
Heavier alkaline earth and heterobimetallic s-block “ate” complexes of a di(amido)siloxane ligand: solid-state structure and dynamic solution-phase behaviour†
 ‡a
Andrea
O'Reilly,
‡b
Alice J. M.
Poole,
‡a
Stefan
Thum,
c
Louis J.
Morris,
a
Martyn P.
Coles,
*b
J. Robin
Fulton,
*b
Sjoerd
Harder,
c
Zoë R.
Turner
*a
and
Dermot
O'Hare
*a
‡a
Andrea
O'Reilly,
‡b
Alice J. M.
Poole,
‡a
Stefan
Thum,
c
Louis J.
Morris,
a
Martyn P.
Coles,
*b
J. Robin
Fulton,
*b
Sjoerd
Harder,
c
Zoë R.
Turner
*a
and
Dermot
O'Hare
*a
Abstract
The diverse solid-state structures and solution-phase dynamics of both neutral and heterometallic s-block “ate” complexes of the heavier alkaline earth metals (Ae; Ca–Ba) supported by a chelating and flexible di(amido)siloxane ligand ([NON-DippL]2− = [O(SiMe2NDipp)2]2−) are described, enabling comparison with those of closely related di(amido) ligands based on either flexible aliphatic or rigid xanthene-based backbones. Three dimeric alkaline earth complexes [(NON-DippL)Ae]2 (Ae = Ca (2), Sr (3) and Ba (4)) which feature a κ3-N,O,N′-κ1-N′-tridentate coordination mode were prepared from protonolysis reactions between NON-DippLH2 with 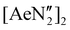 (Ae = Ca, Sr and Ba); N′′ = [N(SiMe3)2]−. In tetrahydrofuran, these complexes were readily converted into the monomeric adducts [(NON-DippL)Ae(thf)n] (n = 2, Ae = Ca (5); n = 3, Ae = Sr (6) and Ba (7)). Heterometallic Ae/K amide “ate” complexes were afforded through two routes: reaction of previously reported [(NON-DippL)Mg]2 (1) with two equivalents of KN′′ at elevated temperatures resulted in [(NNO-DippL)Mg(μ-N′′)K]n (8; NNO-DippL = [OSiMe2NDippSiMe2NDipp]2−), whereas the equimolar reaction of NON-DippLH2 with
(Ae = Ca, Sr and Ba); N′′ = [N(SiMe3)2]−. In tetrahydrofuran, these complexes were readily converted into the monomeric adducts [(NON-DippL)Ae(thf)n] (n = 2, Ae = Ca (5); n = 3, Ae = Sr (6) and Ba (7)). Heterometallic Ae/K amide “ate” complexes were afforded through two routes: reaction of previously reported [(NON-DippL)Mg]2 (1) with two equivalents of KN′′ at elevated temperatures resulted in [(NNO-DippL)Mg(μ-N′′)K]n (8; NNO-DippL = [OSiMe2NDippSiMe2NDipp]2−), whereas the equimolar reaction of NON-DippLH2 with  led to [(NON-DippL)Ae(μ-N′′)K]n (Ae = Ca (9), Sr (10) and Ba (11)). Complexes 8–11 exist as one-dimensional coordination polymers propagated by K+–aryl π-facial interactions in the solid-state. The mixed amide/siloxide “NNO” ligand in 8 results from a 1,3-silyl retro-Brook rearrangement of the original di(amido)siloxane ligand, while the larger Ae2+ congeners readily accommodate the coordination of KN′′ with the di(amido)siloxane ligand retaining a κ3-N,O,N′-tridentate motif in 9–11. Finally, the solution-phase behaviour of 8–11 in both toluene and thf were investigated indicating the reversible dissociation of KN′′ from 9–11 and the thermodynamic parameters of this process were elucidated.
led to [(NON-DippL)Ae(μ-N′′)K]n (Ae = Ca (9), Sr (10) and Ba (11)). Complexes 8–11 exist as one-dimensional coordination polymers propagated by K+–aryl π-facial interactions in the solid-state. The mixed amide/siloxide “NNO” ligand in 8 results from a 1,3-silyl retro-Brook rearrangement of the original di(amido)siloxane ligand, while the larger Ae2+ congeners readily accommodate the coordination of KN′′ with the di(amido)siloxane ligand retaining a κ3-N,O,N′-tridentate motif in 9–11. Finally, the solution-phase behaviour of 8–11 in both toluene and thf were investigated indicating the reversible dissociation of KN′′ from 9–11 and the thermodynamic parameters of this process were elucidated.

 Please wait while we load your content...
Please wait while we load your content...

