
Computational discovery of stable Na-ion sulfide solid electrolytes with high conductivity at room temperature†

Abstract
The search for inorganic solid electrolytes suitable for the realization of solid-state batteries with structural stability and high ion conductivity at room temperature remains a significant challenge. In this study, we employed a multi-stage density functional theory molecular dynamics (DFT-MD) sampling workflow, focusing on Na-ion sulfides 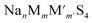 with trivalent (M) and pentavalent (M′) metal ions and an expanded selection of parent structures (Ω). This led to the identification of two promising sampling spaces (M,M′,Ω) = (Ga,P,Na4SiS4) and (Si,Ta,Na4SiS4). The predictions were validated through multi-temperature DFT-MD calculations, wherein σNa,300K ≳ 10−3 S cm−1 are attained within a thermodynamic phase stability range of 9 < Ehull < 25 meV per atom (Ehull is convex hull decomposition energy): Na4Ga0.5P0.5S4, Na3.75Ga0.375P0.625S4, Na4.25Ga0.625P0.375S4, Na3.75Si0.75Ta0.25S4, Na3.625Si0.625Ta0.375S4, and Na3.5Si0.5Ta0.5S4. These compounds are highly suggested for experimental synthesis and investigation. Moreover, our brute-force and highly generalized sampling technique is expected to be applicable in uncovering other solid electrolyte classes, thus potentially contributing to the advancement of solid-state battery technology.
with trivalent (M) and pentavalent (M′) metal ions and an expanded selection of parent structures (Ω). This led to the identification of two promising sampling spaces (M,M′,Ω) = (Ga,P,Na4SiS4) and (Si,Ta,Na4SiS4). The predictions were validated through multi-temperature DFT-MD calculations, wherein σNa,300K ≳ 10−3 S cm−1 are attained within a thermodynamic phase stability range of 9 < Ehull < 25 meV per atom (Ehull is convex hull decomposition energy): Na4Ga0.5P0.5S4, Na3.75Ga0.375P0.625S4, Na4.25Ga0.625P0.375S4, Na3.75Si0.75Ta0.25S4, Na3.625Si0.625Ta0.375S4, and Na3.5Si0.5Ta0.5S4. These compounds are highly suggested for experimental synthesis and investigation. Moreover, our brute-force and highly generalized sampling technique is expected to be applicable in uncovering other solid electrolyte classes, thus potentially contributing to the advancement of solid-state battery technology.

 Please wait while we load your content...
Please wait while we load your content...

