
Data-driven design of double-atom catalysts with high H2 evolution activity/CO2 reduction selectivity based on simple features†
 *c
Tiancheng
Mu
*a
*c
Tiancheng
Mu
*a
Abstract
Double-atom catalysts (DACs) with superior characteristics, including extreme atomic utilization and high activity, have attracted scholars in the field of fundamental electrocatalytic reactions such as the hydrogen evolution reaction (HER) and CO2 reduction reaction (CO2RR). Despite recent developments in computational power, designing a DAC with density functional theory (DFT) is still a challenging and time-consuming process for the large design space of DACs. In the present study, high-throughput DFT calculations and machine learning (ML) were combined to develop ML models and predict the activity of the HER (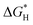 ) of ∼17 000 boron-doped graphene DAC systems with high accuracy. The origin of HER activity was also investigated through DFT and ML analysis. Based on the obtained results, it is found that the number of d electrons of transition metals plays a key role in the binding behavior of hydrogen on DACs, and boron doping on DACs can effectively regulate the electron distribution of DACs to modify the binding strength of adsorbates on DACs. Besides, the Gibbs free energy of CO absorption (
) of ∼17 000 boron-doped graphene DAC systems with high accuracy. The origin of HER activity was also investigated through DFT and ML analysis. Based on the obtained results, it is found that the number of d electrons of transition metals plays a key role in the binding behavior of hydrogen on DACs, and boron doping on DACs can effectively regulate the electron distribution of DACs to modify the binding strength of adsorbates on DACs. Besides, the Gibbs free energy of CO absorption (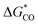 ) and the binding energy of DACs (ΔEbinding) were predicted by our ML models with simple features to evaluate the CO2RR activity and stability of DACs. The present study offers theoretical insight into the electrocatalytic activity of DACs. Most importantly, by screening appropriate energies with ML, either DACs with high HER activity or DACs with high CO2RR activity and low competitive HER activity can be obtained, and the catalytic activity of DACs can be adjusted on purpose.
) and the binding energy of DACs (ΔEbinding) were predicted by our ML models with simple features to evaluate the CO2RR activity and stability of DACs. The present study offers theoretical insight into the electrocatalytic activity of DACs. Most importantly, by screening appropriate energies with ML, either DACs with high HER activity or DACs with high CO2RR activity and low competitive HER activity can be obtained, and the catalytic activity of DACs can be adjusted on purpose.

- This article is part of the themed collection: 2023 Journal of Materials Chemistry A HOT Papers
 Please wait while we load your content...
Please wait while we load your content...

