
Stability and electronic structure of hydrogen vacancies in ADP: hybrid DFT with vdW correction
 *a
*a
Abstract
The formation energies, charge transition levels, and electronic structures of positively charged, neutral, and negatively charged hydrogen vacancies in the NH4H2PO4 (ADP) crystal are investigated in the framework of density functional theory with local and hybrid exchange–correlation functionals. The inclusion of nonlocal exchange opens the ADP fundamental band gap by nearly 1 eV and well reproduces the experimental value. The van der Waals (vdW) interaction is found to have a major influence on the energetics of charged hydrogen vacancies in ADP. The calculated relative stability of 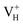 and
and 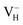 with vdW interaction could well explain the break point on the measured conductivity curve of the ADP crystal in the high temperature region. On the other hand, a missing H atom in the (H2PO4)− group is found to be more energetically preferable than NH4+. It could capture a hole carrier to form a molecular-type polaron with its adjacent two O atoms, and be responsible for the optical absorption under irradiation by a high-intensity laser beam.
with vdW interaction could well explain the break point on the measured conductivity curve of the ADP crystal in the high temperature region. On the other hand, a missing H atom in the (H2PO4)− group is found to be more energetically preferable than NH4+. It could capture a hole carrier to form a molecular-type polaron with its adjacent two O atoms, and be responsible for the optical absorption under irradiation by a high-intensity laser beam.

 Please wait while we load your content...
Please wait while we load your content...

