
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Biobased triblock thermoplastic elastomer with betulin- or carvacryl-methacrylate end-blocks by RAFT polymerization†
Aniello Vittore a,
Pauline Shamraienko*b,
Ilka Hermesb,
Qiong Lib,
Brigitte Voitbc and
Lorella Izzo*a
a,
Pauline Shamraienko*b,
Ilka Hermesb,
Qiong Lib,
Brigitte Voitbc and
Lorella Izzo*a
aDipartimento di Biotecnologie e Scienze della Vita, Università degli Studi dell'Insubria, via J.H. Dunant 3, 21100 Varese, Italy. E-mail: lorella.izzo@uninsubria.it
bLeibniz-Institut für Polymerforschung Dresden e.V., 01069 Dresden, Germany. E-mail: shamraienko@ipfdd.de
cOrganic Chemistry of Polymers, TUD Dresden University of Technology, 01062 Dresden, Germany
First published on 15th July 2025
Abstract
In this work, fully biobased acrylic ABA triblock copolymers were synthesized via reversible addition-fragmentation chain-transfer (RAFT) polymerization using VISIOMER® Terra C13 (ET13) as the “soft” midblock and two terpenoid-derived methacrylates, betulin methacrylate (BetuMA) and carvacryl methacrylate (CaMA), as the glassy blocks. An “R-linked” bifunctional chain transfer agent (bis-CTA) enabled the formation of ET13 macro-CTAs with controlled molecular weights and narrow dispersity (Mn = 84–229 kg mol−1, Đ ≈ 1.1). RAFT homo-polymerizations of BetuMA and CaMA yielded well-defined homopolymers (Mn = 20–33 kg mol−1, Đ < 1.4) with selective methacrylate reactivity. Chain extension of ET13 macro-CTAs produced a series of ABA triblocks featuring 8–39 mol% glassy content. GPC confirmed molecular weights in the range Mn = 97–415 kg mol−1 (Đ < 1.7), while DSC and TGA analyses showed distinct glass transitions for soft block, close to −50 °C, and good thermal stability. AFM evidenced clear microphase separation. Mechanical testing revealed that BetuMA-based copolymers (BEB series) achieved tensile strengths up to 3.9 MPa and elongations up to 760%, outperforming CaMA-based analogs (CEC series: σ ≤ 1.2 MPa, ε ≤ 710%). These results demonstrate the efficacy of RAFT polymerization of terpenoid methacrylates in producing high-performance, sustainable thermoplastic elastomers, offering a viable alternative to petroleum-derived thermoplastic elastomers (TPEs).
Introduction
Among thermoplastic elastomers (TPEs), styrenic block copolymers (SBCs), like poly(styrene-butadiene-styrene) (SBS) and poly(styrene-isoprene-styrene) (SIS), are widely used but face challenges such as oxidation and UV degradation due to unsaturated bonds in their soft blocks.1 In this framework, acrylic-based copolymers represent a promising alternative overcoming degradation stability and with advantages like excellent performance as pressure-sensitive adhesives. Commercially available acrylic TPEs are ABA triblock copolymers based on oil-derivatives, with a soft-middle part made of poly(butyl acrylate) and the external glassy blocks of poly(methyl methacrylate)s.2 In the pursuit of more sustainable materials, considerable efforts are being dedicated to substituting oil-derived components with biobased feedstocks.3–5 To this end, fatty acid derivatives (e.g. poly(lauryl methacrylate)6,7) as well as sugar or lignin derivatives (e.g. isosorbide,8 itaconic acid imides,9 rosin10) were investigated as acrylic monomers for the soft or glassy blocks, respectively. Acrylic biobased monomers offer a wide range of possibility to find a balance between sustainability and performance for diverse applications.11 However, the final copolymers generally exhibit lower performance compared to fossil-based acrylic materials and further investigation appears worthy.In this work, Reversible Addition Fragmentation Chain Transfer (RAFT), a reversible-deactivation radical polymerization (RDRP), was applied for the synthesis of the ABA triblock copolymers using acrylic monomers from renewable feedstocks. Aiming to evaluate different external blocks while keeping the same middle chain, the following research was conducted using a commercially available biobased monomer for the middle block and two different monomers as the glassy components. Plant oils and their fatty acids, due to their low toxicity, availability, and relatively low price, are a particularly attractive building block sources.12 A methacrylate alcohol mixture of average C13 is produced by Evonik, under the name VISIOMER® Terra C13-MA (ET13), with 76% of certified bio-content. The relative polymer has a low glass transition temperature (Tg = ca. −50 °C), resulting in a promising soft-middle block. It is worth noting that the ET13 structure is very similar to that of lauryl methacrylate, in which the alkyl side chain consists of twelve carbon atoms, already reported as a middle elastomeric block.6,7,13 ET13 has been used by Milan Marić's research group as a soft polymer to produce sustainable statistical and block copolymers.14 They also report examples of triblock structures yet with few characterization details, since their work focuses more on the synthesis.15
As biobased monomers for the external blocks A, carvacryl methacrylate (CaMA) and betulin methacrylate (BetuMA) were selected. Carvacrol is a monoterpenoid phenol derivable from the essential oil of Origanum vulgare,16 while betulin is a triterpenoid, with a pentacyclic ring structure and three potential reactive sites. Betulin is found in large quantities in the bark of white birch (Betula papyrifera).17 Materials like branches, bark, leaves, and roots are often waste materials from the wood industry and discarded or incinerated, despite containing valuable elements such as lignin and betulin.
Before the copolymer synthesis, the investigation of homopolymers was conducted by their synthesis and characterization. Two series of ABA copolymers incorporating betulin or carvacrol derivatives were synthesized and characterized employing chemical and material analyses.
Experimental
Materials and methods
All chemicals were of reagent grade and used as received unless otherwise indicated. VISIOMER Terra C13-MA (ET13, Evonik) was passed through a column of basic alumina to remove inhibitors prior to use. S-4-Cyano-4-(dodecylthiocarbonothioylthio)pentanoic acid (CDP) was purchased from Agene Chemicals. Azobisisobutyronitrile (AIBN, Merck, 98%) was recrystallized from methanol and dried in vacuum prior to use. Betulin (Betu, 98.3%) was supplied biobased from Nature Science Technologies chemicals and dried under vacuum at 40 °C overnight before usage. Toluene, 1,4-dioxane, dichloromethane (DCM), anhydrous magnesium sulfate (MgSO4) and 1,4-butanediol were purchased from Merck. 4-Dimethylaminopyridine (DMAP), dicyclohexyl carbodiimide, methacrylic anhydride, 1,3,5-trimethylbenzene and carvacrol (>98.0%) were purchase from TCI. 2-Propanol (99.8%) and THF (99.6%) stabilized with BHT was purchased from Thermo Fischer Scientific. 4-Hydroxy-2,2,6,6-tetramethylpiperdinyloxyl (TEMPOL) and phenothiazine were provided by Hilti Entwicklungsgesellschaft mbH.Synthetic procedures
Characterization methods
Results and discussion
The ABA triblock copolymers were synthesized via RAFT polymerization by a two-step procedure, as shown in Scheme 1. In the first step, the B block was obtained through RAFT polymerization using an “R-linked” bifunctional chain transfer agent (bis-CTA). The bifunctional macro-CTA was then employed in the second step for a RAFT chain extension of the termini. | ||
| Scheme 1 Synthetic pathway of the ABA triblock. | ||
The bis-CTA has been synthesized by esterification of the monofunctional CTA S-4-cyano-4-(dodecylthiocarbono thioylthio)pentanoic acid (CDP), reported to be a suitable CTA for methacrylates.22
Since the macro-CTA would represent the B block of the ABA structure, meaning the elastomeric part of the material, VISIOMER Terra C13-MA (ET13) has been chosen as monomer. Indeed, ET13 is a mixture of methacrylate alcohols having an average of C13 and thus it was considered suitable to provide polymers with low Tg. In the second step, the chain extension of the macro-CTA has been carried out with either BetuMA or CaMA. Such terpenes would confer rigidity to the B blocks, with the consequent formation of “hard” domains.
Overall, the study reported is organized as follow: preliminary investigations were conducted on the RAFT of terpene methacrylates BetuMA and CaMA, and on ET13 to determine the conditions of the process to control the chain growth at both low and high molecular weights. The best experimental conditions, in term of yield and kinetic control over the monomer conversion, have been employed in the second step for the synthesis of the final ABA structure.
RAFT polymerization of terpene methacrylates
The biobased compounds used for monomer synthesis were procured with high purity, first converted into methacrylates and purified before use – CaMA by washing and BetuMA by recrystallization. Only the soft block monomer ET13 (containing ∼5% MMA) was used without further procedure and according to NMR analysis there were no impurities (Fig. S3, S4, and S10†) to impair the radical polymerization.Betulin, an abundant natural triterpenoid, presents three potential reactive sites: two hydroxyl groups (a primary and a secondary one) and an alkenyl group. The primary alcohol was selectively functionalized by a transesterification reaction to insert the polymerizable methacrylic group and form BetuMA (further details in ESI, Fig. S1–S3†). RAFT polymerization of BetuMA was carried out using CDP as the monofunctional CTA (Scheme 2). Results are reported in Table 1.
 | ||
| Scheme 2 (A) Betulin and carvacrol chemical structures and origins; (B) RAFT polymerization of betulin methacrylate (BetuMA) and carvacryl methacrylate (CaMA). | ||
| Entry | Solvent | T (°C) | CTA/AIBN ratio | t (h) | Conversiona (%) |
|---|---|---|---|---|---|
| Reaction conditions: molar equivalent BetuMA/CTA/AIBN 100/1/0.5, 0.2 or 0.1 in dioxane (76 wt%).a Determined by 1H-NMR (CDCl3) of the crude reaction mixture.b Toluene 90 wt%.c Polymer only partially soluble in toluene.d Determined using mesitylene as internal standard. | |||||
| 1 | Tolueneb | 80 | 2 | 8 | >40c |
| 2 | Dioxane | 80 | 2 | 2 | 94 |
| 3 | Dioxane | 80 | 5 | 4 | 97d |
| 4 | Dioxane | 80 | 10 | 6 | 89d |
BetuMA was reported to be slightly soluble in toluene20 thus, a polymerization was first conducted in such a solvent. However, despite using a large excess of toluene, precipitation occurred during the reaction (entry 1). Since dioxane was found to be a more appropriate solvent for both the monomer and polymer, the following polymerizations were performed in dioxane. CTA/initiator ratio was also adjusted to achieve a good conversion in a reasonable time. Albeit with a ratio CTA/AIBN = 2 or 5 an almost full conversion was reached in about 2 or 4 h (entries 2 and 3, respectively), increasing the ratio to 10 (entry 4), a pseudo first kinetic order (Fig. 1A, triangles) and a linear growth of the molecular masses (Fig. 1B, triangles) evidenced a better control over the process. Nevertheless, deviations from ideal RAFT behaviour were observed, such as an induction period and a final number-average molecular weight (Mn = 58.8 kg mol−1), determined by GPC, that exceeded the theoretical value (Mn(th) = 46 kg mol−1). Furthermore, the dispersity (Đ = 1.4–1.7) remained higher than expected for a well-controlled system. These deviations may arise from non-ideal initiation associated with an incomplete CTA consumption.23
 | ||
| Fig. 1 RAFT polymerization of BetuMA: kinetic plot (A) and graph of Mn and Đ versus monomer conversion (B) of entry 3 (circle) and entry 4 (triangle) of Table 1. | ||
In conclusion, at 80 °C and with a CTA/AIBN ratio of 10, BetuMA exhibited a good polymerization kinetic, achieving nearly a complete conversion within 6 h. Besides, 1H-NMR spectroscopy confirmed that the alkenyl group remained unreactive during the polymerization, indicating the high selectivity of RAFT for the methacrylic group. Contrarywise, when a free radical polymerization was performed in similar conditions (BetuMA 100 equiv., AIBN 0.2 equiv., Dioxane, 80 °C, 1 h), the formation of an insoluble microgel was observed.
RAFT polymerization of carvacryl methacrylate (CaMA) (Scheme 2) was carried out using CDP as monofunctional CTA and different targeted degrees of polymerization (DPtarget). As reported in Table 2, good conversions were obtained at lower DP targets (entries 1 and 2), with kinetic plots showing a first order kinetics (Fig. 2A). GPC analyses confirmed a linear molecular mass growth, close to the theoretical one, and a quite good polydispersity index (Đ < 1.3) (Fig. 2B).
 | ||
| Fig. 2 RAFT polymerization of CaMA (A) kinetic plot and (B) Mn and Đ versus conversion of entry 1 (square) and entry 2 (triangle) of Table 2. | ||
Both PBetuMA and PCaMA were characterized by TGA and DSC analyses (Fig. 3). PBetuMA exhibited outstanding thermal stability with a starting point of degradation above 300 °C, while PCaMA degradation began at around 160 °C, with the maximum loss at around 290 °C (Fig. 3A). DSC calorimetry evidenced a glass transition temperature (Tg) of about 79 °C and 220 °C for PCaMA and PBetuMA, respectively (Fig. 3B).
 | ||
| Fig. 3 Thermal analyses of BetuMA and CaMA homo-polymers (A) TGA curves and the relative derivative (inset) and (B) DSC second heating curve. | ||
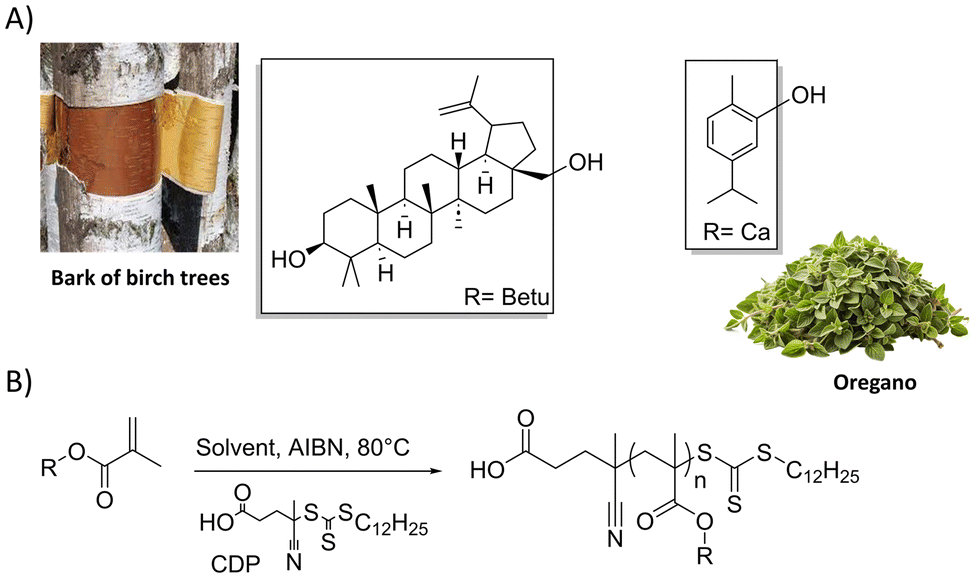
Finally, a kinetic study was carried out on the RAFT polymerization of ET13 with two different degrees of polymerization: DP 500 or DP 1500. In both cases, a first order kinetics, a linear growth of molecular masses vs. monomer conversion and low dispersities were observed, as shown in Fig. 4 (further details in ESI, Fig. S11–S14†).
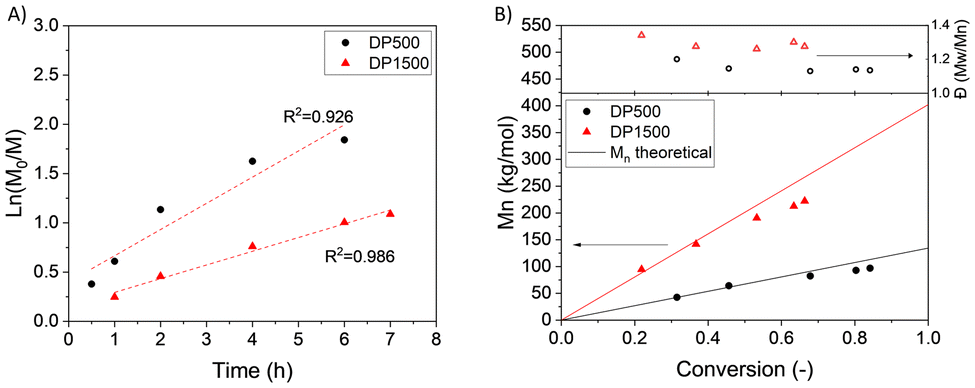 | ||
| Fig. 4 RAFT polymerization of ET13 with two different degrees of polymerization (DP500 or DP1500): kinetic plot (A) and Mn and Đ versus monomer conversion (B). Reaction conditions: ET13/bis-CTA/AIBN molar equivalent 500 or 1500/1/0.5 in 50 wt% toluene at 80 °C. | ||
ABA triblock copolymers
ET13 bis-macro-CTA was synthesized by RAFT with an almost full conversion. Three macro-CTAs, named E1, E2 and E3, with Mn 84.1 kg mol−1, 108 kg mol−1 and 229 kg mol−1, respectively, have been then used for the following chain extension with BetuMA or CaMA (B and C, respectively in Table 3). Aiming to have ABA triblock copolymers with different fractions of the “hard” component, the reaction conditions have been changed using different monomer/macro-CTA ratios (meaning DPtarget) or polymerization times. It is worth noting that generally in an ABA structure, TPEs are characterized by low amounts of the “hard” component; thus, the design of the final copolymer composition provided a percentage of BetuMA or CaMA in the range 15–30 wt%. Polymerization conditions and experimental results are reported in Table 3; the copolymer composition was estimated by 1H NMR spectroscopy and representative spectra are reported in Fig. 5 (further details in ESI, Fig. S15–17†). | ||
| Fig. 5 Representative 1H-NMR spectra of macro-CTA (E1) and the relative copolymers with BetuMA (BE1B 3) or CaMA (CE1C 3). | ||
| ABA polymer | DPtarget | Time (h) | Molar/weight fractiona (%) | Mtotn (th)b (kg mol−1) | Mn/Đc (kg mol−1) |
|---|---|---|---|---|---|
Macro-CTA: E1 (Mn = 84.1 kg mol−1, Đ = 1.10), E2 (Mn = 108 kg mol−1, Đ = 1.11), E3 (Mn = 229 kg mol−1, Đ = 1.25). Reaction conditions: BEB = macro-CTA/BetuMA/AIBN 1/DPtarget/0.5 in dioxane (80 wt%) at 80 °C; CEC = macro-CTA/CaMA/AIBN 1/DPtarget/0.5 in toluene (50 wt%) at 80 °C.a Relative glassy block amount determined by 1H NMR spectroscopy (further details in Fig. S15†).b Total theoretical molecular weight:  where XB and MMB are the molar fraction and molecular mass respectively.c Determined by GPC in THF.d Toluene–dioxane mixture used as reaction solvent. where XB and MMB are the molar fraction and molecular mass respectively.c Determined by GPC in THF.d Toluene–dioxane mixture used as reaction solvent. |
|||||
| BE1B 1 | 50 | 3 | 8/14 | 97 | 105/1.12 |
| BE2B 2 | 80 | 3 | 10/17 | 122 | 127/1.21 |
| BE1B 3 | 100 | 5 | 17/28 | 116 | 138/1.17 |
| BE2B 4d | 100 | 5 | 13/22 | 132 | 165/1.30 |
| BE3B 5d | 200 | 1.5 | 9/17 | 275 | 207/1.55 |
| BE3B 6d | 400 | 5 | 21/34 | 345 | 415/2.00 |
| CE1C 1 | 230 | 7 | 25/21 | 106 | 101/1.18 |
| CE1C 2 | 300 | 15 | 26/22 | 106 | 97/1.17 |
| CE1C 3 | 400 | 24 | 39/34 | 126 | 113/1.22 |
| CE3C 4 | 600 | 20 | 13/11 | 257 | 200/1.45 |
| CE3C 5 | 600 | 30 | 20/17 | 276 | 176/1.76 |
Triblock compositions (Table 3) spanned from 8 to 21 mol% for PBetuMA (BEB copolymers) and from 13 to 39 mol% for PCaMA (CEC copolymers). Since the macro-CTA with higher Mn, namely E3, was only partially soluble in dioxane, a mixture of toluene and dioxane was used as solvent (see BE3B 5 and BE3B 6 in Table 3) and for comparison purpose, BE2B 4 was synthesized using the same solvent mixture.
Refractive index SEC chromatograms showed a time downshift of the copolymers’ curves compared to that of the macro-CTA; moreover, all the final copolymers presented a narrow and monomodal dispersity clearly indicating the formation of copolymeric structures. Finally, the calculated absolute molecular weights were in good accordance with the theoretical ones (Fig. 6 and Fig. S18†). It should be noted that the extension of macro-CTA E3 appeared less effective than E1 or E2. Indeed, for the relative copolymers (CE3C 4, CE3C 5 and BE3B 6) the chromatogram traces show tailing which increases with the hard block ratio, indicating a not ideal macro-CTA extension.
 | ||
| Fig. 6 Characteristic refractive index GPC traces of (A) macro-CTA E1 and of its relative copolymer and (B) macro-CTA E3 and its relative copolymer, with BetuMA (BEB) or CaMA (CEC). | ||
TPE characterization
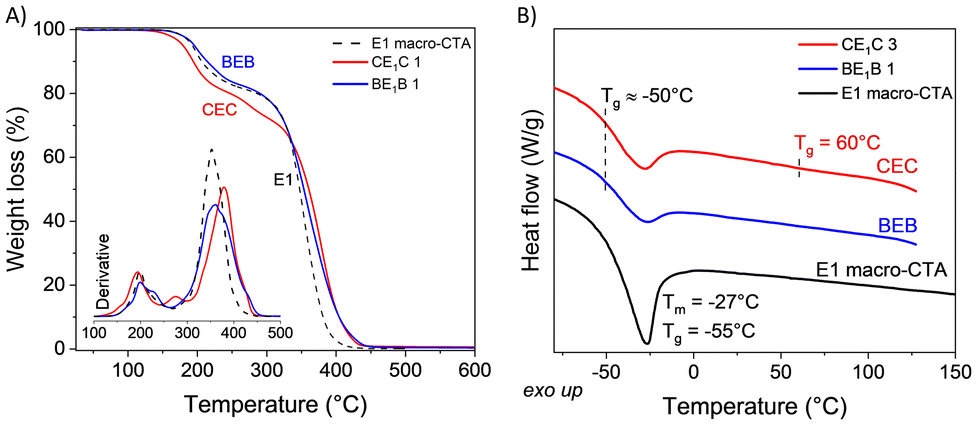 | ||
| Fig. 7 (A) TGA scans and the relative derivative (inset), and (B) representative DSC second heating scans of E1 macro-CTA and its relative copolymers BEB and CEC. | ||
 | ||
| Fig. 8 AFM phase image of triblock copolymer with PCaMA (CE3C 5, CE1C 2 and CE1C 3) or PBetuMA (BE3B 5, BE1B 3 and BE3B 6) as external blocks. Corresponding topography images in ESI, Fig. S19.† | ||
Copolymers with PBetuMA at 17 wt% (BE3B 5) presented an assembly of bright PBetuMA spheres (29 nm) in a dark PET13 matrix. As PBetuMA content increased (BE2B 4 and BE3B 6), samples exhibited a phase separation with a predominant lamellar structure. The domains of the two phases were larger for the sample with PBetuMA at 34 wt% (BE3B 6 = 90 nm vs. BE1B 3 = 30 nm).
Considering the significant difference in the structure of the two glassy block monomers, the morphological change is rather comparable with the different ratios. The low concentration of PBetuMA gives a very similar pattern to the low concentration of PCaMA, as do the medium and high concentrations. The difference is greatest for the latter, but the trend is clearly towards greater domain formation.
To investigate how the differences in the microdomain distribution translate into the mechanical properties of the polymer, the stress–strain behaviour was examined in the mechanical tests.
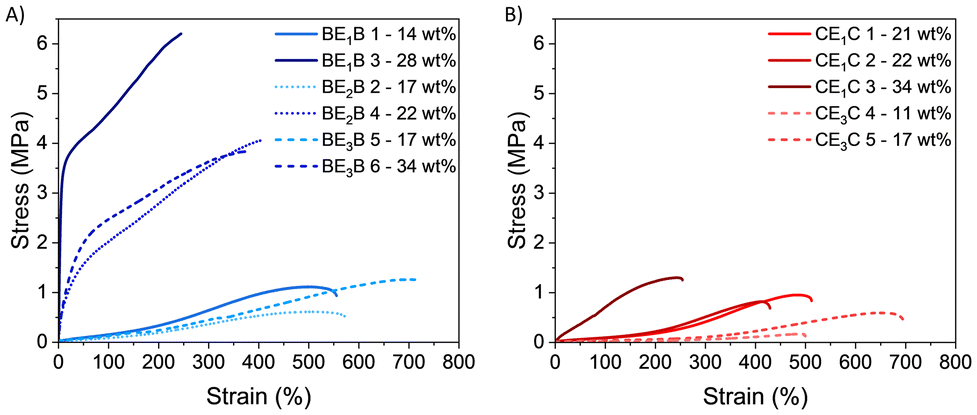 | ||
| Fig. 9 Stress–strain graph of BEBs (A) and CECs copolymers (B) (solid lines indicates E1, dotted lines E2 and dashed lines E3 as macro-CTAs). | ||
| Sample | Glassy block, weight (%) | Modulus (MPa) | Ultimate strength, σ (MPa) | Elongation at break, ε (%) |
|---|---|---|---|---|
| BE1B 1 | 14 | 0.43 | 1.1 | 524 |
| BE2B 2 | 17 | 0.17 | 0.7 | 532 |
| BE1B 3 | 28 | 132 | 6.2 | 238 |
| BE2B 4 | 22 | 8.0 | 3.9 | 363 |
| BE3B 5 | 17 | 0.32 | 1.4 | 759 |
| BE3B 6 | 34 | 11 | 3.9 | 359 |
| CE1C 1 | 21 | 0.24 | 1.0 | 526 |
| CE1C 2 | 22 | 0.22 | 0.8 | 428 |
| CE1C 3 | 34 | 1.7 | 1.2 | 274 |
| CE3C 4 | 11 | 0.07 | 0.2 | 437 |
| CE3C 5 | 17 | 0.14 | 0.7 | 710 |
All samples show elastomeric behaviour with quasi linear stress–strain relationships and without any detectable yield point (Fig. 9).
An evident fast increase of initial modulus, which reached a remarkable final strength (σ) higher than 3 MPa and elongation at break (εb) between 240% and 360%, can be seen for samples containing more than 20% of PBetuMA, namely BE1B 3, BE2B 4 and BE3B 6. Samples with a lower content of glassy blocks (BE1B 1, BE2B 2 and BE3B 5) evidenced an elastomeric behaviour at low strain with a following hardening region, starting from about ε = 200%, typical of rubbers and TEPs (Fig. 9A). These samples exhibited a strength close to 1 MPa and an elongation at break εb above 500%. Noteworthy, the latter εb was higher when compared to the εb of samples with higher glassy amount.
Comparing copolymers with 17 wt% of PBetuMA but with different length of the middle block, the sample with the longest PET13 (BE3B 5) presented higher elongation (εb = 760%) than the one with the shortest middle chain (BE2B 2, εb = 530%). Looking at the tensile test results of CEC copolymers (Fig. 9B), all the samples analysed evidenced an elastomeric behaviour with strength close to or below 1 MPa and an elongation at break decreasing as PCaMA content increased. For example, CE1C 3, containing the highest amount (34 wt%), exhibited a rapid increase of strength up to 1.2 MPa and the lowest εb = 270%. When comparing samples with similar length of PET13 (E1 and E2), CECs copolymers showed slightly lower elongation (εb ≈ 420–500%) than BEB samples (εb ≈ 530%), but a comparable ultimate strength in the range 0.7–1.1 MPa.
Finally, all the samples with E3 as the middle block showed a different behaviour depending on the nature of the hard blocks; indeed, copolymers containing CaMA showed the lowest strength (σ < 0.7 MPa). This can be evidenced by comparing samples with 17 wt% of glassy block content: CE3C 5 showed εb = 710% and σ = 0.65 MPa, while BE3B 5 had εb = 760%, and σ = 1.35 MPa (Table 4).
Generally, as the glassy content increased, the modulus and the ultimate tensile stress increased as well, while the elongation at break decreased; however, the magnitude of this behaviour depended on the nature of the glassy blocks. A comparison of elongation and strength versus the nature and the amount of the glassy component is reported in Fig. 10. By increasing the glassy content, copolymers with BetuMA (BEB) showed a more marked tensile strength increase (Fig. 10, upper panel, blue circle) compared to copolymers with CaMA (CEC) (Fig. 10, red circle). Finally, similar properties are feasible with far less molar percent of BetuMA compared to CaMA: for example, a tensile strength around 1 MPa and an elongation around 500% were shown by samples with 8–10 mol% of BetuMA (14–17 wt%), while similar properties are achievable with about 25–26 mol% of CaMA (21–22 wt%). The correlation of the mechanical characteristics with the glassy block behaviour depending on the monomer structure was further investigated with X-ray analysis to determine possible crystalline domains.
 | ||
| Fig. 10 Comparison of copolymers elongation (lower panel) and tensile strength (upper panel) versus glassy block (Red symbols represent CECs while blue ones BEBs; full symbols represent copolymers made with E1 or E2 as macroCTA, while empty symbols are for copolymers made by E3). | ||
 | ||
| Fig. 11 WAXS diffractograms of selected ABA copolymers. | ||
The peak with an equivalent Bragg spacing of about 0.5 nm corresponds to the van der Waals (VDW) contacts of atoms and is known as the VDW peak. Samples with PCaMA as external blocks, even at high content of CaMA (CE1C 3, 34 wt%), show evidence only of reflexes associated with the middle block. On the other hand, samples containing PBetuMA showed a shoulder with a maximum at about 15° that became more evident as the PBetuMA content increased in the copolymer. This reflex could be due to a partial organization of the betulin rings in a stacked structure, forming crystalline domains. Indeed, betulin itself was reported to form crystalline structures with the more intense peak showing a maximum around 14–15°.29 This finding supports the AFM results on the formation of larger hard block domains with increasing PBetuMa content (Fig. 8), which also impacts the stress–strain behaviour. Leading to the highest tensile strength, but the lowest strain and elongation values (Fig. 9 and 10).
Conclusions
This study demonstrates the successful synthesis of biobased acrylic ABA triblock copolymers via RAFT polymerization, emphasizing the potential of renewable materials in thermoplastic elastomer (TPE) applications. The use of biobased monomers ET13, BetuMA, and CaMA resulted in ABA block copolymers with control of molar mass and a well-defined block structure.Thermal analysis confirmed good stability and a well distinct phase separation, essential for TPE materials, was observed via AFM and supported by DSC and WAXS analysis. Tensile tests showed the presence of elastomeric behaviour for all the block copolymers, with better performance of BEBs compared to CECs. Furthermore, the BEB block copolymers, incorporating BetuMA, achieved high tensile strength and elongation at break with a lower glassy block content, reflecting the superior properties of BetuMA (high Tg = 220 °C, formation of crystalline domains) compared to CaMA as hard block forming monomer and a different influence of the glassy block nature on the mechanical properties. BEB copolymers, particularly those with moderate glassy block content (e.g. BE3B 5), provide an optimal combination of tensile strength and elongation at break. Higher glassy block content increases stiffness but sacrifices elongation. The macro-CTA molecular weight influences the elongation and strength significantly, with E3-based samples showing superior elongation.
Overall, BEB and CEC samples provide a competitive and sustainable alternative to similar oil-based triblock copolymers reported in the literature. Besides, BEBs have only slightly lower performance when compared to an all-acrylic commercial TPE (Kurarity LA2140e, poly(MMA-butyl acrylate-MMA), 23% MMA, σ = 8.1 MPa, ε = 580%).24
These findings underscore the viability of biobased TPEs as sustainable alternatives to fossil-derived counterparts, paving the way for further research into optimizing their performance and expanding their industrial applications.
Author contributions
A. V. investigation, data curation, validation, writing – original draft. P. S. conceptualization, supervision, investigation, writing – original draft. I. H. data curation, writing – review & editing. Q. L. AFM data acquisition and formal analysis. B. V. supervision, writing – review & editing. L. I. supervision, writing – review & editing, funding acquisition.Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the ESI.† All raw and meta data of the measurements are saved in the institute server and are available on request.Acknowledgements
We want to thank our supporting colleagues at IPF, especially Regine Boldt, for the WAXS measurements and Hartmut Komber for the help with the NMR investigations. Also, we want to acknowledge Karsten Scheibe for the mechanical measurements on our delicate samples and Kerstin Arnhold for the TGA & DSC measurements. Evonik, we are grateful to for providing the free sample of VISIOMER® Terra C13. AV and LI are thankful for the support of ESF (European Social Fund) – REACTEU Programme (DM 1061/2021 ).References
- J. G. Drobny, in Handbook of thermoplastic elastomers, PDL(Plastics Design Library)/William Andrew Pub, Norwich, NY, 2014 Search PubMed.
- Advances in thermoplastic elastomers: challenges and opportunities, ed. N. K. Singha and S. C. Jana, Elsevier, Amsterdam London Cambridge, 2024 Search PubMed.
- J. H. Jeon, J. H. Jung and C. Choi, J. Polym. Sci., 2023, 62, 662 CrossRef.
- A. Vittore, O. Santoro, M. Candida, S. Vaghi, S. Pragliola, M. Mella and L. Izzo, Polymer, 2025, 324, 128228 CrossRef CAS.
- F. Molinari, A. Salini, A. Vittore, O. Santoro, L. Izzo, S. Fusco, L. Pollegioni and E. Rosini, Bioresour. Technol., 2024, 408, 131190 CrossRef CAS PubMed.
- K. Y. Cho, S. S. Hwang, H. G. Yoon and K.-Y. Baek, J. Polym. Sci., Part A:Polym. Chem., 2013, 51, 1924–1932 CrossRef CAS.
- S. Wang, W. Ding, G. Yang and M. L. Robertson, Macromol. Chem. Phys., 2016, 217, 292–303 CrossRef CAS.
- M. Nasiri, D. J. Saxon and T. M. Reineke, Macromolecules, 2018, 51, 2456–2465 CrossRef CAS.
- K. Satoh, D. Lee, K. Nagai and M. Kamigaito, Macromol. Rapid Commun., 2014, 35, 161–167 CrossRef CAS PubMed.
- W. Ding, S. Wang, K. Yao, M. S. Ganewatta, C. Tang and M. L. Robertson, ACS Sustainable Chem. Eng., 2017, 5, 11470–11480 CrossRef CAS.
- C. Veith, F. Diot-Néant, S. A. Miller and F. Allais, Polym. Chem., 2020, 11, 7452–7470 RSC.
- Y. Xia and R. C. Larock, Green Chem., 2010, 12, 1893–1909 RSC.
- D. P. Chatterjee and B. M. Mandal, Macromolecules, 2006, 39, 9192–9200 CrossRef CAS.
- F. Asempour, E. Laurent, Y. Ecochard and M. Maric, ACS Appl. Polym. Mater., 2024, 6, 956–966 CrossRef CAS.
- S. Tajbakhsh, F. Hajiali and M. Marić, Ind. Eng. Chem. Res., 2020, 59, 8921–8936 CrossRef CAS.
- N. Leyva-López, E. Gutiérrez-Grijalva, G. Vazquez-Olivo and J. Heredia, Molecules, 2017, 22, 989 CrossRef PubMed.
- S. A. Kuznetsova, G. P. Skvortsova, I. N. Maliar, E. S. Skurydina and O. F. Veselova, Russ. J. Bioorg. Chem., 2014, 40, 742–747 CrossRef CAS.
- T. M. Hinton, C. Guerrero-Sanchez, J. E. Graham, T. Le, B. W. Muir, S. Shi, M. L. V. Tizard, P. A. Gunatillake, K. M. McLean and S. H. Thang, Biomaterials, 2012, 33, 7631–7642 CrossRef CAS PubMed.
- P. Voigt, N. Kiriy, K. Jähnichen and B. Voit, ACS Sustainable Chem. Eng., 2024, 12, 1984–1996 CrossRef CAS.
- D. V. Orekhov, A.P. Sivokhin, A. S. Simagin, O. A. Kazantsev, K. V. Otopkova, A. D. Ovchinnikov, I. N. Makhov and N. B. Melnikova, Polym. Adv. Technol., 2023, 34, 2529–2540 CrossRef CAS.
- J. F. Stanzione, J. M. Sadler, J. J. La Scala and R. P. Wool, ChemSusChem, 2012, 5, 1291–1297 CrossRef CAS PubMed.
- RAFT Polymerization, ed. G. Moad and E. Rizzardo, Wiley, 1st edn, 2021, vol. 1 Search PubMed.
- S. Boner, K. Parkatzidis, N. D. A. Watuthanthrige and A. Anastasaki, Eur. Polym. J., 2024, 205, 112721 CrossRef CAS.
- S. Wang, L. Shuai, B. Saha, D. G. Vlachos and T. H. Epps, ACS Cent. Sci., 2018, 4, 701–708 CrossRef CAS PubMed.
- J. J. Gallagher, M. A. Hillmyer and T. M. Reineke, ACS Sustainable Chem. Eng., 2016, 4, 3379–3387 CrossRef CAS.
- A. Rasmont, P. Leclère, C. Doneux, G. Lambin, J. D. Tong, R. Jérôme, J. L. Brédas and R. Lazzaroni, Colloids Surf., B, 2000, 19, 381–395 CrossRef CAS PubMed.
- W. Wang, R. Schlegel, B. T. White, K. Williams, D. Voyloy, C. A. Steren, A. Goodwin, E. B. Coughlin, S. Gido, M. Beiner, K. Hong, N.-G. Kang and J. Mays, Macromolecules, 2016, 49(7), 2646–2655 CrossRef CAS.
- G. Floudas and P. Štepánek, Macromolecules, 1998, 31, 6951–6957 CrossRef CAS.
- X. Niu, E. J. Foster, B. O. Patrick and O. J. Rojas, Adv. Funct. Mater., 2022, 32, 2206058 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental, spectroscopic and characterisation details. See DOI: https://doi.org/10.1039/d5py00540j |
| This journal is © The Royal Society of Chemistry 2025 |