
Re-evaluating the Cu K pre-edge XAS transition in complexes with covalent metal–ligand interactions†
Abstract
Three [Me2NN]Cu(η2-L2) complexes (Me2NN = HC[C(Me)NAr]2; L2 = PhNO (2), 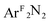 (3), PhCH
(3), PhCH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2 (4); Ar = 2,6-Me2-C6H3; ArF = 3,5-(CF3)2-C6H3) have been studied by Cu K-edge X-ray absorption spectroscopy, as well as single- and multi-reference computational methods (DFT, TD-DFT, CASSCF, MRCI, and OVB). The study was extended to a range of both known and theoretical compounds bearing 2p-element donors as a means of deriving a consistent view of how the pre-edge transition energy responds in systems with significant ground state covalency. The ground state electronic structures of many of the compounds under investigation were found to be strongly influenced by correlation effects, resulting in ground state descriptions with majority contributions from a configuration comprised of a Cu(II) metal center anti-ferromagentically coupled to radical anion O2, PhNO, and
CH2 (4); Ar = 2,6-Me2-C6H3; ArF = 3,5-(CF3)2-C6H3) have been studied by Cu K-edge X-ray absorption spectroscopy, as well as single- and multi-reference computational methods (DFT, TD-DFT, CASSCF, MRCI, and OVB). The study was extended to a range of both known and theoretical compounds bearing 2p-element donors as a means of deriving a consistent view of how the pre-edge transition energy responds in systems with significant ground state covalency. The ground state electronic structures of many of the compounds under investigation were found to be strongly influenced by correlation effects, resulting in ground state descriptions with majority contributions from a configuration comprised of a Cu(II) metal center anti-ferromagentically coupled to radical anion O2, PhNO, and 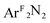 ligands. In contrast, the styrene complex 4, which displays a Cu K pre-edge transition despite its formal d10 electron configuration, exhibits what can best be described as a Cu(I):(styrene)0 ground state with strong π-backbonding. The Cu K pre-edge features for these complexes increase in energy from 1 to 4, a trend that was tracked to the percent Cu(II)-character in the ground state. The unexpected shift to higher pre-edge transition energies with decreasing charge on copper (QCu) contributed to an assignment of the pre-edge features for these species as arising from metal-to-ligand charge transfer instead of the traditional Cu1s → Cu3d designation.
ligands. In contrast, the styrene complex 4, which displays a Cu K pre-edge transition despite its formal d10 electron configuration, exhibits what can best be described as a Cu(I):(styrene)0 ground state with strong π-backbonding. The Cu K pre-edge features for these complexes increase in energy from 1 to 4, a trend that was tracked to the percent Cu(II)-character in the ground state. The unexpected shift to higher pre-edge transition energies with decreasing charge on copper (QCu) contributed to an assignment of the pre-edge features for these species as arising from metal-to-ligand charge transfer instead of the traditional Cu1s → Cu3d designation.

 Please wait while we load your content...
Please wait while we load your content...

