
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDevelopment of dual acid–visible light-degradable core-crosslinked nanogels with extended conjugate aromatic imines for enhanced drug delivery†
Kadambari Bairagi a,
Mehdi Shamekhibc,
Ioanna Tountasd,
Natasha Letourneaud,
Gilles H. Peslherbeabc,
Alisa Pieknyd and
Jung Kwon Oh*a
a,
Mehdi Shamekhibc,
Ioanna Tountasd,
Natasha Letourneaud,
Gilles H. Peslherbeabc,
Alisa Pieknyd and
Jung Kwon Oh*a
aDepartment of Chemistry and Biochemistry, Concordia University, Montreal, Quebec H4B 1R6, Canada. E-mail: john.oh@concordia.ca
bDepartment of Physics, Concordia University, Montreal, Quebec H4B 1R6, Canada
cCenter for Research in Molecular Modeling, Concordia University, Montreal, Quebec H4B 1R6, Canada
dDepartment of Biology, Concordia University, Montreal, Quebec H4B 1R6, Canada
First published on 25th June 2025
Abstract
The development of stimuli-responsive amphiphilic block copolymers and their nanoassemblies/nanogels integrated with degradable covalent chemistry undergoing chemical transitions has been extensively explored as a promising platform for tumor-targeting controlled/enhanced drug delivery. The conjugate aromatic imine bond is unique in responding to acidic pH through acid-catalyzed hydrolysis and visible light through photo-induced E/Z isomerization, thus allowing for a dual acid–light response via a single conjugate aromatic imine bond. Herein, we report a robust strategy for fabricating well-defined core-crosslinked nanogels bearing extended conjugate aromatic imine linkages that exhibit controlled degradation in response to acidic pH and visible light. This approach utilizes the pre-crosslinking of a poly(ethylene glycol)-based block copolymer bearing reactive imidazole pendants with a diol crosslinker bearing an extended conjugate aromatic imine, followed by the mechanical dispersion of the formed crosslinked polymers in an aqueous solution. The fabricated core-crosslinked nanogels with a hydrodynamic diameter of 119 nm are non-cytotoxic, colloidally stable, and capable of encapsulating cancer drug curcumin. They exhibit controlled/enhanced release of encapsulated curcumin at pH = 5 (acidic) or upon irradiation with visible light (λ = 420 nm) as well as exhibit promisingly accelerated and synergistic release under the combination of the above conditions. Furthermore, curcumin-loaded nanogels reduce cell viability in a controlled manner, unlike free drugs. This simplified yet efficient synthetic approach paves the way for the development of smart nanocarriers with potential applications in controlled drug release and cancer therapy.
Introduction
The development of well-defined amphiphilic block copolymers (ABPs) and their nanoassemblies has emerged as a promising platform for the effective intracellular delivery of anticancer therapeutics to tumors.1–6 They consist of hydrophobic cores, which enable the encapsulation of hydrophobic drugs, and hydrophilic corona, which endows them with biocompatibility and structural stability.7 After being intravenously administered, the nanoassemblies extravasate into tumor tissues primarily through the enhanced permeability and retention (EPR) effect.8–15 Following endocytosis into cancer cells, they are expected to control the release of anticancer drugs. However, conventional nanoassemblies present a critical drawback of the lack of control over the release of therapeutics from conventional nanocarriers at tumor sites.To overcome this drawback, stimuli-responsive degradation (SRD) has been integrated into the design of ABPs and their nanoassemblies.16–21 SRD-exhibiting nanoassemblies have been designed with degradable (or labile) covalent bonds, which are cleaved in response to stimuli, preferably endogenous stimuli found in cellular environments.22–24 The degradation causes the disintegration of nanoassemblies, leading to controlled/enhanced release of encapsulated drugs, which improves the biodistribution of small drug molecules, thereby enhancing therapeutic efficacy and minimizing off-target cytotoxicity. Acidic pH is a typical endogenous stimulus because of pH 6.5–6.9 in the tumoral extracellular cellular compartment and pH 4.5–5.5 in endo-/lysosomes.25–27 In addition to the endogenous acidic pH stimulus, light is considered a promising exogenous stimulus to achieve on-demand delivery. Typically, imine, acetal and ketal groups as acid-labile linkages28–35 and o-nitrobenzyl and coumarin dimer groups as photo-cleavable linkages36–38 have been incorporated into ABP-based nanoassemblies for single stimulus acid- and light-responsive degradation. Moreover, a combination of two distinct linkages, typically including the o-nitrobenzyl group (light-responsive) with β-thioester,39 acetal,40 and imine41 groups (acid-responsive), has been formulated to achieve a dual acid/light-response. Despite these advances, conventional strategies could increase synthetic complexity, limit scalability, or cause instability issues. Our recent proof-of-concept studies demonstrate the versatility of our approach by exploring single conjugate benzoic imine chemistry that enables a dual response to acidic pH through acid-catalyzed hydrolysis and UV light through photo-induced E/Z isomerization.42 However, the approach could be limited for tumor-targeting drug delivery because the conjugate benzoic imine bond has its main absorption in the UV region, which is harmful to tissues. We envisioned that extending the conjugation of the benzoic imine bond could enhance its light absorption toward the visible light region. To prove this, we explore an advanced approach that centers on the integration of an extended conjugate benzoic imine bond into SRD-ABP-based nanoassemblies to achieve a dual acidic pH/visible light response.
Another drawback of conventional nanoassemblies involves their nature of self-assembly through the physical entanglement of ABPs in aqueous solutions. Upon dilution in blood (4 L), they are destabilized or disintegrated into the corresponding unimers, causing undesired premature release of drug molecules during blood circulation. To circumvent this drawback, stimuli-responsive degradable core-crosslinked nanoassemblies have been developed.43–45 They endow structural stability upon dilution, while they facilitate the enhanced release of encapsulated drugs in response to endogenous or exogenous stimuli. A prevalent approach for fabricating core-crosslinked nanoassemblies (or nanogels) is called in situ crosslinking. This approach involves the fabrication of aqueous nanoassemblies of reactive ABPs bearing pendant functional groups, followed by crosslinking induced with external crosslinkers. Click-type reactions, such as azide–alkyne cycloaddition,46–48 conjugation,49–52 and in situ imine formation,53–56 allowed for the fabrication of reduction or acid-degradable core-crosslinked nanogels labeled with disulfide,47,48,51,52 imine,53–56 and ketal/acetal46,49,50 linkages. This approach has been proven to be effective; however, when hydrophobic crosslinkers are required for use, this approach remains a critical challenge owing to their limited solubility in the aqueous dispersion of nanoassemblies. We recently explored an alternative but robust approach that centers on the pre-crosslinking of a reactive ABP with a crosslinker in organic solvent, followed by the mechanical dispersion of the formed crosslinked polymers in aqueous solution, yielding well-defined core-crosslinked nanogels. This approach has allowed for precise control over the sizes and functionalities of SRD-exhibiting nanoassemblies with varying ratios of ABP/solvent and reactive functional groups.
In this work, we explore the robust pre-crosslinking dispersion approach to fabricate colloidally stable, core-crosslinked nanogels exhibiting a dual acid/visible light response with a single extended conjugate aromatic imine bond (Scheme 1). A diol bearing an extended conjugate benzoic imine bond (M3) was newly synthesized as an effective crosslinker for a well-defined PEG-based ABP bearing reactive imidazole pendants (PEG-b-PCIMA). The formed crosslinked polymers (P-Im3-DMSO) were mechanically dispersed in aqueous solution, yielding well-defined core/shell nanogels (P-Im3-Aq) with hydrophobic crosslinked cores surrounded by hydrophilic PEG coronas. They were characterized for dual acid/visible light-responsive degradation through acid-catalyzed hydrolysis and photo-induced E/Z isomerization, confirmed by spectroscopic and computational studies. Further, curcumin-loaded nanogels (P-Im3-Aq/Cur) were evaluated for the uptake and release of encapsulated curcumin, which is an anticancer drug, in HeLa cells.
 | ||
| Scheme 1 A robust approach exploring pre-crosslinking and dispersion techniques that allow for the fabrication of colloidally stable, core-crosslinked nanogels exhibiting dual acid/visible light response with a single extended conjugate aromatic imine bond, exhibiting controlled/enhanced release of the encapsulated Cur drug inside cancer cells. The nanogels are expected to enter cells via endocytosis and Cur can be released in acidic pH environments and upon exposure to light. | ||
Experimental
Instrumentation
1H NMR spectra were recorded using a 500 MHz Varian spectrometer. CDCl3 singlet at 7.26 ppm and DMSO-d6 quintet at 2.5 ppm were selected as reference standards. The exact mass of M3 was analyzed by High Resolution Mass Spectroscopy using a Thermo LTQ Orbitrap Velos mass spectrometer equipped with a heated electrospray ion source. A full MS spectrum (m/z 150–600) was acquired in the Orbitrap in positive mode at a resolution of 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000. Fourier-transform infrared (FT-IR) spectra were collected using a Thermo Scientific Nicolet iS5 spectrometer with an iD5 attenuated total reflection (ATR) accessory. UV/vis spectra were obtained using an Agilent Cary 60 spectrometer with a 1-cm quartz cuvette. The hydrodynamic diameter and size distribution of aqueous nanogels were analyzed by dynamic light scattering (DLS) using a Malvern Nano S ZEN1600 system equipped with a 633-nm He–Ne laser at a fixed scattering angle of 175° and a temperature of 25 °C.
000. Fourier-transform infrared (FT-IR) spectra were collected using a Thermo Scientific Nicolet iS5 spectrometer with an iD5 attenuated total reflection (ATR) accessory. UV/vis spectra were obtained using an Agilent Cary 60 spectrometer with a 1-cm quartz cuvette. The hydrodynamic diameter and size distribution of aqueous nanogels were analyzed by dynamic light scattering (DLS) using a Malvern Nano S ZEN1600 system equipped with a 633-nm He–Ne laser at a fixed scattering angle of 175° and a temperature of 25 °C.
Light-responsive experiments were conducted with a PR160L-440 nm Gen 2 LED lamp (Kessil, USA) at λ = 440 nm (60 mW cm−2) and a MAX-302 xenon arc lamp (Asahi Spectra) at λ = 420 nm (1.7 mW cm−2) at a distance of 15 cm from the light sources.
Transmission electron microscopy (TEM) images were acquired using an FEI Tecnai G2 F20 200 kV Cryo-STEM with Gatan Ultrascan 4000 4k × 4k CCD Camera System Model 895. For negative staining, a 1% phosphotungstic acid solution was prepared by dissolving 5 mg of phosphotungstic acid in 0.5 mL of deionized water, followed by the addition of 5 μL of 1 M sodium hydroxide and filtration through a 0.45 μm PES membrane. Aqueous dispersions (5 μL) were drop-cast onto a 300-mesh copper TEM grid and left undisturbed for 2 min. Excess liquid was blotted with filter paper before applying a drop of stain. The grid was blotted again and dried under a fume hood before imaging.
Differential scanning calorimetry (DSC) analysis was conducted to investigate thermal properties, including glass transition temperature (Tg) of crosslinked polymers using a TA Instruments DSC Q20 Differential Scanning Calorimeter. Polymer samples were dried in a vacuum oven for 24 h to remove residual solvents. The temperature ranged from −80 to 200 °C with heating and cooling cycles at a rate of 10 °C min−1 (cycles: cool to −80 °C and heat up to 200 °C (1st run), cool to −80 °C and heat up to 200 °C (2nd run), and cool to 25 °C). Tg values were determined from the 2nd heating run. Thermogravimetric analysis (TGA) was conducted using a TA Instruments Q50 Analyzer. Dried polymer samples (10 mg) were placed in a platinum pan inside a programmable furnace and then heated from 25 to 800 °C at a heating rate of 20 °C min−1 under nitrogen flow.
Materials
2-Hydroxyethyl methacrylate (HEMA, 99%, purified by passing through a column filled with basic aluminum oxide to remove the inhibitor), 1,1′-carbonyldiimidazole (CDI, 97%), 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU, 98%), 4-(methylamino)pyridine (DMAP, 99%), 4-cyano-4 (phenylcarbonothioylthio) pentanoic acid (CTA, 97%), 4-aminobenzyl alcohol (ABA, 98%), terephthalaldehyde (TDA, 98%), curcumin (Cur, 99%), methoxy-terminated poly(ethylene glycol) (PEG, MW = 5000 g mol−1, dried by an azeotropic distillation with anhydrous toluene prior to use), and 2,2′-azobis(2-methylpropionitrile) (AIBN, 98%) from Sigma-Aldrich and 1-ethyl-3-(3 dimethylaminopropyl) carbodiimide·HCl (EDC) from Matrix Innovation were purchased and used as received. A carbonyl imidazole-bearing methacrylate (CIMA)40 and a CTA-functionalized PEG (PEG-RAFT)57 were synthesized, as described elsewhere.Synthesis of M3
ABA (0.73 g, 6.0 mmol) was mixed with TDA (0.40 g, 3.0 mmol) in DMF (7 mL) in an oil-bath preset at 100 °C under stirring for 18 h. The resulting mixture was purified by precipitation from cold diethyl ether. The precipitate was dried in a vacuum oven at room temperature for 24 h to yield a yellow solid. Yield = 0.70 g (67%). 1H NMR (DMSO-d6, ppm): 8.7 (s, 2H, –N![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif)
![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) C6H4N–), 8.06 (m, 4H, NCH
C6H4N–), 8.06 (m, 4H, NCH![[6 with combining low line]](https://www.rsc.org/images/entities/char_0036_0332.gif) 4CHN), 7.32–7.25 (m, 8H, –
4CHN), 7.32–7.25 (m, 8H, –![[4 with combining low line]](https://www.rsc.org/images/entities/char_0034_0332.gif) NCH CHN –), 5.22 (s, 2H, –CH2
NCH CHN –), 5.22 (s, 2H, –CH2![[O with combining low line]](https://www.rsc.org/images/entities/char_004f_0332.gif) ), 4.5 (s, 4H, –2OH). 13C NMR (DMSO-d6, ppm): 159.8, 150.1, 141.3, 138.9, 129.3, 127.8, 63.0. Mass calculated for C22H20N2O2 [M+H]+: 345.1598 and found: 345.1600.
), 4.5 (s, 4H, –2OH). 13C NMR (DMSO-d6, ppm): 159.8, 150.1, 141.3, 138.9, 129.3, 127.8, 63.0. Mass calculated for C22H20N2O2 [M+H]+: 345.1598 and found: 345.1600.
Synthesis of PEG-b-PCIMA via RAFT polymerization
CIMA (1 g, 4.5 mmol), PEG-RAFT (0.24 g, 45.6 μmol), and AIBN (3.7 mg, 22.8 μmol) were mixed with anisole (1.7 mL) in a 10 mL Schlenk flask. The mixture was purged with nitrogen for 45 min and then placed in an oil-bath preset at 70 °C to start polymerization. After 3 h, the mixture was cooled to room temperature to stop polymerization. For purification, as-synthesized polymers were precipitated from cold hexane and isolated by vacuum filtration. The procedure was repeated three times; then, the polymer was dried in a vacuum oven at 40 °C for 24 h.CDI-mediated crosslinking to fabricate P-Im3-DMSO crosslinked polymers
M3 (26 mg, 74.3 μmol, 149 μmol equivalent of OH groups) was mixed with PEG-b-PCIMA (52.5 mg, 3.6 μmol, 149 μmol equivalent of CIMA units) and DBU (4.5 mg, 30 μmol) in DMSO (6 mL) under stirring at room temperature for 24 h. The formed crosslinked polymers were precipitated from cold diethyl ether and then dried in a vacuum oven at 40 °C for 24 h.Fabrication of aqueous P-Im3-Aq nanogels by pre-crosslinking dispersion approach
Typically, to fabricate P-Im3-Aq at 0.31 mg mL−1, M3 (1.7 mg, 5 μmol) was mixed with PEG-b-PCIMA (3.5 mg, 0.2 μmol) and DBU (0.3 mg, 2 μmol) in DMSO (2 mL) under stirring at room temperature for 24 h. Deionized water (10 mL) was then added dropwise at 60 mL h−1 using a syringe pump under stirring. The resulting dispersion was dialyzed against water (1 L) using a dialysis tubing with MWCO = 11–13 kDa for 24 h and then filtered with a 0.45 μm PES filter.A similar procedure was examined with varying amounts of DMSO and water to investigate DMSO/water ratio (called P-Im3-Aq-A, B, and C) as well as varying amounts of PEG-b-PCIMA and M3 to investigate CI/OH mole equivalent ratio (called P-Im3-Aq-D and E). The recipes are summarized in Table S1 (ESI†).
Conventional in situ crosslinking approach in attempt to fabricate aqueous crosslinked nanoassemblies
Typically, deionized water (10 mL) was added dropwise to an organic solution consisting of M3 (4.8 mg, 14.0 μmol) and PEG-b-PCIMA (10 mg, 0.7 μmol) in DMSO (2 mL) at a rate of 60 mL h−1 using a syringe pump under magnetic stirring at room temperature. The resulting dispersion was dialyzed against water (1 L) using dialysis tubing with MWCO = 11–13 kDa for 24 h. DBU (0.8 mg, 5.6 μmol) was added, and the resulting mixture was stirred at room temperature for another 24 h.Acid-responsive degradation of aqueous nanoassemblies
Aqueous P-Im3-Aq nanogels (2 mL, 0.31 mg mL−1) were mixed with 0.2 M sodium acetate buffer solution at pH = 5.0 (2 mL) under stirring at room temperature. Aliquots were taken for DLS and TEM analyses.Acid-responsive degradation of M3 in DMSO using 1H NMR spectroscopy
To investigate the acid-catalyzed hydrolysis of benzoic imine bonds, M3 (10 mg, 30 μmol) was dissolved in DMSO-d6 (1 mL) and then mixed with HCl (5 μL, 60 μmol) at a 1/1 mole equivalent ratio of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N/H+ (equivalent to pH = 1.2 or pD = 1.6, calculated using pD = pH + 0.4). 1H NMR spectra of the resulting mixtures were recorded for given time intervals. A similar procedure was used for the P-Im3-DMSO polymer, except with the use of P-Im3-DMSO (10 mg) swollen in DMSO-d6 (1 mL).
N/H+ (equivalent to pH = 1.2 or pD = 1.6, calculated using pD = pH + 0.4). 1H NMR spectra of the resulting mixtures were recorded for given time intervals. A similar procedure was used for the P-Im3-DMSO polymer, except with the use of P-Im3-DMSO (10 mg) swollen in DMSO-d6 (1 mL).
Light-responsive degradation of aqueous nanoassemblies
Aqueous P-Im3-Aq nanogels (3 mL) in a quartz cuvette were exposed to irradiation with visible light with λ = 440 nm (60 mW cm−2). Their UV/vis spectra were recorded to follow the change in absorbance at λ = 355 nm.Light-responsive degradation of M3 in DMSO via UV/vis spectroscopy
A solution of M3 (31 μg) dissolved in DMSO (3 mL) at 30 μM in a quartz cuvette was exposed to irradiation with visible light with λ = 440 nm (60 mW cm−2). Their UV/vis spectra were recorded to monitor changes in absorbance at λ = 355 nm over irradiation time.Computational details
Density functional theory (DFT) calculations with the range-separated hybrid exchange–correlation (XC) functional based on B97 exchange (ωB97XD) are performed to evaluate the structural and electronic properties of the molecular structures.58 The molecular geometries were fully optimized at the ωB97XD level of theory along with the 6-311++G(d,p) split-valence triple-ζ Pople basis set.59 Vibrational frequency analysis was carried out to confirm that each configuration was a minimum on the potential energy surface (zero imaginary frequencies), and thermodynamic properties were evaluated within the rigid-rotor harmonic oscillator approximation. The STQN method was used to locate the transition states for the interconversion of the structures. Three structure specifications are required in this method: the reactants (EE for EE → EZ and EZ for EZ → ZZ), the products (EZ for EE → EZ and ZZ for EZ → ZZ), and an initial guess for the transition state structure. The energies of the highest occupied molecular orbital (HOMO), lowest unoccupied molecular orbital (LUMO), and electronic bandgap (Eg) between these orbitals were determined by analyzing frontier molecular orbitals (FMOs). The energies of the singlet-excited state transitions were calculated using a time-dependent DFT (TD-DFT) for optimized molecular structures. We note that ωB97XD is a reliable XC functional to accurately capture the UV/vis light absorption spectra for the structures owing to the partitioning of short and long range components, compared to traditional hybrid XC functionals, such as the Becke three-parameter hybrid methods with the Lee–Yang–Parr correlation functional (B3LYP).60 For all the calculations, the Gaussian 16 (revision C.01) software package of programs was used along with the GaussView6 interface for structure and orbital manipulations.61 The enthalpy and free energy of the reaction were calculated, as summarized in Table S2 (ESI†).Preparation of Cur-loaded nanogels
An organic solution of Cur (1 mg) dissolved in DMSO (2 mL) was added dropwise to aqueous P-Im3-Aq nanogel dispersion (0.31 mg mL−1, 12 mL) under stirring for 1 h. The resulting mixture was transferred to dialysis tubing with MWCO = 11–13 kDa for dialysis against water (1 L) for 24 h. The dispersion was filtered through a 0.45 μm PES filter to remove unencapsulated (free) Cur, yielding aqueous Cur-loaded nanogels (P-Im3-Aq/Cur) at 0.27 mg mL−1.To determine the loading level of Cur in nanogels, a series of solutions containing different amounts of Cur in a mixture of THF and water (1/1 v/v) were prepared, and their UV/vis spectra were recorded to determine the extinction coefficient of Cur at λ = 430 nm. Then, aliquots of P-Im3-Aq/Cur (1 mL) were mixed with THF (1 mL) and filtered through a 0.45 μm PTFE filter. UV/vis spectrum of the filtrate was recorded to quantify encapsulated Cur using the pre-determined extinction coefficient.
Stimuli-responsive release of Cur from P-Im3-Aq/Cur nanogels
For light-responsive release, aliquots of P-Im3-Aq/Cur (2 mL) were irradiated with visible light with λ = 420 nm (1.7 mW cm−2). Other aliquots were kept in the dark as controls. For acidic pH-responsive release, aliquots of P-Im3-Aq/Cur (2 mL) were mixed with 0.2 M sodium acetate buffer solution (2 mL) at pH = 5.0 and 7.4 under stirring. For dual acidic pH/light-responsive release, aliquots of P-Im3-Aq/Cur (2 mL) mixed with 0.2 M sodium acetate buffer solution (2 mL) at pH = 5.0 were exposed to visible light with λ = 420 nm (1.7 mW cm−2) at room temperature under stirring. To determine the %release of Cur from nanogels, UV/vis spectra of supernatants were recorded for given time intervals, and the absorbance at λ = 430 nm was monitored.Cell culture
HeLa cervical cancer cells were cultured in DMEM (Dulbecco's modified Eagle's medium) containing 10% CCS (cosmic calf serum) and maintained at 37 °C with 5% CO2 in an HERAcell Vios 160i incubator (ThermoFisher Scientific).Viability assay
HeLa cells were plated at 2.5 × 103 cells per well in a 96-well plate and incubated for 24 h in 100 μL of DMEM with 10% CCS for 24 h at 37 °C with 5% CO2. Various concentrations of aqueous P-Im3-Aq/Cur nanogels as well as aqueous P-Im3-Aq nanogels (empty, e.g. Cur-free) and free Cur as controls were added to the cells for 72 h. Owing to the poor solubility of Cur in aqueous media, aliquots of Cur dissolved in DMSO were diluted with cell culture media to maintain DMSO as low as 1% for all control samples. Cell viability was measured using the WST-8 proliferation assay kit, as per the manufacturer's instructions (Cayman Chemicals). Briefly, the medium containing the samples was carefully removed, and 100 μL of the prepared colorimetric reagent WST-8 (0.5 mM, 5-(2,4-disulfophenyl)-3-(2-methoxy-4-nitrophenyl)-2-(4-nitrophenyl)-2H-tetrazolium, inner salt, monosodium salt) was added to each well. After 2 h, a Tecan Infinite M200 Pro plate reader was used to measure the absorbance at λ = 450 nm for each well. Wells with untreated cells were measured as a positive control, while the WST-8 reagent alone was measured to have a ‘blank’ reference. Each treatment was replicated 3 times. Cell viability was calculated as the percentage ratio of the absorbance of mixtures with nanogels to the control (untreated cells).Live cell imaging by fluorescence microscopy
HeLa cells were plated in DMEM with 10% CCS at 2.5 × 105 cells per mL in a μ-slide 4-well dish. The cells were treated with 240 μg mL−1 of P-Im3-Aq/Cur nanogels, 2.72 μg mL−1 free Cur solution (2.72 μg mL−1) or 240 μg mL−1 of P-Im3-Aq and SYTO Deep Red Nucleic Acid Stain (Thermo Fisher) for 2 h at 37 °C with 5% CO2. Live cells were visualized using a Nikon Ti2 microscope equipped with a Yokogawa CSU-X1 spinning disk, a Gataca Live-SR unit, an Andor Zyla 4.2 (Oxford Instruments, UK), 60×/1.45NA PlanApo objective lens and DIC optics. Cur was excited with a λ = 488 nm laser (18% power, 200 ms exposure time), and SYTO Deep Red was excited with a 638 nm laser (10% power, 200 ms exposure time). Images were acquired using Micro-Manager. Images were viewed and analyzed using ImageJ.Results and discussion
Synthesis and UV/vis absorption characteristics of extended conjugate benzoic imine-bearing diol crosslinker
Fig. 1a depicts our approach for synthesizing a new diol crosslinker bearing two conjugate benzoic imine bonds (called extended conjugate benzoic imine bonds), labelled M3. This approach utilizes a facile condensation reaction of the amine group of ABA with two aldehyde groups of TDA at an elevated temperature through the formation of benzoic imine bonds. 1H NMR spectrum in Fig. 1b shows the new peak at 8.72 ppm (a) corresponding to imine proton, along with the peak at 4.53 ppm (e) corresponding to methylene protons in benzyl moieties and the peaks at 7.35 ppm (b) and 8.07 ppm (c) corresponding to aromatic protons. Their integrals were quantitative to the number of protons. This result, combined with the 13C NMR spectrum (Fig. S1, ESI†) and HR-MS (Fig. S2, ESI†), confirms the successful synthesis of M3. | ||
| Fig. 1 Synthesis of M3 (a diol crosslinker bearing an extended conjugate benzoic imine) through the condensation reaction of ABA with TDA in DMF (a), its 1H NMR spectrum in DMSO-d6 (b), and its UV/vis spectrum in DMSO, compared with those of M2 (a methacrylate bearing a conjugate benzoic imine bond) and M–HX (a methacrylate bearing a benzoic imine bond) in DMF (c). | ||
Given the synthesis of M3, its UV/vis absorption characteristics were investigated in DMSO and compared with M–HX (a methacrylate bearing a benzoic imine bond)62 and M2 (a methacrylate bearing a conjugate benzoic imine bond).42 As depicted in Fig. 1c, M3 exhibits an extended absorption of up to 450 nm, with a distinct absorption at λ = 355 nm corresponding to n → π* transition and a secondary absorption at λ = 310 nm attributed to π → π* transition. Its extinction coefficient at λ = 355 nm was determined to be 32000 M−1 cm−1 (Fig. S3, ESI†). Promisingly, its absorption was red-shifted because of the extended benzoic imine conjugation, compared with M2 having its maximum absorption at λ = 285 nm (ε = 20100 M−1 cm−1 in DMF) and further to M–HX having its maximum absorption at λ = 255 nm (ε = 2200 M−1 cm−1 in DMF).
Synthesis of reactive PEG-b-PCIMA copolymer bearing imidazole pendant
As illustrated in Fig. 2a, RAFT polymerization was examined for CIMA in the presence of PEG-RAFT, a PEG-based macro-RAFT mediator, initiated with AIBN in anisole at 70 °C, to synthesize a well-defined PEG-based block copolymer bearing reactive imidazole pendants (PEG-b-PCIMA). The conditions include the initial mole ratio of [CIMA]0/[PEG-RAFT]0/[AIBN]0 = 100/1/0.5 with the targeted degree of polymerization (DP) = 100 at complete monomer conversion. The synthesized copolymer was purified by precipitation from hexane to remove unreacted CIMA at 40% conversion. 1H NMR spectrum in Fig. 2b shows the presence of PEG at 3.6 ppm and pendant imidazole rings at 7.0–8.2 ppm. Their integral ratio, with the DP of PEG = 113, allows for the determination of the DP of the PCIMA block to be 41, thus forming PEG113-b-PCIMA41. | ||
| Fig. 2 Synthesis via RAFT polymerization (a) and 1H NMR spectrum in DMSO-d6 of the PEG-b-PCIMA block copolymer. Conditions: [CIMA]0/[PEG-RAFT]0/[AIBN]0 = 100/1/0.5; CIMA/anisole (w/w) = 1/0.6. | ||
Studies of CDI-mediated crosslinking reaction to fabricate crosslinked polymers
Given the synthesis of well-defined PEG-PCIMA and M3 diol crosslinker, their CDI-mediated coupling reaction between pendant CI groups with OH groups was investigated in an organic solvent (Fig. 3a). DMSO was chosen because it was found to be a good solvent for both PEG-PCIMA and M3 precursors. With a choice of CI/OH mole equivalent ratio = 1/1, aliquots of PEG-PCIMA and M3 were mixed in DMSO at 50 mg mL−1 to induce CDI-mediated crosslinking. The formed PEG-PCIMA/M3 crosslinked polymer (called P-Im3-DMSO) was isolated by precipitation from diethyl ether and then analyzed by solubility, 1H NMR and FT-IR spectroscopies for chemical structure, and DSC and TGA for thermal properties. | ||
| Fig. 3 Schematic presentation of the CDI-medicated crosslinking reaction of PEG-b-CIMA with the M3 diol crosslinker to form a cross-linked P-Im3-DMSO polymer network (a) and its FT-IR spectrum compared with those of M3 and PEG-b-PCIMA precursors (b). | ||
The polymer turned out to be insoluble in any organic solvent, including DMSO, and its gel content to measure insoluble species in DMSO was greater than 84%. Given that the polymer is swollen in DMSO-d6, the 1H NMR spectrum in Fig. S4 (ESI†) shows no peaks corresponding to protons in the copolymer. In addition, the 1H NMR spectrum of the supernatant in Fig. S5 (ESI†) shows two peaks at 7.0 and 7.6 ppm, presenting imidazole, which could be generated as a side product during CDI-mediated crosslinking. Moreover, its FT-IR spectrum in Fig. 3b shows the characteristic CN stretching vibration at 1620 cm−1, along with C–H stretching vibration at 2800–3000 cm−1 and carbonyl vibration at 1700–1750 cm−1. These results confirm the formation of a crosslinked polymer network through the formation of carbonate bonds by the CDI-medicated crosslinking reaction of PEF-b-PCIMA with M3. The crosslinked polymer had a single glass transition at −21 °C and a single melting transition at 55.2 °C by DSC analysis (Fig. S6, ESI†). It also had the temperature at which its mass loses half to be 407 °C with 15% residue by TGA analysis (Fig. S7, ESI†).
Pre-crosslinking dispersion approach to fabricate aqueous crosslinked nanogels
A new approach centered on pre-crosslinking in a homogeneous solution was explored to fabricate colloidally stable crosslinked nanogels dispersed in an aqueous solution. The approach consists of two steps: (i) the synthesis of P-Im3-DMSO crosslinked polymer from a mixture of PEG-b-PCIMA and M3 at CI/OH = 1/1 in DMSO and (ii) its spontaneous self-assembly in water to yield nanogel dispersion. Because of the amphiphilic nature of the P-Im3-DMSO polymer, the formed nanogels consist of crosslinked hydrophobic cores surrounded by hydrophilic PEG corona in water.After being purified by dialysis, the resulting dispersion was determined to be 0.31 mg mL−1, which is close to that (0.36 mg mL−1) in the recipe, suggesting that the loss of polymeric species during the pre-crosslinking/dispersion process could be negligible. The purified aqueous nanogels were characterized for size and size distribution as well as morphology. As shown in Fig. 4a, the nanogels had a diameter of 119 nm, and their size distribution appeared to be narrow and monomodal based on DLS analysis. No visible aggregates were observed. TEM analysis reveals that they had a diameter of 87 ± 21 nm in the dried state, which is smaller than that determined by the DLS analysis (inset in Fig. 4a). Furthermore, the nanogels were characterized for UV/vis absorption characteristics. As compared in Fig. 4b, it had an absorption at λ = 355 nm, which is similar to the M3 crosslinker, indicating that extended conjugate benzoic imine bonds are intact during the fabrication of the nanogels. These results confirm that the pre-crosslinking/dispersion approach is robust and enables the fabrication of colloidally stable nanogels in an aqueous solution.
 | ||
| Fig. 4 For P-Im3-Aq nanogels in aqueous solution, DLS diagram with the TEM image (scale bar = 200 nm) as the inset (a); UV/vis spectrum at 0.3 mg mL−1, compared with that of M3 in DMSO (30 μM) (b); and evolution of diameter with varying amounts of DMSO and CI/OH mole equivalent ratio (c). | ||
Important parameters that significantly influence colloidal stability and size were systematically investigated by DLS analysis. Fig. 4c shows the change in the diameter of the P-Im3-Aq nanogels in water. One parameter is the CI/OH mole equivalent ratio. When the ratio increased (e.g. decreasing the number of crosslinks), the diameter increased. Another parameter is the amount of PEG-b-PCIMA in DMSO as DMSO/PEG-b-PCIMA v/wt ratio for the CDI-mediated crosslinking reaction. The diameter increased with an increasing ratio (e.g. amount of DMSO). These results and trends of change in sizes indicate that the size of core-crosslinked nanogels greatly relies on crosslinking density; thus, highly crosslinked networks are likely to produce more compact hydrophobic cores.
In our separate experiment, a conventional in situ crosslinking approach was examined in an attempt to fabricate aqueous crosslinked nanoassemblies. This approach involves the fabrication of aqueous nanoassemblies consisting of PEG-b-PCIMA and M3 through self-assembly, followed by the addition of water-soluble DBU as a catalyst in an aqueous solution. The resulting dispersion was white-colored with visible aggregates (Fig. S8, ESI†). The dispersion was subjected to dialysis and filtration to remove a significant amount of white aggregates. The resulting dispersion turned out to be blue-tinted with an average diameter of 125 nm by DLS. When characterized by UV-vis spectroscopy, the dispersion showed no absorption at λ = 355 nm corresponding to the n → π* transition, characteristic of M3, in its UV/vis spectrum. Our attempts with variations in the amounts of PEG-b-PCIMA and M3 over DMSO and water were not straightforward toward the fabrication of colloidally stable aqueous dispersions of crosslinked nanoassemblies using this approach.
Studies of acid-responsive degradation and disassembly
Aqueous P-Im3-Aq nanogels contain extended conjugate benzoic imine bonds in the crosslinks of hydrophobic cores, which can be cleaved in an acidic environment. Aliquots of the nanogels were incubated in an acidic buffer at pH = 5.0, and their acid-responsive degradation was investigated using DLS and TEM techniques. DLS analysis reveals that their size distribution became multimodal with the appearance of large aggregates in 6 h of incubation (Fig. 5a). TEM analysis further confirms the occurrence of large aggregates (Fig. 5b and c), and the digital images of the aqueous nanogel dispersion show the change in color from turbid white to visibly clear after 24 h of incubation in acidic buffer (Fig. 5d and e). Further, precipitates were visible at the bottom of the vials. These results could be attributed to the disintegration of the P-Im3-Aq nanogel cores via the cleavage of benzoic imine bonds to the corresponding aldehyde and amine under acidic conditions.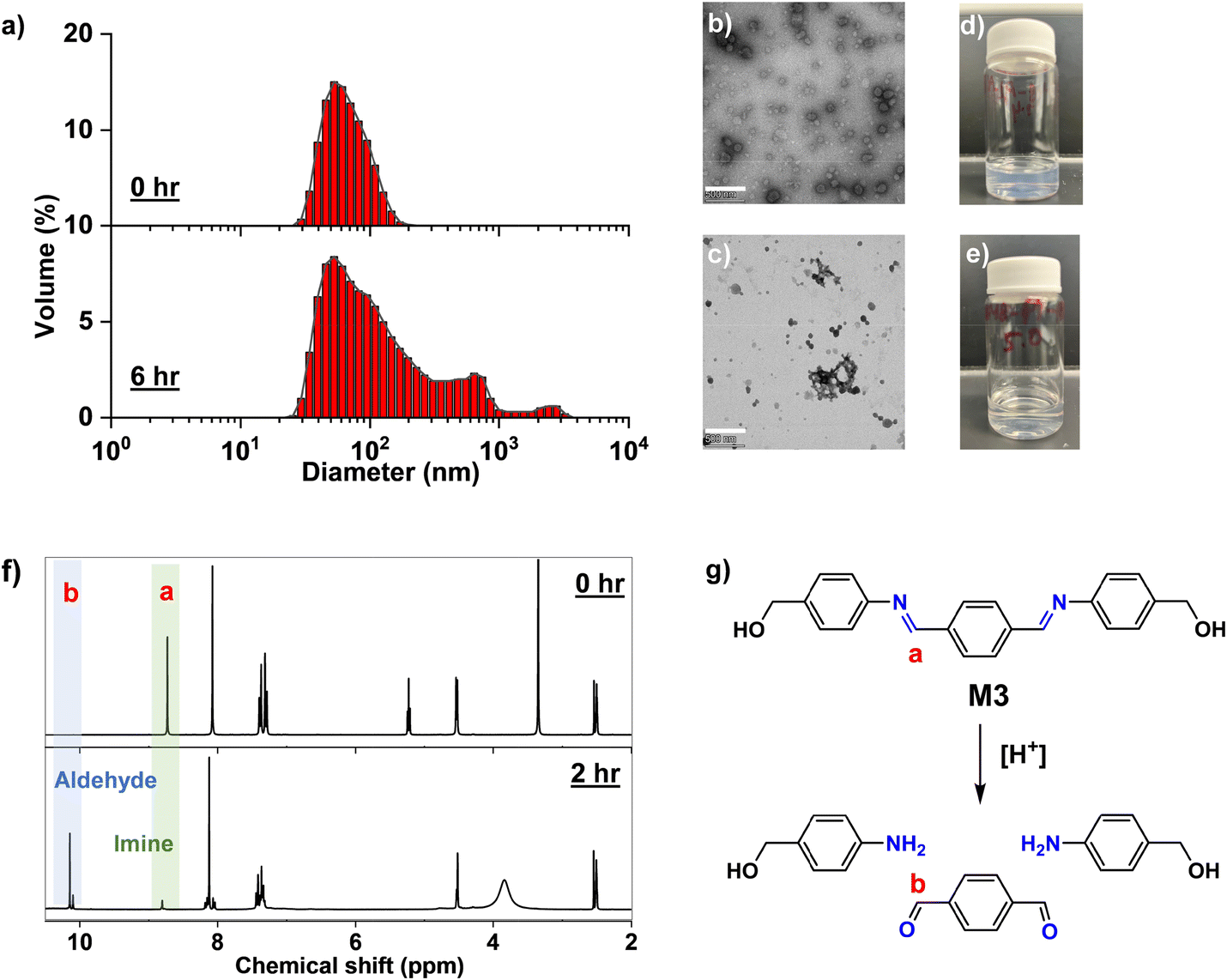 | ||
| Fig. 5 DLS diagrams of aqueous P-Im3-Aq nanogels incubated at pH 5.0 over 6 h (a); their TEM images (scale bar = 500 nm) (b and c), and digital images (d and e); and overlaid 1H NMR spectra of M3 in DMSO-d6/HCl (f) and acid-catalyzed hydrolysis of benzoic imine bond in M3 (g). | ||
To gain further insights, the acid-responsive degradation of M3 and crosslinked polymer (P-Im-DMSO) through acid-catalyzed hydrolysis of benzoic imine bonds was quantitatively investigated using 1H NMR spectroscopic analysis. First, M3 dissolved in DMSO-d6 was incubated with HCl (an acid) for 2 h. The 1H NMR spectrum in Fig. 5g shows the disappearance of the peak at 8.6 ppm corresponding to the imine proton (a) and the appearance of a new peak at 9.2 ppm corresponding to the aldehyde proton (b). These changes confirm the cleavage of imine bonds to the corresponding aldehyde and amine precursors through acid-catalyzed hydrolysis (Fig. 5f). Their integral ratio allows the %hydrolysis to be 82%. Next, P-Im3-DMSO swollen in DMSO-d6 was treated with HCl. As compared in Fig. S9 (ESI†), the 1H NMR spectrum of P-Im3-DMSO/HCl mixture shows peaks greater than 7 ppm, which could be attributed to the cleavage of partial imine bonds in the presence of acid, which could improve the relaxation of protons in degraded network polymers.
Light-responsive degradation and disassembly
The light response of aqueous P-Im3-Aq nanogels was investigated using UV/vis spectroscopy upon irradiation with visible light at λ = 440 nm. As shown in Fig. 6a, the absorption gradually decreased over the irradiation time. The absorbance at λ = 355 nm was monitored to determine %degradation. It decreased to 40% over 24 h. Additionally, the DLS diagrams and TEM images show no change in size distribution over irradiation time, suggesting that only cores could be disintegrated through the E/Z isomerization of conjugate benzoic imines, with no chemical cleavages (Fig. S10, ESI†). | ||
| Fig. 6 UV/vis spectra of aqueous P-Im3-Aq nanogels (a) and M3 in DMSO (b) under visible light irradiation at 420 nm over 24 hours, with inset plots showing absorbance changes at 355 nm. | ||
To gain insight into the plausible mechanism of the decrease in absorption, M3 dissolved in DMSO (30 μM) was examined for the light response of the conjugate benzoic imine bonds. As shown in Fig. 6b, absorbance at λ = 355 nm decreased to >90% in 24 h. Interestingly, the maximum absorption wavelength was blue-shifted over irradiation time, which could be attributed to the photochemical E/Z isomerization of two CN bonds under visible light.42,63 Notably, the degradation of P-Im3-Aq nanogels appeared to be slower than that of M3, which is attributed to the delayed penetration of visible light into crosslinked cores in nanogels. Our results obtained from UV/vis spectroscopic analysis of M3 and P-Im3-Aq suggest that the destabilization of nanogels upon exposure to visible light could be attributed to a change in UV absorption due to photo-induced E/Z isomerization.
Computational studies of E/Z isomerization of M3 by DFT calculations
M3 labeled with two conjugate benzoic imine bonds could be presented with three ground state configurations, as shown in Fig. 7a (e.g. EE, EZ, and ZZ). Their transitions as EE → EZ → ZZ are assumed to be achieved through a photo-induced E/Z isomerization of benzoic imine bonds. We scan the potential energy surface to characterize the ground states of EE, EZ and ZZ configurations of M3 and the transition states TS1 and TS2 for EE → EZ and EZ → ZZ isomerization pathways, respectively. DFT calculations show that EE is the most stable configuration compared to EZ and ZZ, while ZZ is the least stable configuration. The ground state energy of EE calculated through DFT is 3 kcal mol−1 and 11 kcal mol−1 lower compared to the EZ and ZZ configurations, respectively (Fig. S11, ESI†). The STQN method implemented in Gaussian 16 was used to locate these transition states. The estimated activation energies for EE → EZ (ΔG‡EZ) and EZ → ZZ (ΔG‡ZZ) were calculated to be 19 and 21 kcal mol−1, respectively. | ||
| Fig. 7 Three M3 configurations as EE, EZ, and ZZ (a) and absorption spectra of EE (black), EZ (red) and ZZ (blue) configurations calculated via TD-DFT (b). | ||
We used TD-DFT calculations to predict the absorption spectra of EE, EZ and ZZ configurations. The vertical excitation energy for the three configurations of M3 was calculated. The excitation energies, oscillator strength and possible electron transitions are summarized in Table S3 and Fig. S12 (ESI†). Fig. 7b shows the absorption spectra of EE (black), EZ (red) and ZZ (blue) configurations calculated via TD-DFT. The calculated λmax = 337 nm for the EE configuration is in good agreement with the experimental result (λmax = 355 nm). The EZ and ZZ configurations had progressively lower absorbance values. The EZ demonstrates a decrease in absorbance and a blue shift, while the ZZ exhibits the lowest absorbance, signifying substantial disruption in the conjugate structure. The similarity in spectral features between experimental and computational studies indicates that the light-responsive degradation of M3 (shown in Fig. 6b) could be attributed to stepwise transformation to EZ and ZZ forms upon exposure to light.
Frontier molecular orbital (FMO) is crucial for electrical and optical properties and chemical reactions. Fig. S13 (ESI†) shows the HOMO−1, HOMO, LUMO and LUMO+1 of EE configuration. The yellow and cyan regions show an FMO with opposite phases. The positive phase of the molecule is represented in yellow, and the negative phase is represented in cyan. The HOMO is localized mainly on aromatics, while the LUMO is relocated to the whole system. Moreover, electron orbital delocalization can be observed across imine bonds for HOMO. The LUMO is an anti-bonding orbital (π*) with respect to the CN, which justifies the π → π* excitation according to the FMO of the EE configuration.
Encapsulation and dual acidic pH/light-responsive release of Cur
Cur is a potent bioactive agent with therapeutic effects against various diseases, including cancer.64,65 Because it is hydrophobic, Cur could be encapsulated in the hydrophobic cores of P-Im3-Aq nanogels mainly through hydrophobic-hydrophobic interaction. Here, Cur-loaded aqueous nanogels (P-Im3-Aq/Cur) in colloidally stable yellow suspensions were fabricated by adding a solution of Cur in DMSO to the aqueous P-Im3-Aq dispersion, followed by purification with dialysis and filtration. With the pre-determined extinction coefficient of Cur (ε = 55000 M−1 cm−1) in THF/water (1/1 v/v) (Fig. S14, ESI†) and UV/vis spectrum of aqueous P-Im3-Aq/Cur dispersion (Fig. S15, ESI†), the loading level of Cur and its loading efficiency were determined to be 4.7 wt% and 24.5%, respectively. DLS analysis reveals that it had an average diameter of 130 nm (Fig. 8b).
 | ||
| Fig. 8 Digital images (upon irradiation with UV light at 365 nm) and schematic representation of Cur encapsulated in crosslinked nanogels (P-Im3-Aq) to form P-Im3-Aq/Cur nanogels (a); DLS diagram of aqueous P-Im3-Aq/Cur (b); and %release of Cur from P-Im3-Aq/Cur upon irradiation with visible light at 420 nm, compared with that in the dark as the control (c), and in acidic pH 5.0, compared with in pH 7.4, as well as under both conditions (d), measured via UV/vis spectroscopy. Note that the light-responsive release experiment (Fig. 8c) was performed at a greater concentration of P-Im3-Aq/Cur nanogels than that used for acid- and dual-acid–light responsive release experiments. | ||
The fabricated P-Im3-Aq/Cur was next evaluated for in vitro release of Cur in acidic pH upon irradiation with visible light, and both, using UV/vis spectroscopy. For light responsive release, aliquots in quartz cuvettes were exposed to visible light at λ = 420 nm. Owing to poor solubility in water, Cur released from the nanogels was precipitated from the solution. UV/vis spectra of supernatants were recorded for given time intervals (Fig. S16, ESI†). The absorbance at λ = 430 nm was used to determine the amount of released Cur and thus the %release. As illustrated in Fig. 8c, the %release significantly increased to 80% within 8 h. Compared with the negligible release of Cur under dark conditions, this enhanced release of Cur under visible light could be attributed to the structural change in the benzoic imine bonds through photo-induced E/Z isomerization, causing the destabilization of Cur-loaded cores. For pH-responsive release, aliquots were incubated at endo/lysosomal pH = 5.0 and physiological pH = 7.4 for comparison. UV/vis spectra of supernatant were recorded at given time intervals (Fig. S17, ESI†). As compared in Fig. 8d, the %release was less than 10% at pH = 7.4 for up to 25 h, which could be attributed to natural loss. At pH = 5.0, %release increased to 51% in 20 h, which could be attributed to acid-responsive degradation of nanogels through acid-catalyzed hydrolysis of benzoic imine bonds. More promisingly, when P-Im3-Aq/Cur was irradiated by visible light at acidic pH = 5.0, the % release was synergistically accelerated, reaching 70% within 15 h (Fig. S18, ESI†). This rapid release results from the dual-responsive degradation mechanism, involving both the cleavage of conjugate benzoic imine linkages within the crosslinked core and the destabilization of the core structure through benzoic imine isomerization.
Intracellular trafficking and anti-tumor activity
Given the promising results of dual acid/light-responsive degradation and Cur release, the P-Im3-Aq/Cur nanogels were evaluated as a mechanism for effective intracellular drug delivery of Cur. Fig. 9a shows the viability of HeLa (human cervical adenocarcinoma) cells to be >90% when incubated with empty nanogels up to 240 μg mL−1, which suggests that they are not toxic to cells and are biocompatible. As shown in Fig. 9b, the viability of the HeLa cells decreased to 50% when incubated with increasing concentrations of P-Im3-Aq/Cur nanogels up to 240 μg mL−1 (equivalent to 11.2 μg mL−1). The viability of HeLa cells dropped to below 10% with >5 μg mL−1 of free Cur, and the IC50 was calculated to be 2.72 μg mL−1 (equivalent to 7.39 μM), which is in the range of literature values (3.36–13.8 μM for HeLa cells).66,67 The linear reduction in the viability of HeLa cells with increasing concentrations of P-Im3-Aq/Cur nanogels suggests that there is controlled release of Cur. | ||
| Fig. 9 Viability of HeLa cells (%, y-axis) after incubation for 72 h with different concentrations of empty P-Im3-Aq nanogels (μg mL−1, x-axis) is shown in the bar graph (a). Line graphs show the change in HeLa cell viability (%, y-axis) after incubation with different concentrations of P-Im3-Aq/Cur nanogels (μg mL−1, top x-axis, and red line) compared with free Cur (μg mL−1, bottom x-axis, and black line) (b). The averages are shown with error bars as standard deviations for all replicates (n = 9). | ||
Next, the cellular uptake of P-Im3-Aq/Cur nanogels was evaluated in HeLa cells using live-cell imaging with a spinning disk confocal microscope. Fig. 10 shows fluorescence images of HeLa cells after incubation with P-Im3-Aq/Cur nanogels for 2 h (bottom), compared with empty P-Im3-Aq nanogels (middle) and free Cur (top). The nuclei shown in magenta were stained with SYTO Deep Red Nucleic Acid Stain, and Cur fluorescence was shown in green. Free Cur and P-Im3-Aq/Cur nanogels exhibited distinct perinuclear accumulation, indicating efficient cellular uptake by the endomembrane network. In contrast, cells with empty nanogels showed no detectable fluorescence for Cur. These results demonstrate that P-Im3-Aq/Cur nanogels can enter cells, allowing for the delivery of Cur.
 | ||
| Fig. 10 Images show HeLa cells 2 hours after incubation with free Cur (2.72 μg mL−1) (top), empty P-Im3-Aq (240 μg mL−1; middle), and P-Im3-Aq/Cur nanogels (240 μg mL−1; bottom). DIC shows a field of cells, magenta shows the nuclei, green shows Cur, and the merged image shows the three channels together. The images for each channel were collected using the same optical settings and similarly processed. The scale bar is 20 μm. | ||
Conclusion
We developed a robust approach by exploring pre-crosslinking dispersion to fabricate colloidally stable, core-crosslinked nanogels exhibiting a dual acid/visible light response with a single extended conjugate aromatic imine bond. A well-defined PEG-b-PCIMA block copolymer synthesized by RAFT polymerization reacted with a novel M3 diol crosslinker bearing an extended conjugate benzoic imine bond through a CDI-mediated coupling reaction in an organic solvent. The mechanical dispersion of the formed crosslinked polymers in aqueous solution allows for the fabrication of well-defined core/shell nanogels with hydrophobic cores crosslinked through the formation of carbonate linkages surrounded by hydrophilic PEG corona. The nanogels were spherical with a monomodal distribution and were non-cytotoxic. When exposed to acidic pH and visible light, the nanogels (e.g., crosslinked cores) degraded, as confirmed by DLS and TEM analyses. Such degradation could be attributed to acid-catalyzed hydrolysis at acidic pH, while photo-induced E/Z isomerization of aromatic imine bonds upon irradiation with visible light, as confirmed by 1H NMR spectroscopy and computational DFT studies. The nanogels were capable of encapsulating up to 4.6% of the Cur anticancer drug. They exhibited the accelerated and synergistic release of Cur in a combination of acidic pH and visible light and entered HeLa cells, reducing their viability. Notably, compared to free Cur, the linear reduction in cell viability suggests that Cur was released in a controlled manner, which could be highly desired as a method to reduce the side effects caused by treatments using free drugs. These findings underscore the potential of core-crosslinked nanogels bearing extended conjugate aromatic imine bonds as a promising platform for dual stimuli-responsive drug delivery, where dual acidic pH and visible light response enable enhanced control for advanced cancer therapy.Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this study are presented and detailed within the main sections of the manuscript and the ESI.†Acknowledgements
This work is supported by the Natural Science and Engineering Research Council (NSERC) in Canada through Discovery Grant, Collaborative Research and Training Experience Training (CREATE) Program entitled Polymer Nanoparticles Drug Delivery (PoND), and Canada Research Chair (CRC) Award. JKO was entitled Tier II CRC in Nanobioscience (2011–2021). KK thanks the School of Graduate Studies and Research at Concordia University for Concordia University Graduate Fellowship to support her PhD study. Authors thank Dr. Nooshin Movahed at the Center for NanoScience Research for TEM measurements and Heng Jiang for HR-MS studies. Authors also thank Calcul Quebec, the Digital Re-search Alliance of Canada and the Center for Research in Molecular Modeling (CERMM) for providing computational resources.References
- M. A. Beach, U. Nayanathara, Y. Gao, C. Zhang, Y. Xiong, Y. Wang and G. K. Such, Chem. Rev., 2024, 124, 5505 CrossRef CAS PubMed.
- A. Harada and K. Kataoka, Prog. Polym. Sci., 2006, 31, 949 CrossRef CAS.
- A. S. Mikhail and C. Allen, J. Controlled Release, 2009, 138, 214 CrossRef CAS.
- N. Nishiyama and K. Kataoka, Adv. Polym. Sci., 2006, 193, 67 CrossRef CAS.
- X.-B. Xiong, A. Falamarzian, S. M. Garg and A. Lavasanifar, J. Controlled Release, 2011, 155, 248 CrossRef CAS PubMed.
- J. Ding, L. Chen, C. Xiao, L. Chen, X. Zhuang and X. Chen, Chem. Commun., 2014, 50, 11274 RSC.
- C. Allen, D. Maysinger and A. Eisenberg, Colloids Surf., B, 1999, 16, 3 CrossRef CAS.
- S. Taurin, H. Nehoff and K. Greish, J. Controlled Release, 2012, 164, 265 CrossRef CAS PubMed.
- L. Zhang, Y. Li and J. C. Yu, J. Mater. Chem. B, 2014, 2, 452 RSC.
- J. W. Nichols and Y. H. Bae, Nano Today, 2012, 7, 606 CrossRef CAS PubMed.
- Y. H. Bae and K. Park, J. Controlled Release, 2011, 153, 198 CrossRef CAS PubMed.
- O. C. Farokhzad and R. Langer, ACS Nano, 2009, 3, 16 CrossRef CAS PubMed.
- A. Prokop and J. M. Davidson, J. Pharm. Sci., 2008, 97, 3518 CrossRef CAS PubMed.
- H. Cabral, Y. Matsumoto, K. Mizuno, Q. Chen, M. Murakami, M. Kimura, Y. Terada, M. R. Kano, K. Miyazono, M. Uesaka, N. Nishiyama and K. Kataoka, Nat. Nanotechnol., 2011, 6, 815 CrossRef CAS PubMed.
- J. Wang, W. Mao, L. L. Lock, J. Tang, M. Sui, W. Sun, H. Cui, D. Xu and Y. Shen, ACS Nano, 2015, 9, 7195 CrossRef CAS.
- S. Mura, J. Nicolas and P. Couvreur, Nat. Mater., 2013, 12, 991 CrossRef CAS PubMed.
- C. J. F. Rijcken, O. Soga, W. E. Hennink and C. F. van Nostrum, J. Controlled Release, 2007, 120, 131 CrossRef CAS PubMed.
- Q. Zhang, N. R. Ko and J. K. Oh, Chem. Commun., 2012, 48, 7542 RSC.
- A. W. Jackson and D. A. Fulton, Polym. Chem., 2013, 4, 31 RSC.
- Y. Wang, H. Xu and X. Zhang, Adv. Mater., 2009, 21, 2849 CrossRef CAS.
- K. Loomis, K. McNeeley and R. V. Bellamkonda, Soft Matter, 2011, 7, 839 RSC.
- A. B. Cook and P. Decuzzi, ACS Nano, 2021, 15, 2068 CrossRef CAS PubMed.
- K. K. Bawa and J. K. Oh, Mol. Pharmaceutics, 2017, 14, 2460 CrossRef CAS.
- H. Liu, H. H. Lu, Y. Alp, R. Wu and S. Thayumanavan, Prog. Polym. Sci., 2024, 148, 101765 CrossRef CAS PubMed.
- I. F. Tannock and D. Rotin, Cancer Res., 1989, 49, 4373 CAS.
- P. Watson, A. T. Jones and D. J. Stephens, Adv. Drug Delivery Rev., 2005, 57, 43 CrossRef CAS PubMed.
- S. Bazban-Shotorbani, M. M. Hasani-Sadrabadi, A. Karkhaneh, V. Serpooshan, K. I. Jacob, A. Moshaverinia and M. Mahmoudi, J. Controlled Release, 2017, 253, 46 CrossRef CAS PubMed.
- S. Binauld and M. H. Stenzel, Chem. Commun., 2013, 49, 2082 RSC.
- G. Kocak, C. Tuncer and V. Butun, Polym. Chem., 2017, 8, 144 RSC.
- M. Huo, J. Yuan, L. Tao and Y. Wei, Polym. Chem., 2014, 5, 1519 RSC.
- Y. Ding, Y. Kang and X. Zhang, Chem. Commun., 2015, 51, 996 RSC.
- X. Qu and Z. Yang, Chem. Asian J., 2016, 11, 2633 CrossRef CAS PubMed.
- A. M. Jazani and J. K. Oh, Polym. Chem., 2020, 11, 2934 RSC.
- X. Hu, A. M. Jazani and J. K. Oh, Polymer, 2021, 230, 124024 CrossRef CAS.
- N. Kongkatigumjorn and D. Crespy, Polym. Chem., 2024, 15, 4491 RSC.
- J. F. Gohy and Y. Zhao, Chem. Soc. Rev., 2013, 42, 7117 RSC.
- H. Zhao, E. S. Sterner, E. B. Coughlin and P. Theato, Macromolecules, 2012, 45, 1723 CrossRef CAS.
- Y. Zhao, Macromolecules, 2012, 45, 3647 CrossRef CAS.
- Q. Jin, T. Cai, H. Han, H. Wang, Y. Wang and J. Ji, Macromol. Rapid Commun., 2014, 35, 1372 CrossRef CAS PubMed.
- A. M. Jazani and J. K. Oh, Polym. Chem., 2022, 13, 4557 RSC.
- L. Meng, W. Huang, D. Wang, X. Huang, X. Zhu and D. Yan, Biomacromolecules, 2013, 14, 2601 CrossRef CAS PubMed.
- K. Bairagi, J. T. Liu, A. Thinphang-nga and J. K. Oh, Macromolecules, 2023, 56, 4307 CrossRef CAS.
- H. S. El-Sawy, A. M. Al-Abd, T. A. Ahmed, K. M. El-Say and V. P. Torchilin, ACS Nano, 2018, 12, 10636 CrossRef CAS PubMed.
- J. K. Oh, Polym. Chem., 2019, 10, 1554 RSC.
- I. Altinbasak, Y. Alp, R. Sanyal and A. Sanyal, Nanoscale, 2024, 16, 14033 RSC.
- J. Hu, J. He, D. Cao, M. Zhang and P. Ni, Polym. Chem., 2015, 6, 3205 RSC.
- Z. Zhang, L. Yin, C. Tu, Z. Song, Y. Zhang, Y. Xu, R. Tong, Q. Zhou, J. Ren and J. Cheng, ACS Macro Lett., 2013, 2, 40 CrossRef CAS PubMed.
- W. Zhu, Y. Wang, X. Cai, G. Zha, Q. Luo, R. Sun, X. Li and Z. Shen, J. Mater. Chem. B, 2015, 3, 3024 RSC.
- B. Liu and S. Thayumanavan, J. Am. Chem. Soc., 2017, 139, 2306 CrossRef CAS PubMed.
- S.-J. Lee, K.-H. Min, H.-J. Lee, A.-N. Koo, H.-P. Rim, B.-J. Jeon, S.-Y. Jeong, J.-S. Heo and S.-C. Lee, Biomacromolecules, 2011, 12, 1224 CrossRef CAS PubMed.
- W. Chen, M. Zheng, F. Meng, R. Cheng, C. Deng, J. Feijen and Z. Zhong, Biomacromolecules, 2013, 14, 1214 CrossRef CAS PubMed.
- Y. Olszowy, J. Wesselmann, S. F. Over, F. Pätzold and R. Weberskirch, Polym. Chem., 2023, 14, 3761 RSC.
- X. Wang, L. Wang, S. Yang, M. Zhang, Q. Xiong, H. Zhao and L. Liu, Macromolecules, 2014, 47, 1999 CrossRef CAS.
- H. Y. Yang, Y. Li, M.-S. Jang, Y. Fu, T. Wu, J. H. Lee and D. S. Lee, Eur. Polym. J., 2019, 121, 109342 CrossRef CAS.
- M. Hartlieb, T. Bus, J. Kubel, D. Pretzel, S. Hoeppener, M. N. Leiske, K. Kempe, B. Dietzek and U. S. Schubert, Bioconjugate Chem., 2017, 28, 1229 CrossRef CAS PubMed.
- K. Raghupathi, L. Li, J. Ventura, M. Jennings and S. Thayumanavan, Polym. Chem., 2014, 5, 1737 RSC.
- X. Hu and J. K. Oh, Macromol. Rapid Commun., 2020, 41, 2000394 CrossRef CAS PubMed.
- J. D. Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10(44), 6615–6620 RSC.
- M. J. Frisch, J. A. Pople and J. S. Binkley, J. Chem. Phys., 1984, 80, 3265 CrossRef CAS.
- D. Tahaoglu, H. Usta and F. Alkan, J. Phys. Chem. A, 2022, 126, 4199 CrossRef CAS PubMed.
- M. Frisch, G. Trucks, H. Schlegel, G. Scuseria, M. Robb, J. Cheeseman, G. Scalmani, V. Barone, G. Petersson and H. J. G. I. W. C. Nakatsuji, Gaussian 16 (revision C.01), Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- X. Hu and J. K. Oh, Macromol. Rapid Commun., 2020, 41, e2000394 CrossRef PubMed.
- P. J. Coelho, M. C. R. Castro and M. M. M. Raposo, J. Photochem. Photobiol., A, 2013, 259, 59 CrossRef CAS.
- N. Dhillon, B. B. Aggarwal, R. A. Newman, R. A. Wolff, A. B. Kunnumakkara, J. L. Abbruzzese, C. S. Ng, V. Badmaev and R. Kurzrock, Clin. Cancer Res., 2008, 14, 4491 CrossRef CAS PubMed.
- V. Zoi, V. Galani, G. D. Lianos, S. Voulgaris, A. P. Kyritsis and G. A. Alexiou, Biomedicines, 2021, 9, 1086 CrossRef CAS PubMed.
- K. K. Gupta, S. S. Bharne, K. Rathinasamy, N. R. Naik and D. Panda, FEBS J., 2006, 273, 5320 CrossRef CAS.
- R. Rashmi, N. Prakash, H. Narayanaswamy, M. N. Swamy, D. Rathnamma, P. Ramesh, U. Sunilchandra, C. Santhosh, H. Dhanalakshmi, N. Nagaraju and H. Vanishree, J. Entomol. Zool. Stud., 2020, 8, 267 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5tb00734h |
| This journal is © The Royal Society of Chemistry 2025 |