
Computational investigation of explicit solvent effects and specific interactions of hydroxypyrene photoacids in acetone, DMSO, and water†
 *a
and
Christof
Hättig
a
*a
and
Christof
Hättig
a
Abstract
This work employs the correlated wavefunction-based methods ADC(2) and CC2 in combination with the implicit solvent model COSMO to calculate the UV/Vis absorption and fluorescence emission energies of particularly strong hydroxypyrene photoacids in acetone. According to the Förster cycle, the electronic transition energies are first used to compute 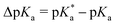 , i.e., the pKa change upon excitation and then the excited-state pKa (labeled
, i.e., the pKa change upon excitation and then the excited-state pKa (labeled 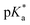 ) with ground-state pKa values based on COSMO-RS as additional inputs. Furthermore, for the strongest photoacid of that class, namely tris(1,1,1,3,3,3-hexafluoropropan-2-yl)-8-hydroxypyrene-1,3,6-trisulfonate, the need to go beyond implicit solvation and to account for explicit solvent effects on the electronic transition energies and the resulting ΔpKa is investigated in the solvents acetone, dimethyl sulfoxide (DMSO), and water. For this, a hybrid implicit–explicit approach is followed by comparing micro-solvated structures that are generated based on Kamlet–Taft considerations. While implicit solvent effects are mostly sufficient for the aprotic solvent acetone, one explicit solvent molecule seems relevant for DMSO due to its stronger hydrogen-bond (HB) acceptance and hence larger interaction with the photoacid OH group as a HB donor. For the protic solvent water, the situation is more complicated, involving at least one water molecule at the OH group and up to three water molecules at the O− group of the corresponding base. Finally, these results are used to rationalize the experimentally observed spectral evolution of the photoacid absorption band in acetone–water solvent mixtures.
) with ground-state pKa values based on COSMO-RS as additional inputs. Furthermore, for the strongest photoacid of that class, namely tris(1,1,1,3,3,3-hexafluoropropan-2-yl)-8-hydroxypyrene-1,3,6-trisulfonate, the need to go beyond implicit solvation and to account for explicit solvent effects on the electronic transition energies and the resulting ΔpKa is investigated in the solvents acetone, dimethyl sulfoxide (DMSO), and water. For this, a hybrid implicit–explicit approach is followed by comparing micro-solvated structures that are generated based on Kamlet–Taft considerations. While implicit solvent effects are mostly sufficient for the aprotic solvent acetone, one explicit solvent molecule seems relevant for DMSO due to its stronger hydrogen-bond (HB) acceptance and hence larger interaction with the photoacid OH group as a HB donor. For the protic solvent water, the situation is more complicated, involving at least one water molecule at the OH group and up to three water molecules at the O− group of the corresponding base. Finally, these results are used to rationalize the experimentally observed spectral evolution of the photoacid absorption band in acetone–water solvent mixtures.

- This article is part of the themed collection: 2023 PCCP HOT Articles
 Please wait while we load your content...
Please wait while we load your content...

