
A theoretical study of M–M′ polar-covalent bonding in heterobimetallic multinuclear organometallic complexes of monovalent group 11 metal centres†‡
 *ac
Dage
Sundholm
*b
and
*ac
Dage
Sundholm
*b
and
Abstract
Complexes with closed-shell (d10–d10) interactions have been studied for their interesting luminescence properties in organic light-emitting diode (OLED) devices. The present computational study aims at understanding the chemical bonding/interactions in a series of molecules with unusually short metal–metal bond distances between monovalent coinage-metal (d10–d10) centres. The investigated molecules include pentanuclear 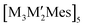 complexes with M or M′ = Cu(I), Ag(I), or Au(I) and Mes = 2,4,6-Me3C6H2. In such complexes, the M–M′ distances are up to 50–100 pm shorter than typical metallophilic bonds in homometallic analogues. Characterization and analysis of the chemical bond strength was performed using ab initio methods, density functional theory methods including a semi-empirical treatment of dispersion interactions (DFT-D3) and semi-empirical calculations at the extended Hückel theory (EHT) level. Population analysis suggests that hybridization occurs by mixing the (n + 1)s and (n + 1)p orbitals of M with the (nd) orbitals of M′. The orbital mixing plays a pivotal role in the polydentated polar-covalency/dative M–M′ bonds that distinguish this bonding from the weaker metallophilic interactions.
complexes with M or M′ = Cu(I), Ag(I), or Au(I) and Mes = 2,4,6-Me3C6H2. In such complexes, the M–M′ distances are up to 50–100 pm shorter than typical metallophilic bonds in homometallic analogues. Characterization and analysis of the chemical bond strength was performed using ab initio methods, density functional theory methods including a semi-empirical treatment of dispersion interactions (DFT-D3) and semi-empirical calculations at the extended Hückel theory (EHT) level. Population analysis suggests that hybridization occurs by mixing the (n + 1)s and (n + 1)p orbitals of M with the (nd) orbitals of M′. The orbital mixing plays a pivotal role in the polydentated polar-covalency/dative M–M′ bonds that distinguish this bonding from the weaker metallophilic interactions.

 Please wait while we load your content...
Please wait while we load your content...

