
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Choice of photo-excitation conditions in xanthate-supported photo-iniferter (XPI) RAFT polymerization†
Vitalii
Tkachenko
 a,
Tzu-Chien
Wu
a,
Anahita
Heraji Esfahani
a,
Johannes
Gurke
ab and
Matthias
Hartlieb
*ab
a,
Tzu-Chien
Wu
a,
Anahita
Heraji Esfahani
a,
Johannes
Gurke
ab and
Matthias
Hartlieb
*ab
aInstitute of Chemistry, University of Potsdam, Karl-Liebknecht-Straße 24-25, 14476, Potsdam, Germany. E-mail: mhartlieb@uni-potsdam.de
bFraunhofer Institute for Applied Polymer Research (IAP), Geiselbergstraße 69, 14476, Potsdam, Germany
First published on 5th June 2025
Abstract
The use of photo-controlled reversible deactivation radical polymerization (RDRP) techniques offers several advantages regarding the control of polymerization processes and material synthesis. Reversible addition–fragmentation chain-transfer (RAFT) polymerization is particularly interesting in this context as the chain transfer agent (CTA) used in these processes can be activated by light directly in a photo-iniferter (PI) process. We have recently introduced a method where a xanthate (a particularly active iniferter) is combined with a second CTA resulting in a xanthate supported (X)PI-RAFT process. Herein conducted DFT calculations suggest a significant difference in orbital contributions to the β-scission process when comparing xanthates and trithiocarbonates (TTC), offering an explanation for the improved performance of xanthate. Experimentally, we investigate how the choice of wavelength, light intensity and irradiation setup influences the control over the polymerization and in particular its livingness (end group fidelity). This is probed via the synthesis of multiblock and diblock copolymers where livingness is essential and can be accessed via deconvolution of the SEC traces. While a change in wavelength to more selectively activate the xanthate while not exciting the π–π* transition of the TTC does not seem to improve livingness, the light intensity was found to have a tremendous impact, with oversaturation being detrimental for chain end fidelity. Also using a flow-chemistry setup did not help to overcome this limitation. As such, the choice of light intensity was isolated as a highly important factor in XPI-RAFT polymerization.
Introduction
Since the advent of reversible deactivation radical polymerization (RDRP), the design of macromolecules with defined molar mass and architecture has become increasingly accessible. In particular, reversible addition–fragmentation chain transfer (RAFT) polymerization1 offers robustness and straightforward polymerization conditions.2 Many chain transfer agents (CTAs) are commercially available and enable control over the polymerization of different monomer families.3 Moreover, the use of light to activate such polymerizations is a versatile method with many advantages.4The photo iniferter (PI) technique developed by Otsu in the 1980's5 has many similarities with the RAFT process, as both use thiocarbonylthio compounds. In a photo-iniferter polymerization, light is used to excite a thiocarbonylthio molecule (that can also act as CTA), which subsequently fragments into a persistent thiocarbonylthio radical and a transient R-radical.6,7 The latter can propagate in the presence of monomers and also interact with other CTA molecules to engage in chain transfer equilibria, that provide control over molar mass and dispersity similar to the RAFT methodology. (Note: Due to this similarity, in recent years this process was often referred to as PI-RAFT polymerization,6 whereas it can also be argued that the absence of an exogenous radical source distinguishes the method from RAFT-polymerization and it should be termed PI polymerization.7) In turn, persistent and transient radicals recombine (deactivation), a process which ensures a high livingness in PI-(RAFT) polymerization.8 This is in sharp contrast to conventional RAFT polymerization where (irreversible) termination is inevitable and creates a number of dead chains equal to the number of previously propagating radicals.9 Respectively a share of macromolecules lacks a CTA end group and cannot be reactivated and is hence not available for chain extension in the synthesis of block copolymers.
In PI polymerization, the used wavelength plays a tremendous role in the outcome. While initial efforts were conducted exciting the intense π–π* transition, it was later discovered that the symmetry forbidden n–π* transition is equally suited for photoactivation and radical generation.10,11 While the intensity of this second band is much lower, quantum yields are higher, leading to more efficient polymerization processes.12,13 It also enables the use of visible light for many conventional CTAs. In addition, the π–π* transition is in comparison much more prone to side reactions like CS2 elimination.14 It has been demonstrated, that cutting-off of short UV wavelengths can increase the livingness of a PI-RAFT polymerization.15 The type of CTA has of course also a strong impact on the outcome of the polymerization.
Not only does its chain transfer coefficient with the monomer determine the molar mass control,16 the substitution of the CTA also affects spectral properties and quantum yields.17 Particularly, xanthates, whose n–π* is usually located in the UV region (∼350 nm), have been demonstrated to efficiently produce polymers with high molar masses and good control.18 This is associated with the lower bond dissociation energy of the C–S bond in xanthates compared to other CTAs.19
In the context of livingness, radical stabilization energy is an important parameter. For instance, it has been reported that CTAs with tertiary R-groups are more susceptible toward degradation in photo-iniferter processes when compared to RAFT agents with secondary R-groups.20 This is ascribed to a slower deactivation, leading to a longer time frame of the radical state and increased likelihood for side reactions. Next to the already described CS2 elimination and coupling of transient radicals, also persistent radicals can react to form disulfides, which are hard to reactivate.21 In this context, the choice of monomer should also be considered, as it typically possesses a different radical stabilization energy than the R group of the CTA. Hence the use of methacrylates can lead to increased photodegradation of the end group compared to acrylates.22 This is particularly important when both monomer families are to be incorporated into the same polymer chain.23 Also the rate of polymerization (kp) is important as propagation can be the rate determining step, and additional radical generation only promotes side reactions.24 This phenomenon was also observed in a flow chemistry setup, where at increased light intensities rate acceleration stalls and dispersity increases.25 Hence, lower light intensities are usually connected to an improved control.26
It is noteworthy in this context that additives can improve such processes, as shown by Coote and Anastasaki with the addition of acid in a PI-polymerization drastically increasing reaction velocity and livingness.27
Aiming for efficient and controlled photopolymerization, often there is the issue that CTAs effectively generating radicals are not necessarily suited to control the desired monomers or vice versa. To overcome this problem, we recently developed xanthate-supported PI (XPI) polymerization, where a mixture of CTAs is used (schematically shown in Fig. 1E).28 Mixing CTAs can be utilized to modulate dispersity of the produced macromolecules.29,30 In our case, however the strategy is designed mainly to increase photo activity without compromising control. One CTA is highly photoactive and predominantly generates radicals while the second CTA controls the polymerization. Still, both are introduced as end groups in the respective polymer. As a result, fast and controlled polymerizations and easy reactivation are possible, which can for instance be used for the production of bioactive materials.31,32 However, in such a complex scenario, where also the second CTA is inherently photoactive, a closer look at the irradiation conditions is necessary. The present study uses the established XPI-RAFT methodology,28 to investigate the photo-activation and respective chain end fidelity closer. Specifically, we want to elucidate how different irradiation conditions (change in wavelength or intensity) are impacting the livingness of the respective polymer. Pseudo-diblock and pseudo-multiblock copolymers based on N-acryloyl morpholine (NAM) are used as a model for this investigation. Moreover, batch and flow chemistry are compared.
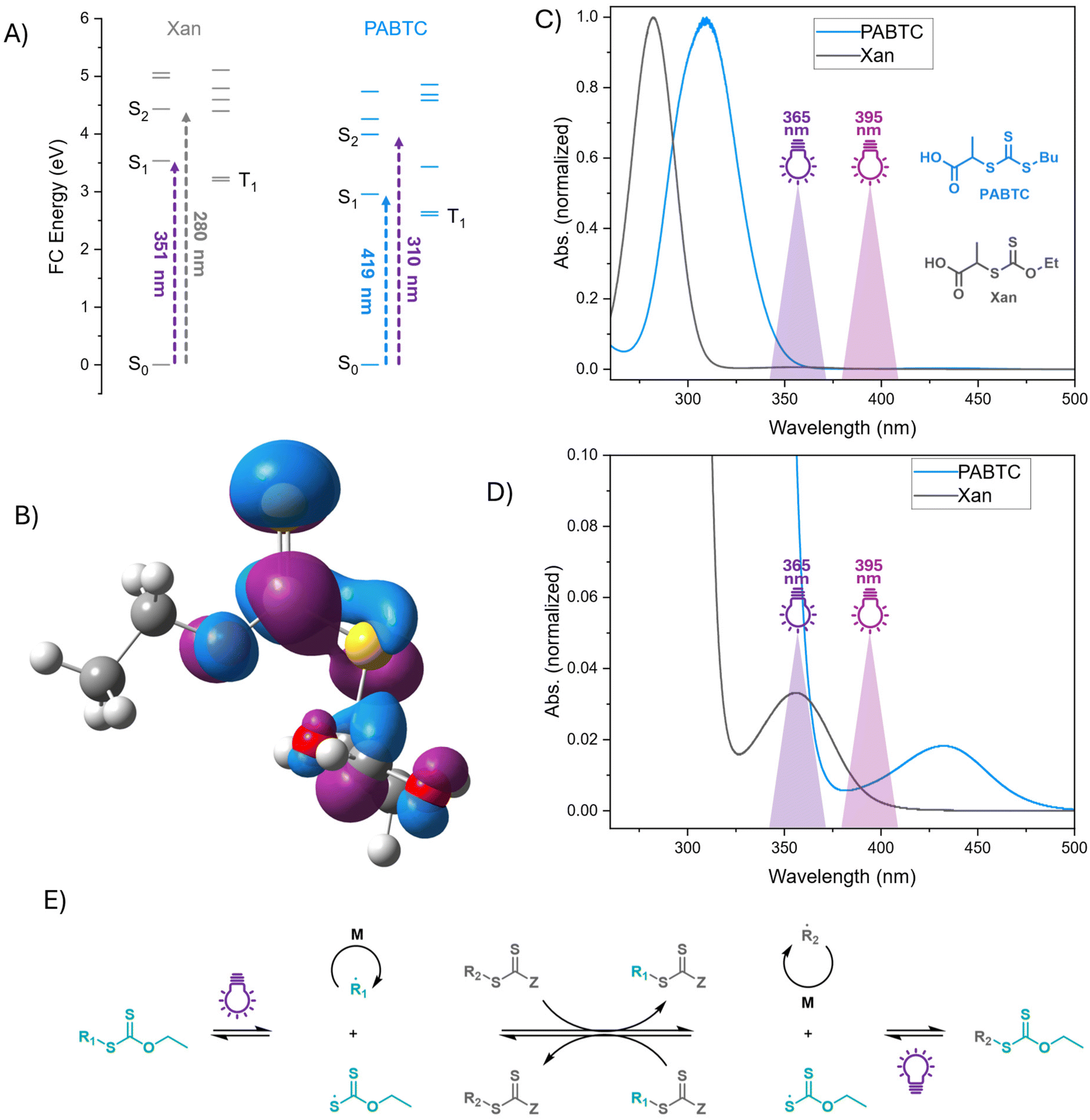 | ||
| Fig. 1 (A) Energy levels of electronic transitions in Xan and PABTC from DFT calculations. (B) Illustration of the NTO of the S2 state for Xan. (C & D) UV Vis spectra of both CTAs in DMSO (c = 0.156 mM (C), c = 1.25 mM (D)) with indications of the light wavelengths used in this work. (E) Schematic representation of the XPI-RAFT concept. | ||
Results and discussion
We first approached the issue from a theoretical side, to gain more insight into actual energy levels of two CTA molecules. The molecular orbital energy diagram, Franck–Condon (FC) energy level diagram and natural transition orbitals33 have been calculated using time-depended density functional theory (TD-DFT) for two CTAs, namely 2-((ethoxycarbonothioyl)thio)propanoic acid (Xan) and 2-(((butylthio)carbonothioyl)thio)propanoic acid (PABTC) on a B3LYP/6-311++g(2df,2p) level of theory. Various functionals have been benchmarked using experimental UV-Vis spectroscopy data (see ESI Fig. S10 and S12–16†).The FC energy level diagram shows consistently that the first and second triplet states lay below the first singlet state (Fig. 1A) For Xan, the calculations show two transitions within the experimentally addressable wavelength window (>265 nm). The transition into the S1 state at 351 nm shows a very small oscillator strength f of 0.0003. Just two canonical molecular orbitals (CMOs, ESI Fig. S11†) participate in the transition; p orbital of the free electron pair at the sulfur in the thiocarbonyl and the anti-bonding π orbital (p → π*). Hence, the resulting natural transition orbitals (NTOs, ESI Table S7†) are identical to the CMOs. The second transition into the S2 state at 280 nm involves four CMOs (two occupied and two virtual) with a significantly increased oscillator strength of 0.1641. We assigned the occupied NTO mainly as non-binding π orbital while the virtual NTO appears to be a mixture of anti-bonding π* orbital and anti-bonding Sigma σ* orbital. Here, the σ* orbital is delocalized into the carboxyl group. The high energy transitions at 249 nm, with an oscillator strength of 0.0023 and at 245 nm, with an oscillator strength of 0.1759, are transiting into NTO dominated by the antibonding Sigma orbital while starting from non-bonding π orbital (nπ → σ*) and free-electron pair p-orbital (p → σ*), respectively.
For PABTC, the calculations show three transitions within the experimentally addressable wavelength window. All of these show two discrete orbitals involved in the transition. Hence, there is no difference between NTO and CMO (ESI Table S9 and Fig. S11†). Moreover, they all end up in the same anti-bonding π* orbital. The most bathochromiclly shifted transition at 419 nm into the S1 state starts from the p-orbital of the free electron pair (p → π*) with a diminutive oscillator strength of 0.0001. The transition into S2 and S3 are significantly hypsochromically shifted compared to the first transition. The transition into S2 is calculated to occur at 311 nm with an oscillator strength of 0.1910. It started from a non-binding π orbital into the anti-bonding π (nπ1 → π*). The transition into S3 is calculated to occur at 291 nm with an oscillator strength of 0.1621. The transition starts from a second non-binding π orbital into the anti-bonding π (nπ2 → π*). The NTO of the high energy transition at 262 nm, with an oscillator strength of 0.0085, is dominated by the antibonding Sigma orbital while starting from the free-electron pair p-orbital (p → σ*).
The low-laying T1 and T2 states in both Xan and PABTC make intersystem crossing after excitation a possible path for both β-scission or slow non-radiative decay. In fact, previous studies by Kwon and coworkers argue that the triplet states are too low in energy to participate in the β-scission and point toward a reaction through a conical intersection of S0 and 1.34
For Xan, the antibonding orbital interaction at the dissociating CS single bond in the NTO of the S2 state (Fig. 1B) makes a β-scission from the singlet state a feasible option, too, as considered for benzodithioate and trithiocarbonate earlier by Falvey and coworker.35 However, our study provides no indication that an antibonding Sigma orbital contributes to the S1 or S2 state of PABTC. This difference in the nature of the orbitals could be a hint at the reason for the significantly improved PI efficiency of Xan compared to PABTC, as observed in experiments.8,28 Both studies by Kwon and coworkers, as well as Falvey and coworkers, do not examine xanthates. Here, further studies are needed to analyze the excited state potential energy surface, including the S2 state.
Following up on our previous experimental work,28 we now aimed to elucidate the impact of light wavelength and intensity in XPI-polymerization further. Our initial investigations on PI and XPI-polymerization were conducted with UV hand lamps (365 nm) at relatively low intensities (∼4 mW cm−2). In the present study, we utilize a photoreactor capable of varying both the intensity and wavelength of irradiation to investigate their effects on the livingness of chain transfer agents (CTAs) and the molecular weight distribution of the resulting polymers. This is particularly interesting as in XPI-polymerization a mixture of CTAs is used, where both components are photoactive. While at 365 nm xanthate is excited in its n–π* transition, the trithiocarbonate PABTC is excited via its π–π* band (Fig. 1C and D). As previously described, this difference could in principle influence the tendency for unwanted side reactions. In addition to a variation in light intensity, a second solution could be the use of light with a different wavelength (395 nm). While the absorption band of the Xan n–π* transition is still excited, there is in principle no contribution of the π–π* band of PABTC, thus potentially limiting side reactions.
To probe these hypotheses, the synthesis of pseudo-multiblock copolymers through XPI-RAFT polymerization was performed (Fig. 2). A sequence NAM was produced with a degree of polymerization (DP) ranging from 25 to 200 for 10 blocks (Tables S1–S3†), using a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :8 mixture of MeXan and PABTC (Fig. 2). The conversion for each step was driven to above 90% to render an intermediate purification unnecessary.
:8 mixture of MeXan and PABTC (Fig. 2). The conversion for each step was driven to above 90% to render an intermediate purification unnecessary.
 | ||
| Fig. 2 Synthesis of pseudo block copolymers using NAM, Xan and PABTC at different irradiation conditions. (A) Schematic overview of the synthesis and target structure. (B) PhotoCube from ThalesNano used to conduct photopolymerizations. (C) SEC traces calibrated (THF, PS calibration) showing chain extension for 10 blocks. (D) Evolution of Mn and Đ upon chain extension, (E) reaction time necessary to reach a conversion of >90% for each block and irradiation condition. | ||
Three different irradiation conditions at 365 nm with low (25 mW cm−2) and high (182 mW cm−2), as well as 395 nm with high (211 mW cm−2) intensity were employed (each measured at sample position) within a PhotoCube photoreactor (Fig. 2B). Reaction rates differed between the three setups (Fig. S1†) in the order 365 nm (high) > 395 nm (high) > 365 nm (low).
The size exclusion chromatography (SEC) curves (Fig. 2C) reveal that different wavelengths and intensities significantly influence the polymerization process and the molecular weight distribution of the synthesized polymers. Each color in the SEC curves corresponds to a different number of blocks, ranging from 1 to 10. Notably, the molecular weight increases with each additional block, indicative of successful chain extension under all conditions. However, significant differences are observed in the width and modality of the molecular weight distributions among the three different irradiation conditions.
At 365 nm with high intensity (top panel), the SEC traces reveal broad molecular weight distribution with significant presence of multiple distributions attributed to dead chains. This suggests a loss of control over the polymerization process, likely due to the degradation of the CTA under high-intensity irradiation. The high energy of the 365 nm light at full intensity can cause photodegradation of the CTAs, leading to premature termination of the polymer chains and reduced livingness. In contrast, at 365 nm with low intensity (middle panel), the SEC traces are narrower compared to those obtained at high intensity. The lower intensity reduces the energy input, mitigating the degradation of the CTAs and maintaining better control over the polymerization process. This condition produces polymers with a more controlled molecular weight distribution, although still with a significant presence of dead chains.
The control over polymer molecular weight at 395 nm with 100% intensity appears similar to that observed at 365 nm with low intensity, as reflected by the SEC traces. While dispersities are slightly lower, also required reaction times are reduced to reach the same result. This wavelength thus strikes a favorable balance between adequate energy to drive the polymerization forward and sufficient control to prevent significant CTA degradation, though it is comparable in performance to the lower-intensity 365 nm condition.
The evolutions of the number-average molecular weight (Mn) as a function of the number of blocks for each irradiation conditions (Fig. 2D) demonstrates a steady increase in Mn with the number of blocks, consistent with a controlled living polymerization process. The increased Đ to 2.13 for later blocks under 365 nm high intensity is indicative of dead chain formation. In contrast, the 365 nm at low intensity condition maintains a dispersity around 1.41, showing improved control and reduced side reactions. The best control over polymerization in terms of Đ is observed under 395 nm irradiation at high intensity, where the Đ = 1.31, maintaining the highest degree of livingness during polymerization. It is noteworthy that the final DP of polymer chain is with 725 relatively high for any of the discussed cases. Still, later blocks will likely possess an increased dispersity when compared to the overall system.36 Given the still considerable time necessary to reach high conversion even for 365 nm with high intensities (Fig. 2E), it is likely that in this case photoactivation outcompetes the rate of polymerization and that the velocity of the overall process is capped by chain growth. However, this also leads to more significant CTA degradation, causing broader molecular weight distributions and loss of control over the polymerization. In contrast, the 395 nm (high intensity) condition seems to offer a more balanced approach, while the polymerization rate is only slightly decreased.
To further investigate the observed differences in polymerization performance, we conducted a series of experiments where we chose to limit the system to one chain extension of PNAM with NAM to create pseudo diblock copolymers. This method enabled precise quantification of dead chain formation via deconvolution of SEC traces. Copolymers of varying DPs (50 + 50, 100 + 100, 250 + 250, and 500 + 500) were produced by sequential addition of monomer. The general irradiation times are provided in Table S4,† highlighting a trend where 365 nm (high intensity) achieved approximately twice the rate of polymerization relative to 365 nm (low intensity) and 395 nm (high intensity).
Fig. 3 presents SEC traces for all three polymerization conditions, with all reactions achieving near-complete monomer conversion (approximately 100%) except for PNAM500–PNAM500 under 395 nm irradiation, which reaches 97% conversion (Table S4†). The dispersity increases consistently with DP across all conditions, from 1.13 for PNAM50–PNAM50 to 1.29 for PNAM500–PNAM500. These findings suggest that dispersity alone may not serve as a straightforward optimization metric, given its sensitivity to various reaction conditions.
 | ||
| Fig. 3 SEC traces of pseudo-diblock copolymers of NAM at different irradiation conditions and DPs (solvent: THF with PS calibration). | ||
To evaluate chain-end fidelity, SEC trace deconvolution was performed on each diblock copolymer's extended block (Fig. 4A). A Gauss deconvolution model was applied, anchoring the first peak's maximum to the elution volume of the initial block and the second peak to the maximum of the extended block (an example is shown in Fig. 4C for PNAM500–PNAM500).
 | ||
| Fig. 4 Livingness of pseudo diblock copolymers based on XPI-RAFT polymerization at different reaction conditions. (A) Livingness as extrapolated from deconvolution of SEC traces for polymerization reactions at different DP values and irradiation conditions. (B) Livingness at constant DP (500 + 500) as a function of monomer (and CTA) concentration. (C) Exemplary deconvolution of SEC traces where peak maxima are based on the maxima of first and second block (performed in Origin software). | ||
Complete deconvolution results are presented in Fig. S2–S4,† with summary data in Table S6.† Notably, the highest livingness is achieved with 365 nm (low intensity), reaching a minimum of 73.5% for DP = 500. In contrast, 395 nm (high) demonstrated only 63.5% livingness. At 365 nm (high intensity), livingness drops to 56.4%, similar to that of the pseudo-multiblock system, where the highest dispersity index is also observed. The more pronounced loss in livingness of 395 nm (high) and 365 nm (low) could also be observed in photobleaching experiments (Fig. S5†). To quantitatively assess the photodegradation (bleaching) of CTAs during irradiation, we adopted the methodology of separating absorption and scattering contributions to extinction using Rayleigh scattering correction.37,38
These results underscore the importance of carefully controlled irradiation conditions to achieve both high livingness and narrow molecular weight distributions in photoiniferter polymerizations. The initial hypothesis, stating that highest livingness would be achieved if PABTC is not excited directly (under 395 nm light) seems unlikely based on these results.
The better performance of 365 nm light at low intensities when compared to irradiation at 395 nm points toward a connection of livingness and oversaturation. More precisely, the livingness decreases when photo excitation surpasses the threshold that is given by the velocity of the polymerization itself (limited by kp and monomer concentration). Every additional photon is only increasing the likelihood for side reactions leading to decreased chain end fidelity. If the excitation of the PABTC π–π* band would have been the main issue, the change in wavelength to 395 nm should have resulted in superior results.
For all irradiation conditions, a decrease in livingness with increasing DP is observed. While monomer concentration was kept constant for all mixtures, the increase in DP is connected to a decrease in the total CTA concentration. It can be assumed that polymerization rate is not changing but that overall radical concentration is lower due to the lower concentration of CTA molecules. Overall, the values achieved for livingness are relatively low with a maximum of ∼80%. However, this might be underestimated by the deconvolution method and the initial synthesis of multiblock copolymers indicates higher end group fidelities, as otherwise dispersities would be far higher for later blocks.
To elucidate whether the decline of livingness is a result of the increasing polymer length or rather is connected to the lowered CTA concentration under otherwise identical conditions, we conducted further experiments at varying monomer concentrations (Table S5 and corresponding SEC curves in Fig. S7†). The DP of 500 + 500 and 365 nm at high intensity were chosen for these experiments as these conditions are the most challenging investigated so far and should show the lowest chain end fidelity. The overall concentration was varied to match the CTA concentration at different DPs from the previous experiment.
The steep drop in livingness at lowest monomer/CTA concentration coincides with the results from the previous experiment where highest DPs possess lowest end group fidelity (Fig. 4B). It should be noted that overall dispersity is not considered but only the ratio of chain extended and non-extended block.
Thus, it can be concluded that under constant irradiation conditions, a decreased amount of CTA also leads to increased amount of side reactions. This supports the hypothesis of oversaturation as with lower concentrations of CTA, the ratio of photons to CTA is also increased drastically. Moreover, at the coinciding lower concentrations of monomer, the polymerization cannot progress as fast, limiting achievable velocity and making it easier to reach a condition where additional irradiation is not productive anymore. This is consistent with work on PI polymerization using visible light.22
A further way to precisely control the absorbed photon flux density39 in chemical reactions is the use of a flow reactor as here the flow speed, and hence the residence time within the photo-reactor can be tuned.25 Photoreactions occurring in dynamic flow setups have been reported to benefit from shorter pathway of light exposure and uniform delivery of energy to the system, which permits minimized side reactions and elevated reaction efficiency.40,41 Furthermore, flow setups facilitate the adjustment of parameter settings and the integration of inline analysis systems, leading to more reliable and reproducible data observations.42,43
As our reactor setup permits the introduction of a reactor coil, we attempted to produce pseudoblock copolymers by flow chemistry (Fig. 5A). The XPI polymerization performed in a dynamic flow coil reactor was expected to diminish Lambert's penalty while providing a flexible method to manipulate polymerization process via altering the residence time to avoid light oversaturation.
 | ||
| Fig. 5 Flow chemistry experiment using an 8 mL coil within a photo reactor to produce PNAM500 first block to be chain extended with a second block of NAM in batch reaction. (A) Schematic overview over the reaction. (B) Conversion as a function of time within the coil. (C) SEC traces of initial PNAM and after chain extension including peak deconvolution (deconvolution data presented in Fig. S8†). | ||
Herein, we conducted XPI RAFT polymerization to produce NAM homopolymer in the first block (targeted DP 500 at a monomer concentration of 1 mol L−1) at 395 nm (100%) in an 8 mL coil reactor. The conversion rate increases with longer retention time, reaching up to 93% at 64 minutes (Fig. 5B).
To then probe the livingness of the first block under conditions most comparable to the polymerizations carried out before, chain extension was performed in batch. This was also necessary as the viscosity of the solution made a chain extension in flow challenging. The first segment (retention time (Tr): 64 min) was used as the macroCTA under otherwise identical conditions as utilized in flow (targeted DP 500, monomer concentration = 1 mol L−1, 395 nm, 100%). Still the result should be indicative about the chain end fidelity of the macro-CTA as after chain extension any chain ends degraded during flow reaction would not be chain extended and present themselves as a low molar mass shoulder (Fig. S9†).
Indeed, as shown in Fig. 5C, the livingness is with about 60% relatively low and also lower as the comparable experiment that was conducted entirely in batch (where a livingness of 66% was achieved). As such even with precise control over the reaction and exposure time chain end degradation is pronounced. The decrease in livingness could be explained by the higher surface area of the reaction solution in flow when compared to batch experiments, which again could lead to local overexposure and hence loss in chain end fidelity.
Conclusions
This contribution investigates the role of wavelength and light intensity in XPI-RAFT polymerization and its influence on polymer livingness. An NTO analysis by TD-DFT calculations has been conducted, showing a significant difference in the nature of the S2 excited state of Xan and PABTC. The xanthate shows the contribution of the σ* orbital relevant for β-scission, which can explain the improved PI performance. Experimentally, long irradiation times can lead to CTA degradation, especially in multiblock syntheses involving longer sequences. A change in wavelength to activate the Xan more exclusively did not lead to the anticipated increase in livingness, while a reduction in intensity did (without substantially increased reaction times). As such over-saturation, as also shown for PI polymerization,22 is likely to cause loss in end group fidelity as it leads to unproductive excitation. Hence in an XPI process aiming for multiblock copolymers or other complex structures that require chain extension, a very low UV light intensity (as also used for multiblock production in PI polymerization8) is recommended. Still, for homopolymer synthesis high intensities are suitable and enable fast polymerization.The concentration of CTA is also a factor to be considered and similar to multiblock synthesis using RAFT polymerization,44 a high monomer and CTA concentration is beneficial. Since precise reaction time control in a flow chemistry setup did not lead to any improvements either, it can be concluded that photon flux density (in relation to CTA) is the most important parameter to optimized for highly living XPI polymerizations. While the aspect hasn't been tested in this study, a link to the type of monomer is likely. The maximum productive photon flux density is likely capped by how fast the respective monomer can propagate. As such oversaturation would be easier to reach for e.g. methacrylates (which is consistent with findings about the monomer scope in XPI-polymerization28).
Data availability
The raw data has been uploaded as ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge the financial support by the German Research Foundation (DFG) within the framework of the Emmy Noether program (T. W., M. H., project number 445804074) and the collaborative research center (CRC) 1636, project B03 (A. H. E., M. H., Project number 51094393). V. T. gratefully acknowledges the Alexander von Humboldt Foundation for providing a scholarship. J. G. acknowledges the support from the German Federal Ministry of Education and Research (Bundesministerium für Bildung und Forschung (BMBF) via the NanoMatFutur Program Gz. 13XP5237). We acknowledge the fruitful and critical discussion with Dr Christopher Penschke concerning our methodology and results of the computational studies. This work was performed using the compute cluster for High Performance Computing operated by the Center for Information Technology and Media Management of the University of Potsdam (https://www.uni-potsdam.de/en/zim/angebote-loesungen/hpc). The University Research Focus “Sustainable Materials Design” of the University of Potsdam is gratefully acknowledged for supporting this research. The authors thank the NMR core facility of the Institute of Chemistry (University of Potsdam) of Prof. Dr Heiko Möller, Dr Ruslan Nedielkov, and Angela Krtitschka. Prof. Dr Helmut Schlaad and Sascha Prentzel from the Institute of Chemistry (University of Potsdam) are gratefully acknowledged for providing the facility to perform SEC measurements.References
- J. Chiefari, Y. K. Chong, F. Ercole, J. Krstina, J. Jeffery, T. P. T. Le, R. T. A. Mayadunne, G. F. Meijs, C. L. Moad, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 1998, 31, 5559–5562 CrossRef CAS
.
- S. Perrier, Macromolecules, 2017, 50, 7433–7447 CrossRef CAS
- M. Destarac, Polym. Chem., 2018, 9, 4947–4967 RSC
- M. A. Beres, C. Boyer, M. Hartlieb, D. Konkolewicz, G. G. Qiao, B. S. Sumerlin and S. Perrier, ACS Polym. Au, 2025 DOI:10.1021/acspolymersau.4c00101
- T. Otsu, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 2121–2136 CrossRef CAS
- M. Hartlieb, Macromol. Rapid Commun., 2022, 43, 2100514 CrossRef CAS PubMed
- R. W. Hughes, M. E. Lott, R. A. Olson S and B. S. Sumerlin, Prog. Polym. Sci., 2024, 156, 101871 CrossRef CAS
- A.-C. Lehnen, J. A. M. Kurki and M. Hartlieb, Polym. Chem., 2022, 13, 1537–1546 RSC
- M. H. Stenzel and C. Barner-Kowollik, Mater. Horiz., 2016, 3, 471–477 RSC
- T. G. McKenzie, Q. Fu, E. H. H. Wong, D. E. Dunstan and G. G. Qiao, Macromolecules, 2015, 48, 3864–3872 CrossRef CAS
-
J. Xu, S. Shanmugam, N. A. Corrigan and C. Boyer, in Controlled Radical Polymerization: Mechanisms, American Chemical Society, 2015, vol. 1187, ch. 13, pp. 247–267 Search PubMed
- J. D. Coyle and H. A. J. Carless, Chem. Soc. Rev., 1972, 1, 465–480 RSC
- R. W. Hughes, M. E. Lott, J. I. Bowman and B. S. Sumerlin, ACS Macro Lett., 2023, 12, 14–19 CrossRef CAS PubMed
- S. R. Turner and R. W. Blevins, Macromolecules, 1990, 23, 1856–1859 CrossRef CAS
- L. Lu, H. Zhang, N. Yang and Y. Cai, Macromolecules, 2006, 39, 3770–3776 CrossRef CAS
- D. J. Keddie, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 2012, 45, 5321–5342 CrossRef CAS
-
R. Chapman, K. Jung and C. Boyer, in RAFT Polymerization, Wiley VCH, 2021, pp. 611–645. DOI:10.1002/9783527821358.ch12
- R. N. Carmean, T. E. Becker, M. B. Sims and B. S. Sumerlin, Chem, 2017, 2, 93–101 CAS
- B. Zhao, J. Li, Y. Xiu, X. Pan, Z. Zhang and J. Zhu, Macromolecules, 2022, 55, 1620–1628 CrossRef CAS
- J. F. Quinn, L. Barner, C. Barner-Kowollik, E. Rizzardo and T. P. Davis, Macromolecules, 2002, 35, 7620–7627 CrossRef CAS
- P. Lambrinos, M. Tardi, A. Polton and P. Sigwalt, Eur. Polym. J., 1990, 26, 1125–1135 CrossRef CAS
- M. A. Beres, J. Y. Rho, A. Kerr, T. Smith and S. Perrier, Polym. Chem., 2024, 15, 522–533 RSC
- I. Kurowska, M. Odnoroh, O. Ivanchenko, M. Guerre and M. Destarac, Polym. Chem., 2024, 15, 4888–4893 RSC
- S. B. Lemich, N. Sobania, N.-F. Meyer, P. Schütz, B. Hankiewicz and V. Abetz, Macromol. Chem. Phys., 2022, 2200355 Search PubMed
- G. D. Ammini, J. P. Hooker, J. Van Herck, A. Kumar and T. Junkers, Polym. Chem., 2023, 14, 2708–2716 RSC
- H. Zhou and J. A. Johnson, Angew. Chem., Int. Ed., 2013, 52, 2235–2238 CrossRef CAS PubMed
- M.-N. Antonopoulou, N. P. Truong, T. Egger, A. A. Kroeger, M. L. Coote and A. Anastasaki, Angew. Chem., Int. Ed., 2025, 64, e202420733 CrossRef CAS PubMed
- A.-C. Lehnen, J. Gurke, A. M. Bapolisi, M. Reifarth, M. Bekir and M. Hartlieb, Chem. Sci., 2023, 14, 593–603 RSC
- R. Whitfield, K. Parkatzidis, N. P. Truong, T. Junkers and A. Anastasaki, Chem, 2020, 6, 1340–1352 CAS
- K. Parkatzidis, N. P. Truong, M. N. Antonopoulou, R. Whitfield, D. Konkolewicz and A. Anastasaki, Polym. Chem., 2020, 11, 4968–4972 RSC
- J. Martin, M. Michaelis, S. Petrović, A.-C. Lehnen, Y. Müllers, P. Wendler, H. M. Möller, M. Hartlieb and U. Glebe, Macromol. Biosci., 2024, 2400316 Search PubMed
- A. M. Bapolisi, A.-C. Lehnen, M. Wolff, J. Kramer, S. Kogikoski Jr., R. Steinbrecher, N. Michler, A. Kiesow, I. Bald, M. Obry, S. Kersting, T. Stensitzki, H. M. Müller-Werkmeister, M. N. Leiske, S. Chiantia and M. Hartlieb, Angew. Chem., Int. Ed., 2025 DOI:10.1002/anie.202507564
- R. L. Martin, J. Chem. Phys., 2003, 118, 4775–4777 CrossRef CAS
- C. Yu, J.-K. Ha, M. Park, J. Lee, J. Choi, B. Y. Park, C. Boyer, S. K. Min and M. S. Kwon, Chem. Sci., 2025 10.1039/D5SC02594J
- M. D. Thum, S. Wolf and D. E. Falvey, J. Phys. Chem. A, 2020, 124, 4211–4222 CrossRef CAS PubMed
- G. Gody, P. B. Zetterlund, S. Perrier and S. Harrisson, Nat. Commun., 2016, 7, 10514 CrossRef CAS PubMed
- E. J. Cornel, S. van Meurs, T. Smith, P. S. O'Hora and S. P. Armes, J. Am. Chem. Soc., 2018, 140, 12980–12988 CrossRef CAS PubMed
- V. Tkachenko, C. Matei Ghimbeu, C. Vaulot, L. Vidal, J. Poly and A. Chemtob, Polym. Chem., 2019, 10, 2316–2326 RSC
- D. Cambié, C. Bottecchia, N. J. W. Straathof, V. Hessel and T. Noël, Chem. Rev., 2016, 116, 10276–10341 CrossRef PubMed
- L. D. Elliott, J. P. Knowles, P. J. Koovits, K. G. Maskill, M. J. Ralph, G. Lejeune, L. J. Edwards, R. I. Robinson, I. R. Clemens, B. Cox, D. D. Pascoe, G. Koch, M. Eberle, M. B. Berry and K. I. Booker-Milburn, Chem. – Eur. J., 2014, 20, 15226–15232 CrossRef CAS PubMed
- T. Junkers and B. Wenn, React. Chem. Eng., 2016, 1, 60–64 RSC
- M. A. Bereś, B. Zhang, T. Junkers and S. Perrier, Polym. Chem., 2024, 15, 3166–3175 RSC
- J. Van Herck, I. Abeysekera, A.-L. Buckinx, K. Cai, J. Hooker, K. Thakur, E. Van de Reydt, P.-J. Voorter, D. Wyers and T. Junkers, Digital Discovery, 2022, 1, 519–526 RSC
- G. Gody, T. Maschmeyer, P. B. Zetterlund and S. Perrier, Nat. Commun., 2013, 4, 2505 CrossRef PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section, materials and methods, supplementary figures and tables. See DOI: https://doi.org/10.1039/d5py00383k |
| This journal is © The Royal Society of Chemistry 2025 |