ethyl Peroxy (C2H5O2), Acetyl Peroxy (CH3C(O)O2), and Acetonyl Peroxy (CH3C(O)CH2O2) Radicals, J. Phys. Chem. A, 2004, 108, 5979–5989 CrossRef CAS.
- M. D. Hurley, J. C. Ball, T. J. Wallington, M. P. S. Andersen, O. J. Nielsen, D. A. Ellis, J. W. Martin and S. A. Mabury, Atmospheric Chemistry of n-CxF2x+1CHO (x = 1, 2, 3, 4): Fate of CxF2x+1C(O) Radicals, J. Phys. Chem. A, 2006, 110, 12443–12447 CrossRef CAS PubMed.
- M. E. Jenkin, M. D. Hurley and T. J. Wallington, Investigation of the radical product channel of the CH3C(O)O2 + HO2 reaction in the gas phase, Phys. Chem. Chem. Phys., 2007, 9, 3149–3162 RSC.
- M. E. Jenkin, M. D. Hurley and T. J. Wallington, Investigation of the radical product channel of the CH3C(O)CH2O2 + HO2 reaction in the gas phase, Phys. Chem. Chem. Phys., 2008, 10, 4274–4280 RSC.
- M. E. Jenkin, M. D. Hurley and T. J. Wallington, Investigation of the radical product channel of the CH3OCH2O2 + HO2 reaction in the gas phase, J. Phys. Chem. A, 2010, 114, 408–416 CrossRef CAS PubMed.
- A. L. Hasson, G. S. Tyndall, J. J. Orlando, S. Singh, S. Q. Hernandez, S. Campbell and Y. Ibarra, Branching Ratios for the Reaction of Selected Carbonyl-Containing Peroxy Radicals with Hydroperoxy Radicals, J. Phys. Chem. A, 2012, 116, 6264–6281 CrossRef CAS PubMed.
- F. A. F. Winiberg, T. J. Dillon, S. C. Orr, C. B. M. Groß, I. Bejan, C. A. Brumby, M. J. Evans, S. C. Smith, D. E. Heard and P. W. Seakins, Direct measurements of OH and other product yields from the HO2 + CH3C(O)O2 reaction, Atmos. Chem. Phys., 2016, 16, 4023–4042 CrossRef CAS.
- G. G. Guilbault, P. J. Brignac Jr and M. Juneau, New Substrates for the Fluorometric Determination of Oxidative Enzymes, Anal. Chem., 1968, 40, 1256–1263 CrossRef CAS PubMed.
- A. S. Hasson, G. Orzechowska and S. E. Paulson, Production of stabilized Criegee intermediates and peroxides in the gas phase ozonolysis of alkenes 1. ethene, trans-2-butene, and 2,3-dimethyl-2-butene, J. Geophys. Res., 2001, 106, 34131–34142 CrossRef CAS.
- J. D. Crounse, K. A. McKinney, A. J. Kwan and P. O. Wennberg, Measurement of Gas-Phase Hydroperoxides by Chemical Ionization Mass Spectrometry, Anal. Chem., 2006, 78, 6726–6732 CrossRef CAS PubMed.
- N. C. Eddingsaas, C. L. Loza, L. D. Yee, J. H. Seinfeld and P. O. Wennberg, α-pinene photooxidation under controlled chemical conditions – part 1: gas-phase composition in low- and high-NOx environments, Atmos. Chem. Phys., 2012, 12, 6489–6504 CrossRef CAS.
- R. H. Schwantes, A. P. Teng, T. B. Nguyen, M. M. Coggon, J. D. Crounse, J. M. St. Clair, X. Zhang, K. A. Schilling, J. H. Seinfeld and P. O. Wennberg, Isoprene NO3 Oxidation Products from the RO2 + HO2 Pathway, J. Phys. Chem. A, 2015, 119, 10158–10171 CrossRef CAS PubMed.
- E. Praske, J. D. Crounse, K. H. Bates, T. Kurtén, H. G. Kjaergaard and P. O. Wennberg, Atmospheric Fate of Methyl Vinyl Ketone: Peroxy Radical Reactions with NO and HO2, J. Phys. Chem. A, 2015, 119, 4562–4572 CrossRef CAS PubMed.
- P. Dagaut, T. J. Wallington and M. J. Kurylo, Temperature Dependence of the Rate Constant for the HO2 + CH3O2 Gas-Phase Reaction, J. Phys. Chem., 1988, 92, 3833–3836 CrossRef CAS.
- P. Dagaut, T. J. Wallington and M. J. Kurylo, Flash Photolysis Kinetic Absorption Spectroscopy Study of the Gas-Phase Reaction HO2 + C2H5O2 over the Temperature Range 228 – 380 K, J. Phys. Chem., 1988, 92, 3836–3839 CrossRef CAS.
- G. K. Moortgat, B. Veyret and R. Lesclaux, Kinetics of the reaction of HO2 with CH3C(O)O2 in the temperature range 253-368 K, Chem. Phys. Lett., 1989, 160, 443–447 CrossRef CAS.
- B. Veyret, R. Lesclaux, M.-T. Rayez, J.-C. Rayez, R. A. Cox and G. K. Moortgat, Kinetics and Mechanism if the Photooxidation of Formaldehyde. 1. Flash Photolysis Study, J. Phys. Chem., 1989, 93, 2368–2374 CrossRef CAS.
- P. D. Lightfoot, B. Veyret and R. Lesclaux, Flash Photolysis Study of the CH3O2 + HO2 Reaction between 248 and 573 K, J. Phys. Chem., 1990, 94, 708–714 CrossRef CAS.
- F. F. Fenter, V. Catoire, R. Lesclaux and P. D. Lightfoot, The Ethylperoxy Radical: Its Ultraviolet Spectrum, Self-Reaction, and Reaction with HO2, Each Studied as a Function of Temperature, J. Phys. Chem., 1993, 97, 3530–3538 CrossRef CAS.
- I. Bridier, B. Veyret, R. Lesclaux and M. E. Jenkin, Flash Photolysis Study of the UV Spectrum and Kinetics of Reactions of the Acetonylperoxy Radical, J. Chem. Soc., Faraday Trans., 1993, 89, 2993–2997 RSC.
- M. M. Maricq and J. J. Szente, A Kinetic Study of the Reaction between Ethylperoxy and HO2, J. Phys. Chem., 1994, 98, 2078–2082 CrossRef CAS.
- V. Catoire, R. Lesclaux, P. D. Lightfoot and M.-T. Rayez, Kinetic Study of the Reactions of CH2ClO2 with Itself and with HO2, and Theoretical Study of the Reactions of CH2ClO, between 251 and 600 K, J. Phys. Chem., 1994, 98, 2889–2898 CrossRef CAS.
- G. D. Hayman, M. E. Jenkin, T. P. Murrels and C. E. Johnson, Tropospheric degradation chemistry of HCFC-123 (CF3CHCl2): a proposed replacement chlorofluorocarbon, Atmos. Environ., 1994, 28, 421–437 CrossRef CAS.
- G. D. Hayman and F. Battin-Leclerc, Kinetic of the Reactions of the HO2 Radical with Peroxyl Radicals derived from Hydrochlorofluorocarbons and Hydrofluorocarbons, J. Chem. Soc., Faraday Trans., 1995, 91, 1313–1323 RSC.
- A. Tomas, E. Villenave and R. Lesclaux, Reactions of the HO2 Radical with CH3CHO and CH3C(O)O2 in the Gas Phase, J. Phys. Chem., 2001, 105, 3505–3514 CrossRef CAS.
- J.-P. Le Crâne, M.-T. Rayez, J.-C. Rayez and E. Villenave, A reinvestigation of the kinetics and the mechanism of the CH3C(O)O2 + HO2 reaction using both experimental and theoretical approaches, Phys. Chem. Chem. Phys., 2006, 8, 2163–2171 RSC.
- T. P. Murrells, M. E. Jenkin, S. J. Shalliker and G. D. Hayman, Laser Flash Photolysis of the UV Spectrum and Kinetics of Reactions of HOCH2CH2O2 Radicals, J. Chem. Soc., Faraday Trans., 1991, 87, 2351–2360 RSC.
- M. E. Jenkin and G. D. Hayman, Kinetics of Reactions of Primary, Secondary and Tertiary β–Hydroxy Peroxyl Radicals, J. Chem. Soc., Faraday Trans., 1995, 91, 1911–1922 RSC.
- A. A. Boyd, P.-M. Flaud, N. Daugey and R. Lesclaux, Rate Constants for RO2 + HO2 Reactions Measured under a Large Excess of HO2, J. Phys. Chem. A, 2003, 107, 818–821 CrossRef CAS.
- T. J. Dillon and J. N. Crowley, Direct detection of OH formation in the reactions of HO2 with CH3C(O)O2 and other substituted peroxy radicals, Atmos. Chem. Phys., 2008, 8, 4877–4889 CrossRef CAS.
- A. C. Noell, L. S. Alconel, D. J. Robichaud, M. Okumura and S. P. Sander, Near-Infrared Kinetic Spectroscopy of the HO2 and C2H5O2 Self-Reactions and Cross Reactions, J. Phys. Chem. A, 2010, 114, 6983–6995 CrossRef CAS PubMed.
- E. Roth, A. Chakir and A. Ferhati, Study of a Benzoylperoxy Radical in the Gas Phase: Ultraviolet Spectrum and C6H5C(O)O2 + HO2 Reaction between 295 and 357 K, J. Phys. Chem. A, 2010, 114, 10367–10379 CrossRef CAS PubMed.
- C. B. M. Groß, T. J. Dillon, G. Schuster, J. Lelieveld and J. N. Crowley, Direct Kinetic Study of OH and O3 Formation in the Reaction of CH3C(O)O2 with HO2, J. Phys. Chem. A, 2014, 118, 974–985 CrossRef PubMed.
- A. O. Hui, M. Fradet, M. Okumura and S. P. Sander, Temperature Dependence Study of the Kinetics and Product Yields of the HO2 + CH3C(O)O2 Reaction by Direct Detection of OH and HO2 Radicals Using 2f-IR Wavelength Modulation Spectroscopy, J. Phys. Chem. A, 2019, 123, 3655–3671 CrossRef CAS PubMed.
- K. Zuraski, A. O. Hui, F. J. Grieman, E. Darby, K. H. Møller, F. A. F. Winiberg, C. J. Percival, M. D. Smarte, M. Okumura, H. G. Kjaergaard and S. P. Sander, Acetonyl Peroxy and Hydro Peroxy Self- and Cross-Reactions: Kinetics, Mechanism, and Chaperone Enhancement from the Perspective of the Hydroxyl Radical Product, J. Phys. Chem. A, 2020, 124, 8128–8143 CrossRef CAS PubMed.
- M. E. Jenkin, R. A. Cox, G. D. Hayman and L. J. Whyte, Kinetic study of the reactions CH3O2+ CH3O2 and CH3O2+ HO2 using molecular modulation spectroscopy, J. Chem. Soc., Faraday Trans. 2, 1988, 84, 913–930 RSC.
- M. E. Jenkin and R. A. Cox, Kinetics of Reactions of CH3O2 and HOCH2CH2O2 Radicals Produced by the Photolysis of Iodomethane and 2-Iodoethanol, J. Phys. Chem., 1991, 95, 3229–3237 CrossRef CAS.
- M. T. Raventós-Duran, M. McGillen, C. J. Percival, P. D. Hamer and D. E. Shallcross, Kinetics of the CH3O2 + HO2 Reaction: A Temperature and Pressure Dependence Study Using Chemical Ionization Mass Spectrometry, Int. J. Chem. Kinet., 2007, 39, 571–579 CrossRef.
- M. T. Raventós-Duran, C. J. Percival, M. R. McGillen, P. D. Hamer and D. E. Shallcross, Kinetics and branching ratio studies of the reaction of C2H5O2 + HO2 using chemical ionisation mass spectrometry, Phys. Chem. Chem. Phys., 2007, 9, 4338–4348 RSC.
- G. S. Tyndall, R. A. Cox, C. Granier, R. Lesclaux, G. K. Moortgat, M. J. Pilling, A. R. Ravishankara and T. J. Wallington, Atmospheric chemistry of small organic peroxy radicals, J. Geophys. Res., 2001, 106, 12157–12182 CrossRef CAS.
- H. Keller-Rudek, G. K. Moortgat, R. Sander and R. Sörensen, The MPI-Mainz UV/VIS spectral atlas of gaseous molecules of atmospheric interest, Earth Syst. Sci. Data, 2013, 5, 365–373 CrossRef.
- G. K. Moortgat, R. A. Cox, G. Schuster, J. P. Burrows and G. S. Tyndall, Peroxy Radical Reactions in the Photo-oxidation of CH3CHO, J. Chem. Soc., Faraday Trans. 2, 1989, 85, 809–829 RSC.
- F. F. Østerstrøm, L. Onel, A. Brennan, J. M. Parr, L. K. Whalley, P. W. Seakins and D. E. Heard, Kinetics of the Cross-reaction of CH3O2 + HO2 in the Highly Instrumented Reactor for Atmospheric Chemistry, Int. J. Chem. Kinet., 2023, 55, 489–500 CrossRef.
- D. Mihelcic, P. Müsgen and D. H. Ehhalt, An Improved Method of Measuring Tropospheric NO2 and RO2 by Matrix Isolation and Electron Spin Resonance, J. Atmos. Chem., 1985, 3, 341–361 CrossRef CAS.
- D. Mihelcic, A. Volz-Thomas, H. W. Pätz, D. Kley and M. Mihelcic, Numerical Analysis of ESR Spectra from Atmospheric Samples, J. Atmos. Chem., 1990, 11, 271–297 CrossRef CAS.
- T. M. Hard, R. J. O'Brien, C. Y. Chan and A. A. Mehrabzadeh, Tropospheric free radical determination by fluorescence assay with gas expansion, Environ. Sci. Technol., 1984, 18, 768–777 CrossRef CAS.
- P. S. Stevens, J. H. Mather and W. H. Brune, Measurement of tropospheric OH and HO2 by laser-induced fluorescence at low pressure, J. Geophys. Res., 1994, 99, 3543–3557 CrossRef CAS.
- W. H. Brune, P. S. Stevens and J. H. Mather, Measuring OH and HO2 in the Troposphere by Laser-Induced Fluorescence at Low Pressure, J. Atmos. Sci., 1995, 52, 3328–3336 CrossRef.
- D. J. Creasey, P. A. Halford-Maw, D. E. Heard, M. J. Pilling and B. J. Whitaker, Implementation and initial deployment of a field instrument for measurement of OH and HO2 in the troposphere by laser-induced fluorescence, J. Chem. Soc., Faraday Trans., 1997, 93, 2907–2913 RSC.
- H. Fuchs, F. Holland and A. Hofzumahaus, Measurement of tropospheric RO2 and HO2 radicals by a laser-induced fluorescence instrument, Rev. Sci. Instrum., 2008, 79, 084104 CrossRef PubMed.
- L. Whalley, M. A. Blitz, M. Desservettaz, P. W. Seakins and D. E. Heard, Reporting the sensitivity of laser-induced fluorescence instruments used for HO2 detection to an interference from RO2 radicals and introducing a novel approach that enables HO2 and certain RO2 types to be selectively measured, Atmos. Meas. Tech., 2013, 6, 3425–3440 CrossRef CAS.
- L. Onel, A. Brennan, P. W. Seakins, L. Whalley and D. E. Heard, A new method for atmospheric detection of the CH3O2 radical, Atmos. Meas. Tech., 2017, 10, 3985–4000 CrossRef CAS.
- C. A. Cantrell and D. H. Stedman, A possible technique for the measurement of atmospheric peroxy radicals, Geophys. Res. Lett., 1982, 9, 846–849 CrossRef CAS.
- C. A. Cantrell, D. H. Stedman and G. J. Wendel, Measurement of Atmospheric Peroxy Radicals by Chemical Amplification, Anal. Chem., 1984, 56, 1496–1502 CrossRef CAS.
- D. R. Hastie, M. Weissenmayer, J. P. Burrows and G. W. Harris, Calibrated Chemical Amplifier for Atmospheric ROx Measurements, Anal. Chem., 1991, 63, 2048–2057 CrossRef CAS.
- C. A. Cantrell, R. E. Shetter, J. A. Lind, A. H. McDaniel and J. G. Calvert, An Improved Chemical Amplifier Technique for Peroxy Radical Measurements, J. Geophys. Res., 1993, 98, 2897–2909 CrossRef CAS.
- C. A. Cantrell, R. E. Shetter and J. G. Calvert, Dual-Inlet Chemical Amplifier for Atmospheric Peroxy Radical Measurements, Anal. Chem., 1996, 68, 4194–4199 CrossRef CAS PubMed.
- T. Reiner, M. Hanke and F. Arnold, Atmospheric peroxy radical measurements by ion molecule reaction-mass spectrometry: a novel analytical method using amplifying chemical conversion to sulphuric acid, J. Geophys. Res., 1997, 102, 1311–1326 CrossRef CAS.
- T. J. Green, C. E. Reeves, N. Brough, G. D. Edwards, P. S. Monks and S. A. Penkett, Airborne measurements of peroxy radicals using the PERCA technique, J. Environ. Monit., 2003, 5, 75–83 RSC.
- Y. Sadanaga, J. Matsumoto, K. Sakurai, R. Isozaki, S. Kato, T. Nomaguchi, H. Bandow and Y. Kajii, Development of a measurement system of peroxy radicals using a chemical amplification/laser-induced fluorescence technique, Rev. Sci. Instrum., 2004, 75, 864–872 CrossRef CAS.
- D. Kartal, M. D. Andrés-Hernández, L. Reichert, H. Schlager and J. P. Burrows, Technical note: characterisation of a DUALER instrument for the airborne measurement of peroxy radicals during AMMA 2006, Atmos. Chem. Phys., 2010, 10, 3047–3062 CrossRef CAS.
- M. Horstjann, M. D. Andrés Hernández, V. Nenakhov, A. Chrobry and J. P. Burrows, Peroxy radical detection for airborne atmospheric measurements using absorption spectroscopy of NO2, Atmos. Meas. Tech., 2014, 7, 1245–1257 CrossRef CAS.
- Y. Liu and J. Zhang, Atmospheric Peroxy Radical Measurements Using Dual-Channel Chemical Amplification Cavity Ringdown Spectroscopy, Anal. Chem., 2014, 86, 5391–5398 CrossRef CAS PubMed.
- Y. Chen, C. Yang, W. Zhao, B. Fang, X. Xu, Y. Gai, X. Lin, W. Chen and W. Zhang, Ultra-sensitive measurement of peroxy radicals by chemical amplification broadband cavity-enhanced spectroscopy, Analyst, 2016, 141, 5870–5878 RSC.
- E. C. Wood, B. L. Deming and S. Kundu, Ethane-Based Chemical Amplification Measurement Techniques for Atmospheric Peroxy Radicals, Environ. Sci. Technol. Lett., 2017, 4, 15–19 CrossRef CAS.
- P. W. Villalta, L. G. Huey and C. J. Howard, A Temperature-Dependent Kinetics Study of the CH3O2 + NO Reaction Using Chemical Ionization Mass Spectrometry, J. Phys. Chem., 1995, 99, 12829–12834 CrossRef CAS.
- P. W. Villalta and C. J. Howard, Direct Kinetics Study of the CH3C(O)O2 + NO Reaction Using Chemical Ionization Mass Spectrometry, J. Phys. Chem., 1996, 100, 13624–13628 CrossRef CAS.
- P. W. Villalta, E. R. Lovejoy and D. R. Hanson, Reaction probability of peroxyacetyl radical on aqueous surfaces, Geophys. Res. Lett., 1996, 23, 1765–1768 CrossRef CAS.
- J. Eberhard, P. W. Villalta and C. J. Howard, Reaction of Isopropyl Peroxy Radicals with NO over the Temperature Range 201–410 K, J. Phys. Chem., 1996, 100, 993–997 CrossRef CAS.
- J. Eberhard and C. J. Howard, Rate Coefficients for the Reactions of Some C3 to C5 Hydrocarbon Peroxy Radicals with NO, J. Phys. Chem. A, 1997, 101, 3360–3366 CrossRef CAS.
- K. W. Scholtens, B. M. Messer, C. D. Cappa and M. J. Elrod, Kinetics of the CH3O2 + NO Reaction: Temperature Dependence of the Overall Rate Constant and an Improved Upper Limit for the CH3ONO2 Branching Channel, J. Phys. Chem. A, 1999, 103, 4378–4384 CrossRef CAS.
- T. Moise, W. Denzer and Y. Rudich, Direct Kinetics of the Reaction of Peroxyacetyl Radical with NO between 218 and 370 K, J. Phys. Chem. A, 1999, 103, 6766–6771 CrossRef CAS.
- D. L. Ranschaert, N. J. Schneider and M. J. Elrod, Kinetics of the C2H5O2 + NO Reactions: Temperature Dependence of the Overall Rate Constant and the C2H5ONO2 Branching Channel of C2H5O2 + NO, J. Phys. Chem. A, 2000, 104, 5758–5765 CrossRef CAS.
- D. Zhang, R. Zhang, C. Church and S. W. North, Experimental study of hydroxyalkyl peroxy radicals from OH-initiated reactions of isoprene, Chem. Phys. Lett., 2001, 343, 49–54 CrossRef CAS.
- J. M. Chow, A. M. Miller and M. J. Elrod, Kinetics of the C3H7O2 + NO Reaction: Temperature Dependence of the Overall Rate Constant and the i-C3H7ONO2 Branching Channel, J. Phys. Chem. A, 2003, 107, 3040–3047 CrossRef CAS.
- A. M. Miller, L. Y. Yeung, A. C. Kiep and M. J. Elrod, Overall rate constant measurements of the reactions of alkene-derived hydroxyalkylperoxy radicals with nitric oxide, Phys. Chem. Chem. Phys., 2004, 6, 3402–3407 RSC.
- D. Hanson, J. Orlando, B. Noziere and E. Kosciuch, Proton transfer mass spectrometry studies of peroxy radicals, Int. J. Mass Spectrom., 2004, 239, 147–159 CrossRef CAS.
- A. Bacak, M. W. Bardwell, M. T. Raventos, C. J. Percival, G. Sanchez-Reyna and D. E. Shallcross, Kinetics of the Reaction of CH3O2 + NO: A Temperature and Pressure Dependence Study with Chemical Ionization Mass Spectrometry, J. Phys. Chem. A, 2004, 108, 10681–10687 CrossRef CAS.
- A. K. Patchen, M. J. Pennino and M. J. Elrod, Overall Rate Constant Measurements of the Reaction of Chloroalkylperoxy Radicals with Nitric oxide, J. Phys. Chem. A, 2005, 109, 5865–5871 CrossRef CAS PubMed.
- A. W. Birdsall, J. F. Andreoni and M. J. Elrod, Investigation of the Role of Bicyclic Peroxy Radicals in the Oxidation Mechanism of Toluene, J. Phys. Chem. A, 2010, 114, 10655–10663 CrossRef CAS PubMed.
- A. W. Birdsall and M. J. Elrod, Comprehensive NO-Dependent Study of the Products of the Oxidation of Atmospherically Relevant Aromatic Compounds, J. Phys. Chem. A, 2011, 115, 5397–5407 CrossRef CAS PubMed.
- M. J. Elrod, Kinetics Study of the Aromatic Bicyclic Peroxy Radical + NO Reaction: Overall Rate Constant and Nitrate Product Yield Measurements, J. Phys. Chem. A, 2011, 115, 8125–8130 CrossRef CAS PubMed.
- T. Jokinen, M. Sipilä, S. Richters, V.-M. Kerminen, P. Paasonen, F. Stratmann, D. Worsnop, M. Kulmala, M. Ehn, H. Herrmann and T. Berndt, Rapid Measurements Forms Highly Oxidized RO2 Radicals in the Atmosphere, Angew. Chem., Int. Ed., 2014, 53, 14596–14600 CrossRef CAS PubMed.
- T. Berndt, S. Richters, R. Kaethner, J. Voigtländer, F. Stratmann, M. Sipilä, M. Kulmala and H. Herrmann, Gas-Phase Ozonolysis of Cycloalkenes: Formation of Highly Oxidized RO2 Radicals and Their Reactions with NO, NO2, SO2, and Other RO2 Radicals, J. Phys. Chem. A, 2015, 119, 10336–10348 CrossRef CAS PubMed.
- P. R. Veres, J. M. Roberts, R. J. Wild, P. M. Edwards, S. S. Brown, T. S. Bates, P. K. Quinn, J. E. Johnson, R. J. Zamora and J. de Gouw, Peroxynitric acid (HO2NO2) measurements during the UBWOS 2013 and 2014 studies using iodide ion chemical ionization mass spectrometry, Atmos. Chem. Phys., 2015, 15, 8101–8114 CrossRef CAS.
- J. Sanchez, D. J. Tanner, D. Chen, L. G. Huey and N. L. Ng, A new technique for the direct detection of HO2 radicals using bromide chemical ionization mass spectrometry (Br-CIMS): initial characterization, Atmos. Meas. Tech., 2016, 9, 3851–3861 CrossRef CAS.
- B. Nozière and D. R. Hanson, Speciated Monitoring of Gas-Phase Organic Peroxy Radicals by Chemical Ionization Mass Spectrometry: Cross-Reactions between CH3O2, CH3(CO)O2, (CH3)3CO2, and c-C6H11O2, J. Phys. Chem. A, 2017, 121, 8453–8464 CrossRef PubMed.
- A. Hansel, W. Scholz, B. Mentler, L. Fischer and T. Berndt, Detection of RO2 radicals and other products from cyclohexene ozonolysis with NH4+ and acetate chemical ionization mass spectrometry, Atmos. Environ., 2018, 186, 248–255 CrossRef CAS.
- B. Nozière and L. Vereecken, Direct Observation of Aliphatic Peroxy Radical Autoxidation and Water Effects: An Experimental and Theoretical Study, Angew. Chem., Int. Ed., 2019, 58, 13976–13982 CrossRef PubMed.
- S. R. Albrecht, A. Novelli, A. Hofzumahaus, S. Kang, Y. Baker, T. Mentel, A. Wahner and H. Fuchs, Measurements of hydroperoxy radicals (HO2) at atmospheric concentrations using bromide chemical ionisation mass spectrometry, Atmos. Meas. Tech., 2019, 12, 891–902 CrossRef CAS.
- D. B. Atkinson and J. L. Spillman, Alkyl Peroxy Radical Kinetics Measured using Near-infrared CW-Cavity Ring-down Spectroscopy, J. Phys. Chem. A, 2002, 106, 8891–8902 CrossRef CAS.
- J. Thiébaud and C. Fittschen, Near infrared cw-CRDS coupled to laser photolysis: spectroscopy and kinetics of the HO2 radical, Appl. Phys. B, 2006, 85, 383–389 CrossRef.
- M. K. Sprague, E. R. Garland, A. K. Mollner, C. Bloss, B. D. Bean, M. L. Weichman, L. A. Mertens, M. Okumura and S. P. Sander, Kinetics of n-Butoxy and 2-Pentoxy Isomerization and Detection of Primary Products by Infrared Cavity Ringdown Spectroscopy, J. Phys. Chem. A, 2012, 116, 6327–6340 CrossRef CAS PubMed.
- L. Onel, A. Brennan, M. Gianella, G. Ronnie, A. L. Aguila, G. Hancock, L. Whalley, P. W. Seakins, G. A. Ritchie and D. E. Heard, An intercomparison of HO2 measurements by fluorescence assay by gas expansion and cavity ring-down spectroscopy within HIRAC (Highly Instrumented Reactor for Atmospheric Chemistry), Atmos. Meas. Tech., 2017, 10, 4877–4894 CrossRef CAS.
- L. Onel, A. Brennan, M. Gianella, J. Hooper, N. Ng, G. Hancock, L. Whalley, P. W. Seakins, G. A. Ritchie and D. E. Heard, An intercomparison of CH3O2 measurements by fluorescence assay by gas expansion and cavity ring-down spectroscopy within HIRAC (Highly Instrumented Reactor for Atmospheric Chemistry), Atmos. Meas. Tech., 2020, 13, 2441–2456 CrossRef CAS.
- C. Wang, W. Zhao, B. Fang, N. Yang, F. Cheng, X. Hu, Y. Chen, W. Zhang, C. Fittschen and W. Chen, Portable cavity ring-down spectrometer for an HO2 radical measurement: instrument's performance and potential improvement using a narrow linewidth laser, Opt. Express, 2022, 30, 37446–37456 CrossRef CAS PubMed.
- M. Shamas, M. Assali, C. Zhang, X. Tang, W. Zhang, L. Pillier, C. Schoemaecker and C. Fittschen, Rate Constant and Branching Ratio for the Reactions of the Ethyl Peroxy Radical with Itself and with the Ethoxy Radical, ACS Earth Space Chem., 2022, 6, 181–188 CrossRef CAS.
- M. Assali and C. Fittschen, Rate Constants and Branching Ratios for the Self-Reaction of Acetyl Peroxy (CH3C(O)O2·) and its Reaction with CH3O2, Atmosphere, 2022, 13, 186 CrossRef CAS.
- M. Assali and C. Fittschen, Self-Reaction of Acetonyl Peroxy Radicals and Their Reaction with Cl Atoms, J. Phys. Chem. A, 2022, 126, 4585–4597 CrossRef CAS PubMed.
- T. J. Johnson, F. G. Wienhold, J. P. Burrows, G. W. Harris and H. Burkhard, Measurements of Line Strengths in the HO2 ν1 Overtone Band at 1.5 μm Using an InGaAsP laser, J. Phys. Chem., 1991, 95, 6499–6502 CrossRef CAS.
- C. A. Taatjes and D. B. Oh, Time-resolved wavelength modulation spectroscopy measurements of HO2 kinetics, Appl. Opt., 1997, 36, 5817–5821 CrossRef CAS PubMed.
- L. E. Christensen, M. Okumura, S. P. Sander, R. R. Friedl, C. E. Miller and J. J. Sloan, Measurements of the Rate Constant of HO2 + NO2 + N2 → HO2NO2 + N2 Using Near-Infrared Wavelength-Modulation Spectroscopy and UV−Visible Absorption Spectroscopy, J. Phys. Chem. A, 2004, 108, 80–91 CrossRef CAS.
- F. J. Grieman, A. C. Noell, C. Davis-Van Atta, M. Okumura and S. P. Sander, Determination of Equilibrium Constants for the Reaction between Acetone and HO2 Using Infrared Kinetic Spectroscopy, J. Phys. Chem. A, 2011, 115, 10527–10538 CrossRef CAS PubMed.
- D. E. Heard and M. J. Pilling, Measurement of OH and HO2 in the Troposphere, Chem. Rev., 2003, 103, 5163–5198 CrossRef CAS PubMed.
- D. E. Heard, Atmospheric field measurements of the hydroxyl radical using laser-induced fluorescence spectroscopy, Annu. Rev. Phys. Chem., 2006, 57, 191–216 CrossRef CAS PubMed.
- D. Stone, L. K. Whalley and D. E. Heard, Tropospheric OH and HO2 radicals: field measurements and model comparisons, Chem. Soc. Rev., 2012, 41, 6348–6404 RSC.
- R. S. Hornbrook, J. H. Crawford, G. D. Edwards, O. Goyea, R. L. Mauldin III, J. S. Olson and C. A. Cantrell, Measurements of tropospheric HO2 and RO2 by oxygen dilution modulation and chemical ionization mass spectrometry, Atmos. Meas. Tech., 2011, 4, 735–756 CrossRef CAS.
- IUPAC Datasheet for Gas Phase Reactions, retrieved from https://iupac.aeris-data.fr/ Search PubMed.
- J. J. Orlando and G. F. Tyndall, Laboratory studies of organic peroxy radical chemistry: an overview with emphasis on recent issues of atmospheric significance, Chem. Soc. Rev., 2012, 41, 6294–6317 RSC.
- M. E. Jenkin, R. Valorso, B. Aumont and A. R. Rickard, Estimation of rate coefficients and branching ratios for reactions of organic peroxy radicals for use in automated mechanism construction, Atmos. Chem. Phys., 2019, 19, 7691–7717 CrossRef CAS.
- J. B. Burkholder, S. P. Sander, J. Abbatt, J. R. Barker, C. Cappa, J. D. Crounse, T. S. Dibble, R. E. Huie, C. E. Kolb, M. J. Kurylo, V. L. Orkin, C. J. Percival, D. M. Wilmouth and P. H. Wine, Chemical Kinetics and Photochemical Data for Use in Atmospheric Studies, Evaluation No. 19, JPL Publication 19-5, Jet Propulsion Laboratory, Pasadena, 2019, https://jpldataeval.jpl.nasa.gov Search PubMed.
- M. E. Jenkin, S. M. Saunders and M. J. Pilling, The tropospheric degradation of volatile organic compounds: a protocol for mechanism development, Atmos. Environ., 1997, 31, 81–104 CrossRef CAS.
- S. M. Saunders, M. E. Jenkin, R. G. Derwent and M. J. Pilling, Protocol for the development of the Master Chemical Mechanism, MCM v3 (part A): tropospheric degradation of non-aromatic volatile organic compounds, Atmos. Chem. Phys., 2003, 3, 161–180 CrossRef CAS.
- J. G. Calvert, R. G. Derwent, J. J. Orlando, G. S. Tyndall and T. J. Wallington, Mechanisms of Atmospheric Oxidation of the Alkanes, Oxford University Press, Oxford, 2008 Search PubMed.
- M. D. King, C. E. Canosa-Mas and R. P. Wayne, Gas-phase reactions between RO2 and NO, HO2 or CH3O2: correlations between rate constants and the SOMO energy of the peroxy (RO2) radical, Atmos. Environ., 2001, 35, 2081–2088 CrossRef CAS.
- D. Johnson, D. W. Price and G. Marston, Correlation-type structure activity relationships for the kinetics of gas-phase RO2 self-reactions and reaction with HO2, Atmos. Environ., 2004, 38, 1447–1458 CrossRef CAS.
- C. S. Kan, J. G. Calvert and J. H. Shaw, Reactive Channels of the CH3O3-CH3O2 Reaction, J. Phys. Chem., 1980, 84, 3411–3417 CrossRef CAS.
- H. Niki, P. D. Maker, C. M. Savage and L. P. Breitenbach, Fourier Transform Infrared Studies of the Self-Reaction of CH3O2 Radicals, J. Phys. Chem., 1981, 85, 877–881 CrossRef CAS.
- N. R. Subbaratnam and J. G. Calvert, The Mechanism of Methyl Hydroperoxide Formation in the Photooxidation of Azomethane at 25°, J. Am. Chem. Soc., 1962, 84, 1113–1118 CrossRef CAS.
- D. F. Dever and J. G. Calvert, Rate Studies of the Oxidation of Methyl Radicals in Oxygen-rich Media at 25°, J. Am. Chem. Soc., 1962, 84, 1362–1368 CrossRef CAS.
- J. J. Orlando, G. S. Tyndall, L. Vereecken and J. Peeters, The Atmospheric Chemistry of the Acetonoxy Radical, J. Phys. Chem. A, 2000, 104, 11578–11588 CrossRef CAS.
- A. Mellouki, M. Ammann, R. A. Cox, J. N. Crowley, H. Herrmann, M. E. Jenkin, V. F. McNeill, J. Troe and T. J. Wallington, Evaluated kinetic and photochemical data for atmospheric chemistry: volume VIII – gas-phase reactions of organic species with four, or more, carbon atoms (≥C4), Atmos. Chem. Phys., 2021, 21, 4797–4808 CrossRef CAS.
- R. Volkamer, B. Klotz, I. Barnes, T. Imamura, K. Wirtz, N. Washida, K. H. Becker and U. Platt, OH-initiated oxidation of benzene part I. Phenol formation under atmospheric conditions, Phys. Chem. Chem. Phys., 2002, 4, 1598–1610 RSC.
- A. S. Hasson, K. T. Kuwata, M. C. Arroyo and E. B. Petersen, Theoretical studies of the reaction of hydroperoxy radicals (HO2) with ethyl peroxy (CH3CH2O2), acetyl peroxy (CH3C(O)O2), and acetonyl peroxy (CH3C(O)CH2O2) radicals, J. Photochem. Photobiol., A, 2005, 176, 218–230 CrossRef CAS.
- H. Hou and B. Wang, A Systematic Computational Study on the Reactions of HO2 with RO2: The HO2 + CH3O2(CD3O2) and HO2 + CH2FO2 Reactions, J. Phys. Chem. A, 2005, 109, 451–460 CrossRef CAS PubMed.
- H. Hou, L. Deng, J. Li and B. Wang, A Systematic Computational Study of the Reactions of HO2 with RO2: The HO2 + CH2ClO2, CHCl2O2, and CCl3O2 Reactions, J. Phys. Chem. A, 2005, 109, 9299–9309 CrossRef CAS PubMed.
- J. P. Anglada, S. Olivella and A. Solé, Mechanistic Study of the HO2 + CH3O2→ CH3O2H + O2 Reaction in the Gas Phase. Computational Evidence for the Formation of a Hydrogen-Bonded Diradical Complex, J. Phys. Chem. A, 2006, 110, 6073–6082 CrossRef CAS PubMed.
- T. L. Nguyen, L. Vereecken and J. Peeters, Theoretical Study of the HOCH2OO + HO2 Reaction: Detailed Molecular Mechanisms of the Three Reaction Channels, Z. Phys. Chem., 2010, 2010, 1081–1093 CrossRef.
- L. Vereecken and J. S. Francisco, Theoretical studies of atmospheric reaction mechanisms in the troposphere, Chem. Soc. Rev., 2012, 41, 6259–6293 RSC.
- M. J. Elrod, D. L. Ranschaert and N. J. Schneider, Direct Kinetics Study of the Temperature Dependence of the CH2O Branching Channel for the CH3O2 + HO2 Reaction, Int. J. Chem. Kinet., 2001, 33, 363–376 CrossRef CAS.
- X.-M. Zhou, Z.-Y. Zhou, Q.-Y. Wu, A. F. Jalbout and N. Zhang, Reaction of CH3O2 and HO2: ab initio characterization of dimer structure and vibrational mode analysis for reaction mechanisms, Int. J. Quantum Chem., 2006, 106, 514–525 CrossRef CAS.
- E. Drougas, Quantum mechanical studies of the CH3O2 + HO2 reaction, Comput. Theor. Chem., 2016, 1093, 98–103 CrossRef CAS.
- A. Novelli, L. Vereecken, B. Bohn, H.-P. Dorn, G. I. Gkatzelis, A. Hofzumahaus, F. Holland, D. Reimer, F. Rohrer, S. Rosanka, D. Taraborrelli, R. Tillmann, R. Wegener, Z. Yu, A. Kiendler-Scharr, A. Wahner and H. Fuchs, Importance of isomerization reactions for OH radical regeneration from the photo-oxidation of isoprene investigated in the atmospheric simulation chamber SAPHIR, Atmos. Chem. Phys., 2020, 20, 3333–3355 CrossRef CAS.
- E. Praske, R. V. Otkjaer, J. D. Crounse, J. C. Hethcox, B. M. Stoltz, H. G. Kjaergaard and P. O. Wennberg, Atmospheric autoxidation is increasingly important in urban and suburban North America, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 64–69 CrossRef CAS PubMed.
- M. J. Newland, D. J. Bryant, R. E. Dunmore, T. J. Bannan, W. J. F. Acton, B. Langford, J. R. Hopkins, F. A. Squires, W. Dixon, W. S. Drysdale, P. D. Ivatt, M. J. Evans, P. M. Edwards, L. K. Whalley, D. E. Heard, E. J. Slater, R. Woodward-Massey, C. Ye, A. Mehra, S. D. Worrall, A. Bacak, H. Coe, C. J. Percival, C. N. Hewitt, J. D. Lee, T. Cui, J. D. Surratt, X. Wang, A. C. Lewis, A. R. Rickard and J. F. Hamilton, Low-NO atmospheric oxidation pathways in a polluted megacity, Atmos. Chem. Phys., 2021, 21, 1613–1625 CrossRef CAS.
- R. J. Yokelson, J. D. Crounse, P. F. DeCarlo, T. Karl, S. Urbanski, E. Atlas, T. Campos, Y. Shinozuka, V. Kapustin, A. D. Clarke, A. Weinheimer, D. J. Knapp, D. D. Montzka, J. Holloway, P. Weibring, F. Flocke, W. Zheng, D. Toohey, P. O. Wennberg, C. Wiedinmyer, L. Mauldin, A. Fried, D. Richter, J. Walega, J. L. Jimenez, K. Adachi, P. R. Buseck, S. R. Hall and R. Shetter, Emissions from biomass burning in the Yucatan, Atmos. Chem. Phys., 2009, 9, 5785–5812 CrossRef CAS.
- M. O. Andreae, Emission of trace gases and aerosols from biomass burning – an updated assessment, Atmos. Chem. Phys., 2019, 19, 8523–8546 CrossRef CAS.
- J. N. Crowley, N. Pouvesle, G. J. Phillips, R. Axinte, H. Fischer, T. Petäjä, A. Nölscher, J. Williams, K. Hens, H. Harder, M. Martinez-Harder, A. Novelli, D. Kubistin, B. Bohn and J. Lelieveld, Insights into HOx and ROx chemistry in the boreal forest via measurement of peroxyacetic acid, peroxyacetic nitric anhydride (PAN) and hydrogen peroxide, Atmos. Chem. Phys., 2018, 18, 13457–13479 CrossRef CAS.
- D. Tan, I. Faloona, J. B. Simpas, W. Brune, P. B. Shepson, T. L. Couch, A. L. Sumner, M. A. Carroll, T. Thornberry, E. Apel, D. Riemer and W. Stockwell, HOx budgets in a deciduous forest: results from the PROPHET summer 19989 campaign, J. Geophys. Res., 2001, 106, 24407–24427 CrossRef CAS.
- A. Hofzumahaus, F. Rohrer, K. Lu, B. Bohn, T. Brauers, C.-C. Chang, H. Fuchs, F. Holland, K. Kita, Y. Kondo, X. Li, S. Lou, M. Shao, L. Zeng, A. Wahner and Y. Zhang, Amplified Trace Gas Removal in the Troposphere, Science, 2009, 324, 1702–1704 CrossRef CAS PubMed.
- D. Kubistin, H. Harder, M. Martinez, M. Rudolf, R. Sander, H. Bozem, G. Eerdekens, H. Fischer, C. Gurk, T. Klüpfel, R. Königsstedt, U. Parchatka, C. L. Schiller, A. Stickler, D. Taraborrelli, J. Willimas and J. Lelieveld, Hydroxyl radicals in the tropical troposphere over the Suriname rainforest: comparison of measurements with the box model MECCA, Atmos. Chem. Phys., 2010, 10, 9705–9728 CrossRef CAS.
- J. Peeters, T. L. Nguyen and L. Vereecken, HOx radical regeneration in the oxidation of isoprene, Phys. Chem. Chem. Phys., 2009, 11, 5935–5939 RSC.
- J. Peeters, J.-F. Müller, T. Stavrakou and V. S. Nguyen, Hydroxyl Radical Recycling in Isoprene Oxidation Driven by Hydrogen Bonding and Hydrogen Tunneling: The Upgraded LIM1 Mechanism, J. Phys. Chem. A, 2014, 118, 8625–8643 CrossRef CAS PubMed.
- D. J. Medeiros, M. A. Blitz, P. W. Seakins and L. K. Whalley, Direct Measurements of Isoprene Autoxidation: Pinpointing Atmospheric Oxidation in Tropical Forests, JACS Au, 2022, 2, 809–818 CrossRef PubMed.
- F. Paulot, J. D. Crounse, H. G. Kjaergaard, A. Kürten, J. M. S. Clair, J. H. Seinfeld and P. O. Wennberg, Unexpected Epoxide Formation in the Gas-Phase Photooxidation of Isoprene, Science, 2009, 325, 730–733 CrossRef CAS PubMed.
- S. Iyer, H. Reiman, K. H. Møller, M. P. Rissanen, H. G. Kjaergaard and T. Kurtén, Computational Investigation of RO2 + HO2 and RO2 + RO2 Reactions of Monoterpene Derived First-Generation Peroxy Radicals Leading to Radical Recycling, J. Phys. Chem. A, 2018, 122, 9542–9552 CrossRef CAS PubMed.
- M. Rolletter, M. Kaminski, I.-H. Acir, B. Bohn, H.-P. Dorn, X. Li, A. Lutz, S. Nehr, F. Rohrer, R. Tillmann, R. Wegener, A. Hofzumahaus, A. Kiendler-Scharr, A. Wahner and H. Fuchs, Investigation of the α-pinene photooxidation by OH in the atmospheric simulation chamber SAPHIR, Atmos. Chem. Phys., 2019, 19, 11635–11649 CrossRef CAS.
- A. W. Rollins, A. Kiendler-Scharr, J. L. Fry, T. Brauers, S. S. Brown, H.-P. Dorn, W. P. Dubé, H. Fuchs, A. Mensah, T. F. Mentel, F. Rohrer, R. Tillmann, R. Wegener, P. J. Wooldridge and R. C. Cohen, Isoprene oxidation by nitrate radical: alkyl nitrate and secondary organic aerosol yield, Atmos. Chem. Phys., 2009, 9, 6685–6703 CrossRef CAS.
- E. Hellpointner and S. Gäb, Detection of methyl, hydroxymethyl and hydroxyethyl hydroperoxides in air and precipitation, Nature, 1989, 337, 631–634 CrossRef CAS.
- A. V. Jackson and C. N. Hewitt, Atmosphere Hydrogen Peroxide and Organic Hydroperoxides: A Review, Crit. Rev. Environ. Sci. Technol., 1999, 29, 175–228 CrossRef CAS.
- M. Lee, B. G. Heikes and D. W. O'Sullivan, Hydrogen peroxide and organic hydroperoxide in the troposphere, Atmos. Environ., 2000, 34, 3475–3494 CrossRef CAS.
- A. Monod, E. Chevallier, R. D. Jolibois, J. F. Doussin, B. Picquet-Varrault and P. Carlier, Photooxidation of methylhydroperoxide and ethylhydroperoxide in the aqueous phase under simulated cloud droplet conditions, Atmos. Environ., 2007, 41, 2412–2426 CrossRef CAS.
- S. A. Epstein, D. Shemesh, V. T. Tran, S. A. Nizkorodov and R. B. Gerber, Absorption Spectra and Photolysis of Methyl Peroxide in Liquid and Frozen Water, J. Phys. Chem. A, 2012, 116, 6068–6077 CrossRef CAS PubMed.
- J. Peeters and J.-F. Müller, HOx radical regeneration in isoprene oxidation via peroxy radical isomerisations. II: experimental evidence and global impact, Phys. Chem. Chem. Phys., 2010, 12, 14227–14235 RSC.
- G. M. Wolfe, J. D. Crounse, J. D. Parrish, J. M. St. Clair, M. R. Beaver, F. Paulot, T. P. Yoon, P. O. Wennbergb and F. N. Keutsch, Photolysis, OH reactivity and ozone reactivity of a proxy for isoprene-derived hydroperoxyenals (HPALDs), Phys. Chem. Chem. Phys., 2012, 14, 7276–7286 RSC.
- J. C. Rivera-Rios, T. B. Nguyen, J. D. Crounse, W. Jud, J. M. S. Clair, T. Mikoviny, J. B. Gilman, B. M. Lerner, J. B. Kaiser, J. de Gouw, A. Wisthaler, A. Hansel, P. O. Wennberg, J. H. Seinfeld and F. N. Keutsch, Conversion of hydroperoxides to carbonyls in field and laboratory instrumentation: observational bias in diagnosing pristine versus anthropogenically controlled atmospheric chemistry, Geophys. Res. Lett., 2014, 41, 8645–8651 CrossRef CAS.
- A.-K. Bernhammer, M. Breitenlechner, F. N. Keutsch and A. Hansel, Technical note: conversion of isoprene hydroxy hydroperoxides (ISOPOOHs) on metal environmental simulation chamber walls, Atmos. Chem. Phys., 2017, 17, 4053–4062 CrossRef CAS.
- J. Sanchez and T. N. Myers, Peroxides and Peroxide Compounds, Organic Peroxides, in Kirk-Othmer Encyclopedia of Chemical Technology, Wiley, 1st edn, 2000 Search PubMed.
- G. L. Vaghjiani and A. R. Ravishankara, Kinetics and Mechanism of OH Reaction with CH3OOH, J. Phys. Chem. A, 1989, 93, 1948–1959 CrossRef CAS.
- H. R. Williams and H. S. Mosher, Peroxides. I. n-Alkyl Hydroperoxides, J. Am. Chem. Soc., 1954, 76, 2984–2987 CrossRef CAS.
- H. R. Williams and H. S. Mosher, Organic Peroxides. II. Secondary Alkyl Hydroperoxides, J. Am. Chem. Soc., 1954, 76, 2987–2990 CrossRef CAS.
- M. Riva, S. H. Budisulistiorini, Y. Chen, Z. Zhang, E. L. D'Ambro, X. Zhang, A. Gold, B. J. Turpin, J. A. Thornton, M. R. Canagaratna and J. D. Surratt, Chemical Characterization of Secondary Organic Aerosol from Oxidation of Isoprene Hydroxy Hydroperoxides, Environ. Sci. Technol., 2016, 50, 9889–9899 CrossRef CAS PubMed.
- P. Mettke, A. Mutzel, O. Böge and H. Herrmann, Synthesis and Characterization of Atmospherically Relevant Hydroxy Hydroperoxides, Atmosphere, 2022, 13, 507 CrossRef CAS.
- X. Zhao, T. Zhang, Y. Zhou and D. Liu, Preparation of peracetic acid from hydrogen peroxide part I: kinetics for peracetic acid synthesis and hydrolysis, J. Mol. Catal. A:Chem., 2007, 271, 246–252 CrossRef CAS.
- P. Heinmöller, H.-H. Kurth, R. Rabong, W. V. Turner, A. Kettrup and S. Gäb, Selective HPLC Analysis of n-Alkyl Hydroperoxides up to C18H38O2, Anal. Chem., 1998, 70, 1437–1439 CrossRef PubMed.
- K. G. Paul, P. I. Ohlsson and S. Wold, Formation of Horseradish Peroxidase Compound I with Alkyl Hydroperoxides, Acta Chem. Scand., Ser. B, 1979, 33, 747–754 CrossRef.
- M. R. McGillen, G. S. Tyndall, J. J. Orlando, A. S. Pimentel, D. J. Medeiros and J. B. Burkholder, Experimentally Determined Site-Specific Reactivity of the Gas-Phase OH and Cl + i-Butanol Reactions Between 251 and 340 K, J. Phys. Chem. A, 2016, 120, 9968–9981 CrossRef CAS PubMed.
- M. R. McGillen, L. Michelat, J. J. Orlando and W. P. L. Carter, The use of the electrotopological state as a basis for predicting hydrogen abstraction rate coefficients: a proof of principle for the reactions of alkanes and haloalkanes with OH, Environ. Sci.: Atmos., 2024, 4, 18–34 CAS.
- B. Ervens, A. Rickard, B. Aumont, W. P. L. Carter, M. McGillen, A. Mellouki, J. Orlando, B. Picquet-Varrault, P. Seakins, W. R. Stockwell, L. Vereecken and T. J. Wallington, Opinion: challenges and needs of tropospheric chemical mechanism development, Atmos. Chem. Phys., 2024, 24, 13317–13339 CrossRef CAS.
- I. Barnes, K. H. Becker and L. Ruppert, FTIR product study of the self-reaction of β-hydroxyethl peroxy radicals, Chem. Phys. Lett., 1993, 203, 295–301 CrossRef CAS.
- B. Nozière and L. Vereecken, Direct Observation of Aliphatic Peroxy Radical Autoxidation and Water Effects: An Experimental and Theoretical Study, Angew. Chem., Int. Ed., 2019, 58, 13976–13982 CrossRef PubMed.
- B. Nozière and L. Vereecken, H-shift and cyclization reactions in unsaturated alkylperoxy radicals near room temperature: propagating or terminating autoxidation?, Phys. Chem. Chem. Phys., 2024, 26, 25373–25384 RSC.
- B. Nozière and F. Fache, Reactions of organic peroxy radicals, RO2, with substituted and biogenic alkenes at room temperature: unsuspected sinks for some RO2 in the atmosphere?, Chem. Sci., 2021, 12, 11676–11683 RSC.
- B. Nozière, O. Durif, E. Dubus, S. Kylington, A. Emmer, F. Fache, F. Piel and A. Wisthaler, The reaction of organic peroxy radicals with unsaturated compounds controlled by a non-epoxide pathway under atmospheric conditions, Phys. Chem. Chem. Phys., 2023, 25, 7772–7782 RSC.
- L. Onel, A. Brennan, F. F. Østerstrøm, E. Cooke, L. Whalley, P. W. Seakins and D. E. Heard, Kinetics and Product Branching Ratio Study of the CH3O2 Self-Reaction in the Highly Instrumented Reactor for Atmospheric Chemistry, J. Phys. Chem. A, 2022, 126, 7639–7649 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2025 |