Average ligand removal enthalpies of 30 differently coordinated mono-nuclear fourth-row transition metal complexes taken from a database recently considered by Johnson and Becke [Can. J. Chem., 2009, 8, 1369] have been computed in the gas phase using unrestricted pseudo-spectral (LACV3P) and fully analytic (qzvp(-g)) B3LYP including a recently developed empirical dispersion correction. Heats of formation of neutral singlet reactants and neutral, potentially high spin, products have been taken from NIST's Organometallic Thermochemistry Database. Comparison of B3LYP-MM//qzvp(-g) and experimental average ligand removal enthalpies reveals a systematic error in the reported experimental enthalpies for manganese-containing complexes which is verified with high-level, CCSD(T)-F12//family of cc-pVTZ, explicitly correlated coupled-cluster methods. Other B3LYP-MM//qzvp(-g) error patterns give rise to a d-block localized orbital correction (DBLOC) scheme containing six transferable parameters that correct the functional's description of metal–ligand bonding, cation-π, and dispersion interactions as well as metal and/or ligand multi-reference effects. Metal–ligand cation-π and dispersion interactions have been fit to the monopole/induced-dipole, 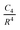 , and induced-dipole/induced-dipole,
, and induced-dipole/induced-dipole, 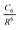 , interaction functions, respectively. This DBLOC model has been built upon a previously determined set of metal atom parameters which are necessary to properly describe the free metal atom reaction products. The final DBLOC model brings the mean unsigned error of B3LYP-MM//qzvp(-g) from 3.74 ± 3.51 kcal/mol to 0.94 ± 0.68 kcal/mol and corrects the functional's under binding in nearly every case. Several important connections among DBLOC parameters have been made.
, interaction functions, respectively. This DBLOC model has been built upon a previously determined set of metal atom parameters which are necessary to properly describe the free metal atom reaction products. The final DBLOC model brings the mean unsigned error of B3LYP-MM//qzvp(-g) from 3.74 ± 3.51 kcal/mol to 0.94 ± 0.68 kcal/mol and corrects the functional's under binding in nearly every case. Several important connections among DBLOC parameters have been made.

You have access to this article
 Please wait while we load your content...
Something went wrong. Try again?
Please wait while we load your content...
Something went wrong. Try again?


