Synthesis of new water-soluble phosphonium salts and their Wittig reactions in water
Matthew G. Russell and Stuart Warren*
University Chemical Laboratories, Lensfield Road, Cambridge, UK CB2 1EW
First published on UnassignedUnassigned3rd February 2000
Abstract
Some new water-soluble alkyltriarylphosphonium salts are reported. Their solubility is due to the presence of either carboxy or hydroxy groups on the phenyl rings attached to phosphorus. Examples of both benzyl- and butylphosphonium salts are presented and investigation into their Wittig reactions with a range of aldehydes using water as the solvent is described.
Introduction
We have previously reported new phosphonium salts designed to be soluble in aqueous base and their Wittig reactions in water with substituted benzaldehydes.1 In this paper, we report more fully on the synthesis and chemistry of these and other new phosphonium salts and discuss the limitations of carrying out Wittig reactions in water. Generally, reactions in water have a lower impact on the environment than reactions carried out in organic solvents. The Wittig reaction is a good candidate for aqueous conditions with the additional benefit that any water-soluble phosphine oxide by-products could be more easily separated from the olefin products.Wittig reactions and Horner–Wadsworth–Emmons reactions have previously been carried out in the presence of water, most typically in either biphasic solutions using strong bases (e.g. aqueous 50% sodium hydroxide)![[hair space]](https://www.rsc.org/images/entities/char_200a.gif) 2–6 or in an organic solvent with trace amounts of water using solid sodium hydroxide or sodium carbonate.7–9 In both these instances, the natural behaviour of phosphonium salts as phase transfer catalysts is used to promote the formation of the ylides used in the reaction. In Horner–Wadsworth–Emmons reactions, tetrabutylammonium iodide is sometimes added as a phase transfer catalyst.4–6
2–6 or in an organic solvent with trace amounts of water using solid sodium hydroxide or sodium carbonate.7–9 In both these instances, the natural behaviour of phosphonium salts as phase transfer catalysts is used to promote the formation of the ylides used in the reaction. In Horner–Wadsworth–Emmons reactions, tetrabutylammonium iodide is sometimes added as a phase transfer catalyst.4–6
There are fewer examples of aqueous Wittig reactions carried out in the absence of organic solvent; the first example is the reaction of (nitrobenzyl)triphenylphosphonium bromides with formaldehyde to give nitrostyrenes.10 The synthesis of p-carboxystyrene from (p-carboxyphenylmethyl)triphenylphosphonium bromide is also known.11 The authors note that in these cases, only formaldehyde could be used in the Wittig reactions; attempts with other aldehydes proved unsuccessful. Vitamin A acetate is synthesised industrially in water.12 In all cases, pre-mixing of phosphonium salt and aldehyde was necessary for the reaction to proceed.
What is perhaps surprising is that fairly dilute solutions of aqueous base (10–20% aqueous sodium hydroxide or sodium carbonate) were used to form the ylides. The proton removed to form the ylide in benzyl- or allylphosphonium salts has a pKa value in the region of 17–18;13 in these weak aqueous bases, complete deprotonation will not occur. As long as the ylide is in equilibrium with the phosphonium salt, however, the Wittig reaction may proceed.
Although these isolated examples of aqueous Wittig reactions are known, we expected some difficulty in developing a more general method. Specifically, side-reactions may compete with Wittig reactions using aqueous base. Enolisable aldehydes easily undergo self-condensation in carbonate or hydroxide solution at room temperature or below (and this may be the reason for pre-mixing the reagents in the reactions above). Deprotonation of the phosphonium salt to give the ylide is also likely to be in competition with the alkaline hydrolysis of the phosphonium salt to give the phosphine oxide.14
Results and discussion
Synthesis of new phosphonium salts
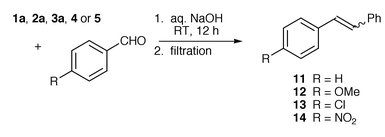
We have designed several phosphonium salts which are soluble in aqueous base; these are derived from alkyltriphenylphosphonium salts where the phenyl rings bear groups to promote solubility in water. Our initial targets were phosphonium bromides 1a and 1b (with one phenyl group bearing a p-COOH group); this substituent was known to promote solubility in the synthesis of p-carboxystyrene and should have a minimal electronic effect on the phosphonium salt. Other phosphonium bromides (2–5) contain one or more phenyl groups with a hydroxy substituent in either a para or meta position relative to phosphorus. These substituents were chosen as they will be deprotonated in aqueous base and the resulting charged species may be soluble in water.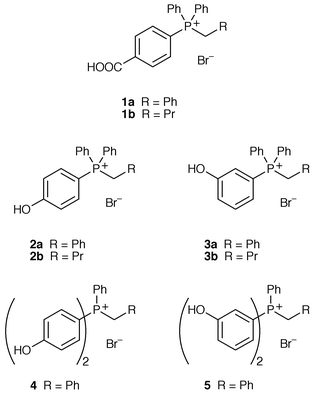
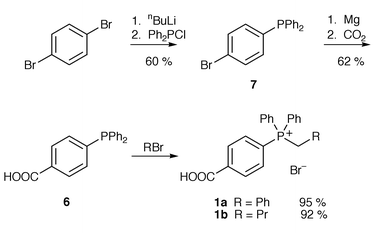
Phosphonium bromides 1a and 1b were synthesised from phosphine 6 and benzyl bromide in refluxing toluene or refluxing bromobutane respectively. Phosphine 6 was obtained by a modified literature procedure;15 we found that the Grignard reagent derived from phosphine 7 when reacted with carbon dioxide gave the required phosphine 6 in an improved 62% yield over the literature method which uses n-butyllithium. Phosphonium bromide 1a was obtained in 37% yield over the three steps; phosphonium bromide 1b was obtained in 36% overall yield (Scheme 1).
 | ||
| Scheme 1 | ||
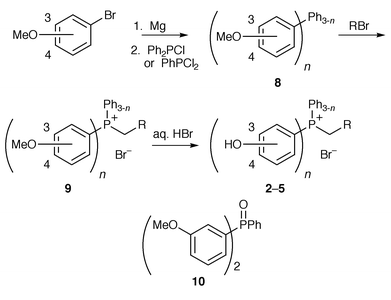
The hydroxy-substituted phosphonium bromides 2–5 were synthesised from the corresponding m- or p-bromoanisoles. The Grignard derivatives were quenched with either chlorodiphenylphosphine (for the series with one hydroxyphenyl substituent) or dichlorophenylphosphine to give (methoxyphenyl)phosphines 8 in good yields.16 Phosphonium bromides 9 were synthesised using similar conditions to those outlined above and were subsequently demethylated in refluxing hydrobromic acid to give the hydroxy-substituted phosphonium salts 2–5 (Scheme 2 and Table 1). We opted for this route as we had difficulties with demethylation of the (methoxyphenyl)phosphines 8, obtaining a mixture of the (hydroxyphenyl)phosphine and its phosphine oxide in each case; alkylation of the phosphine conveniently “protects” the phosphorus against oxidation in the demethylation step.
| Phosphine | Yield (%) | Methoxy-substituted phosphonium salt | Yield (%) | Hydroxy-substituted phosphonium salt | Yield (%) |
|---|---|---|---|---|---|
| a Yield calculated from one-pot reduction–alkylation of phosphine oxide 10. | |||||
| 8 (4-OMe; n = 1) | 86 | 9 (4-OMe; n = 1; R = Ph) | 97 | 2a (4-OH; n = 1; R = Ph) | 99 |
| 9 (4-OMe; n = 1; R = Pr) | 75 | 2b (4-OH; n = 1; R = Pr) | 61 | ||
| 8 (3-OMe; n = 1) | 55 | 9 (3-OMe; n = 1; R = Ph) | 87 | 3a (3-OH; n = 1; R = Ph) | 95 |
| 9 (3-OMe; n = 1; R = Pr) | 81 | 3b (3-OH; n = 1; R = Pr) | 91 | ||
| 8 (4-OMe; n = 2) | 76 | 9 (4-OMe; n = 2; R = Ph) | 87 | 4 (4-OH; n = 2; R = Ph) | 95 |
| 9 (3-OMe; n = 2; R = Ph) | 81a | 5 (3-OH; n = 2; R = Ph) | 40 | ||
 | ||
| Scheme 2 | ||
A fast and reliable method for distinguishing between phosphines, phosphine oxides and phosphonium salts was essential to be certain of the oxidation state of the phosphorus in these reactions. Although a mass spectrum can identify the molecular ion, we discovered that we were able to use the 1JCP and 2JCP coupling constants of the 13C NMR signals of the aryl groups attached to phosphorus. Phosphines show coupling constants of 8–12 and 19–21 Hz respectively, phosphine oxides have coupling constants of 100–110 and 9–12 Hz respectively and phosphonium salts coupling constants of 84–94 and 9–12 Hz respectively, allowing a clear assessment of the nature of the phosphorus compound.
An unexplained oxidation was observed in the reaction of m-bromoanisole with dichlorophenylphosphine; a mixture of the required phosphine 8 (3-OMe; n = 2) and the phosphine oxide 10 was obtained. The mixture was oxidised with hydrogen peroxide to give the phosphine oxide 10 in a quantitative yield. Phosphonium bromide 9 (3-OMe; n = 2; R = Ph) was synthesised directly from the phosphine oxide by a one-pot reduction with polymethylhydrosiloxane and titanium(IV) isopropoxide and subsequent alkylation17 with benzyl bromide in 81% yield.
In spite of these difficulties, we obtained the methoxy-substituted phosphonium salts 9 in good yields. The subsequent demethylation in most cases gave our target phosphonium salts 2–5 in near quantitative yield. Phosphonium salts 2–5 are all insoluble in water at pH 7 but dissolve completely (at approximately 0.1 mol dm−3) in 10% aqueous sodium hydroxide. Stirring in aqueous base at room temperature for up to 24 hours and subsequent addition of aqueous acid caused the phosphonium salts to reprecipitate. They were recovered quantitatively by filtration, showing that alkaline hydrolysis of these phosphonium salts is slow; presumably, the presence of the –COO− or –O− groups inhibits this process. Other phosphonium salts are known to hydrolyse in water; more specifically, water-soluble phosphonium salts bearing sulfonate groups hydrolyse very quickly.18
Wittig reactions with benzylphosphonium salts in aqueous solution
Our initial conditions broadly followed those of previous Wittig reactions carried out in water; phosphonium bromide 1a (with one p-carboxy group) and benzaldehyde were suspended in water and 20% aqueous sodium hydroxide was added dropwise. The phosphonium salt, as expected, dissolved upon addition of base. The reaction mixture was left to stir at room temperature for two hours and then extracted with diethyl ether to give stilbene 11, but only in a low yield of 13% and as a mixture of E- and Z-isomers (approximately 1∶1) (Scheme 3). The phosphine oxide remained in the aqueous basic solution but precipitated on addition of aqueous acid and was subsequently isolated by filtration. No phosphonium salt was recovered.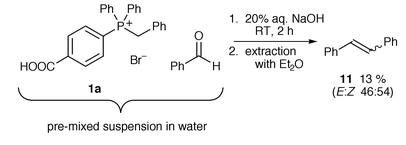 | ||
| Scheme 3 | ||
Addition of ethanol (up to 40% of the solvent content) to the aqueous solutions led to an increase of yield, presumably by promoting dissolution of either starting materials or intermediates. Similar results were observed with phosphonium salt 2a, although lower concentrations of ethanol were required to give acceptable yields of stilbene (Table 2).
We had hoped, however, to be able to recover the olefin products by filtration from the aqueous solution. Stilbene 11 itself is only sparingly soluble in water so we had hoped that it would precipitate from the aqueous basic solution. We felt we could encourage this by increasing the concentration of the reagents. Our initial experiments, requiring extraction with an organic solvent (typically diethyl ether) to recover the olefin, used a concentration of reagents of 0.08 M. We repeated our initial reaction conditions (i.e. no ethanol) using 0.22 M reagents.
Phosphonium bromide 2a (with one p-hydroxy group) and benzaldehyde were suspended in 10 cm3 water. 10% Aqueous sodium hydroxide was added dropwise such that the final concentration of base was 5%. The suspension dissolved and the mixture was stirred overnight at room temperature, although a white precipitate was observed after only one hour. Simple filtration and an aqueous wash gave pure (E)-stilbene in a 40% yield. Addition of dilute hydrochloric acid to the aqueous filtrate gave a second precipitate. This too was isolated in an 86% yield by filtration and was identified as (4-hydroxyphenyl)diphenylphosphine oxide.
The yield of stilbene is lower than that of phosphine oxide; this is probably because only the E-isomer is a solid at room temperature. (Z)-Stilbene is a liquid at room temperature and would not therefore be recovered by filtration. A number of substituted benzaldehydes which would give solid stilbenes were therefore investigated under the same conditions with phosphonium bromides 1a, 2a, 3a, 4 and 5 (Scheme 4 and Table 3).
| Yield (%) (E∶Z) of stilbene | ||||
|---|---|---|---|---|
| Phosphonium salt | 11 | 12 | 13 | 14 |
| a Products recovered by extraction with CH2Cl2. b Only (E)-stilbene 11 is recovered by filtration. c Phosphonium salt 4 recovered. | ||||
| 1a | 91a (46∶54) | 43a (45∶55) | 58a (50∶50) | 100a (50∶50) |
| 2a | 40b | 40 (79∶21) | 91 (72∶28) | 76 (63∶37) |
| 3a | 28b | 72 (68∶32) | 87 (74∶26) | 98 (60∶40) |
| 4 | 17c | 0c | 0c | 6c (91∶9) |
| 5 | 45b | 24 (100∶0) | 81 (86∶14) | 73 (82∶18) |
 | ||
| Scheme 4 | ||
Phosphonium bromide 1a (with one p-carboxy group) showed lower reactivity than the hydroxy-substituted phosphonium bromides 2a, 3a, 4 and 5. The reactions required longer reaction times (days rather than hours) and did not give clean precipitation of olefin products; instead a slurry was observed. Extraction with dichloromethane gave good to excellent yields of olefins, requiring no further purification. The yields of stilbenes 11–14 reflect the reactivity of the benzaldehyde starting materials. In most examples, acidification and filtration of the aqueous residue were carried out to recover (4-carboxyphenyl)diphenylphosphine oxide.
Phosphonium bromides containing hydroxy groups on the whole gave good results (Table 3); the reaction between aldehydes and the phosphonium bromides 2a, 3a, 4 and 5 occurred within hours, but were usually left overnight. In all cases, a white precipitate was observed which was isolated by simple filtration. Again, the phosphine oxide by-products could be recovered by acidification and subsequent filtration of the aqueous filtrate.
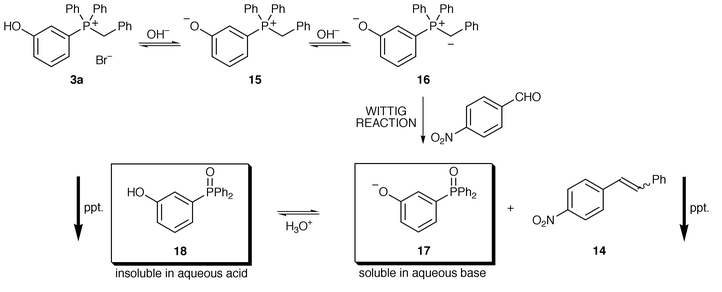
We wish to be precise about the processes occurring in this reaction. Phosphonium bromide 3a (for example) does not dissolve in water but upon addition of aqueous base goes into solution. We presume the ylide 16 is formed via the zwitterionic species 15 (Scheme 5). The ylide can then react with p-nitrobenzaldehyde (for example) to give the stilbene 14, which precipitates out of the reaction mixture as it is insoluble in water. The ionised phosphine oxide 17 remains in solution. After the stilbene has been removed by filtration, aqueous acid is added to the reaction mixture and a second precipitate—the phosphine oxide 18—evolves. Both products of the reaction are therefore recovered by filtration. The yield of p-nitrostilbene in this case is 98%.
 | ||
| Scheme 5 | ||
With the exception of stilbene 11, the olefins were isolated as (E)- and (Z)-alkene mixtures; the yields of stilbene 11 appear lower as no attempt was made to recover the Z-isomer. In one isolated case (the reaction of benzaldehyde with phosphonium bromide 3a), however, an extraction of the aqueous filtrate with diethyl ether recovered an additional 42% yield of stilbene 11 (E∶Z, 72∶28), giving a total yield of 70% of stilbenes.
The number and position of hydroxy groups present in the phosphonium salt have a remarkable effect on the efficacy of the Wittig reaction. The presence of one hydroxy group certainly has a good effect (benzyltriphenylphosphonium bromide did not react and indeed did not even dissolve in aqueous base) but two hydroxy groups seem to have a detrimental effect. The position of the hydroxy group on the phenyl ring is also significant; a meta-hydroxy group seem to promote the Wittig reaction more thoroughly than one in the para position.
Phosphonium bromide 4 (with two para-hydroxy groups) gave low yields of olefins but, in all cases, a precipitate was observed after 1–2 hours of stirring at room temperature in the basic aqueous solution. This precipitate was isolated after an aqueous wash as phosphonium bromide 4. No Wittig reaction took place and presumably one of the deprotonated intermediates is insoluble in water and came out of solution. More importantly, no hydrolysis was detected and no phosphine oxides were isolated.
The high electron-donating potential of the oxido (O−) group may explain why phosphonium salts 3a and 5 (with meta-hydroxy groups) give higher yields of olefins than 2a and 4 respectively; the substituent coefficients σ for para-O− and meta-O− are −0.81 and −0.47 respectively.19 A para-oxido group will therefore have a much greater effect on the phosphonium salt making the deprotonation to form the ylide more difficult and this is reflected in the yields of olefins. Phosphonium salt 3a is deprotonated to give phosphonium salt 15 and, subsequently, ylide 16 (Scheme 5). Presumably, the equilibrium is more towards the ylide than in the para-substituted analogues derived from phosphonium salt 2a. This effect is much more evident in the bis-hydroxy salts 4 and 5.
By comparing the reactions of phosphonium salts 2a and 3a with 4 and 5, it is clear that better yields of olefins are obtained when only one hydroxy group is present. In basic solution, the hydroxy groups are likely to be ionised and will therefore inhibit formation of the phosphorus ylides for two reasons. In addition to the electron-donating potential of the oxido group reducing the positive charge on phosphorus, the phosphorus ylides would be either dianions (for ylides derived from phosphonium salts 2a and 3a) or even trianions (for ylides derived from phosphonium salts 4 and 5, each containing two hydroxy groups); each successive deprotonation will be less favoured. The ylides derived from phosphonium salts 4 and 5 are therefore likely to be present in lower concentrations, slowing down or even stopping the Wittig reaction.
Water-soluble phosphonium salts provide an improved practical synthesis of substituted stilbenes by the Wittig reaction. Although the stilbenes could be recovered by extraction of the basic aqueous solution with an organic solvent (diethyl ether or dichloromethane), in most cases a simple filtration gave good yields, comparable to extraction. Similarly, the phosphine oxide by-product can be recovered either by filtration from an acidic aqueous solution or by extraction with an organic solvent. Both products of the reaction are therefore recovered separately in good to excellent yields in spectroscopically pure form. The reactions require no organic solvent, either during the reaction or in the isolation of the product. Phosphonium bromide 3a (with one hydroxy group meta to phosphorus) gives the best overall reaction with substituted benzaldehydes in water, on the grounds of ease of synthesis, rate of reaction and simplicity of work-up.
We studied the reaction of our best phosphonium bromide 3a with a wider range of aldehydes (Table 4). The results were less encouraging. Attention is drawn to the range of aldehydes listed in the table: entries 1–6 are non-enolisable aldehydes; entries 7–10 are all enolisable. In addition, entries 3–6, and 9 and 10 are all water-soluble aldehydes. 2-Furaldehyde and pyridine-2-carbaldehyde (entries 1 and 2) are water-insoluble and non-enolisable and show similar reactivities to the benzaldehydes. Wittig reactions carried out under the same conditions gave olefin products (by filtration or extraction with dichloromethane) in acceptable yields.
| Entry | Aldehyde | Products and yields (%) |
|---|---|---|
| 1 | 2-Furaldehyde | Alkene – 75 |
| Phosphine oxide – 67 | ||
| 2 | Pyridine-2-carbaldehyde | Alkene – 47 |
| Phosphine oxide – 80 | ||
| 3 | 3-Hydroxybenzaldehyde | Phosphonium salt recovered |
| Aldehyde recovered | ||
| 4 | 4-Hydroxybenzaldehyde | Phosphonium salt recovered |
| Aldehyde recovered | ||
| 5 | Terephthaldehyde | Phosphine oxide – 79 |
| 6 | Aq. formaldehyde | Phosphine oxide – 42 |
| 7 | Butyraldehyde | Phosphine oxide – 54 |
| 8 | Isobutyraldehyde | Phosphonium salt recovered |
| 9 | Chloral | Phosphonium salt recovered |
| 10 | Glutaraldehyde | Phosphonium salt recovered |
Surprisingly, water-soluble aldehydes did not undergo Wittig reactions in water. We expected the hydroxybenzaldehydes (entries 3 and 4) to dissolve in aqueous base to react to give hydroxystilbenes (which should remain in the aqueous basic solution). The aldehydes did indeed dissolve upon addition of base, but only starting materials were recovered—both aldehyde and phosphonium salt—in quantitative yield after acidification and filtration in both cases. The water-soluble dialdehydes (entries 5 and 6) also yielded no olefins but phosphine oxide was recovered after acidification and filtration. The enolisable water-soluble aldehydes (entries 9 and 10) also did not react and the phosphonium bromide 3a was recovered. Other enolisable aldehydes (entries 7 and 8) also led to recovery of phosphonium bromide 3a; base-catalysed self-condensations of aldehyde took place preferentially in these cases.
Attempted aqueous Wittig reactions with butylphosphonium salts
Under the optimal conditions previously determined, phosphonium bromide 1b was added to benzaldehyde in 5% aqueous NaOH. The phosphonium bromide 1b dissolved upon addition of base and the mixture was stirred overnight. No precipitation was observed and extraction with diethyl ether and subsequent evaporation gave butyldiphenylphosphine oxide 17 in a 31% yield. Acidification of the aqueous residue and subsequent extraction gave phosphine oxide 18 in a 67% yield (Scheme 6). This was the first evidence of any hydrolysis having occurred with these compounds; hydrolysis with loss of phenyl anion or p-carboxyphenyl [or possibly p-(COO−)phenyl] anion to give phosphine oxides 17 and 18 occurs in a statistical ratio with almost quantitative conversion. No Wittig reaction took place between the ylide and benzaldehyde.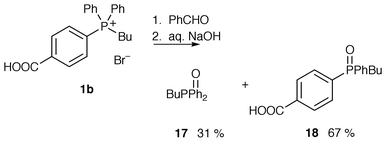 | ||
| Scheme 6 | ||
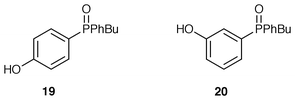
Phosphonium bromides 2b and 3b and benzaldehyde under the same conditions led only to quantitative recovery of starting material. No hydrolysis of the phosphonium salts occurred and it may be that the oxido group makes the phosphonium salts resistant to hydrolysis by deactivation of the phosphorus towards attack by hydroxide. Increasing concentrations of base (up to 50% aqueous NaOH) were used in similar reactions but in all cases only phosphonium salts were recovered. Increasing the temperature to reflux and increasing the reaction time did eventually lead to hydrolysis of the phosphonium salt; (hydroxyphenyl)phenylphosphine oxides 19 or 20 and starting material (to a greater or lesser extent) were recovered after refluxing the reaction mixtures for 1 day. In other related experiments, the addition of ethanol also promoted hydrolysis.
Conclusion
We have shown that our new phosphonium salts dissolve in aqueous base (5–10% aqueous NaOH) and are resistant to hydrolysis at room temperature. Wittig reactions of benzyl-containing phosphonium bromides 1a, 2a, 3a, 4 and 5 with substituted benzaldehydes in aqueous NaOH gave unsymmetrical stilbenes (predominantly E) in good yields. Phosphonium bromide 3a, containing one hydroxy group meta to phosphorus, gave the best results under these conditions.Experimental
Flash chromatography was carried out using Merck Kieselgel 60 (230–400 mesh). Thin layer chromatography (TLC) was carried out on commercially available pre-coated glass plates (Merck silica Kieselgel 60F254). All solvents were distilled before use. Anhydrous solvents were distilled from lithium aluminium hydride in the case of tetrahydrofuran and diethyl ether, and from calcium hydride in the case of toluene.Melting points were determined on a Reichart hot stage microscope or a Stuart SMP1 melting point apparatus and are uncorrected. Infra-red spectra were recorded on a Perkin-Elmer 1600 series FTIR spectrometer. All 1H and 13C NMR spectra were recorded on Bruker WM 200, WM 250 or WM 400 Fourier transform spectrometers, using an internal deuterium lock. The 13C NMR spectra were recorded with broad band decoupling and attached proton test (APT). The symbols + and − after the 13C NMR chemical shifts indicate odd and even numbers respectively of attached protons. δ Values are given in ppm relative to tetramethylsilane. J Values are given in hertz (Hz). In the assignment of the 1H and 13C NMR spectra of phosphorus compounds, the following notation has been used: Ph refers to a phenyl ring attached to phosphorus or a C![[double bond, length half m-dash]](https://www.rsc.org/images/entities/char_e006.gif) C bond and numbering starts from the point of attachment, Ar refers to phenyl rings with more than one substituent and numbering starts from the point of attachment to phosphorus or a CC bond, where appropriate.
C bond and numbering starts from the point of attachment, Ar refers to phenyl rings with more than one substituent and numbering starts from the point of attachment to phosphorus or a CC bond, where appropriate.
Mass spectra were determined by electron impact (EI) or fast atom bombardment (+FAB). All mass spectra were recorded on a Kratos MS890 double-beam mass spectrometer by technical staff. Microanalyses were performed by technical staff using either Carlo Elba 1106 or Perkin Elmer 240 automatic analysers.
(4-Bromophenyl)diphenylphosphine 7
By the method of Ravindar et al.,15 the phosphine was obtained (62%) as white plates, mp 74–77 °C (from MeOH) (lit.,15 79–80 °C). No NMR data were recorded previously: δH(250 MHz; CDCl3) 7.53 (2 H, dd, 3JHP 1.2 and J 8.4, ArH-2), 7.43–7.40 (10 H, m, 2 × Ph) and 7.26 (2 H, dd, 4JHP 6.9 and J 8.4, ArH-3); δC(63 MHz; CDCl3) 136.8− (PhC-1), 136.7− (ArC-1), 135.3+ (d, 2JCP 20.0, ArC-2), 133.9+ (d, 2JCP 19.7, PhC-2), 131.8+ (d, 3JCP 6.8, ArC-3), 128.8+ (PhC-4), 128.7+ (d, 3JCP 8.7, PhC-3) and 123.5− (ArC-4).4-(Diphenylphosphanyl)benzoic acid 6
We report an improved synthesis of phosphine 6.15 Dry magnesium turnings (0.60 g, 25.0 mmol) were stirred in dry THF (20 cm3) under argon. (4-Bromophenyl)diphenylphosphine 7 (8.35 g, 24.48 mmol) was dissolved in dry THF (20 cm3) and approximately one half of this was added to the magnesium. One crystal of iodine was added and the reaction vessel was heated until the Grignard reaction was initiated. The remaining phosphine solution was added at such a rate so as to maintain reflux and the solution was then refluxed until all the magnesium had been consumed, giving a red solution. The solution was allowed to cool and was poured onto crushed CO2 kept at −78 °C. A gum developed which was subsequently covered with CO2 pellets. Dry THF (20 cm3) was added and the solution was allowed to stir overnight giving a yellow suspension. Further CO2 was added and allowed to dissolve. Saturated aqueous Na2CO3 (20 cm3) was added to quench the reaction, followed by water (20 cm3). 3 M HCl was added to pH 1 and the solution was extracted with diethyl ether (3 × 20 cm3). The combined organic extracts were washed with 3 M HCl (20 cm3), dried (MgSO4) and evaporated under reduced pressure to give a yellow residue. This residue was recrystallised from acetic acid to give the phosphine 6 (2.13 g, 29%) as pale yellow needles. The remaining solution was evaporated under reduced pressure and a further recrystallisation gave more phosphine 6 (2.45 g, 33%) again as pale yellow needles, mp 159–162 °C (from AcOH) [lit.,15 156–158 °C (from MeOH–H2O)]. No NMR data were recorded previously: δH(250 MHz; d6-DMSO) 13.04 (1 H, br s, COOH), 7.9 (2 H, dd, 3JHP 0.9 and J 7.9, ArH-2) and 7.5–7.2 (12 H, m, ArH-3 and 2 × Ph); δC(62.9 MHz; d6-DMSO) 168.2− (COOH), 140.2− (d, 1JCP 12.3, ArC-1), 136.5− (d, 1JCP 11.3, PhC-1), 136.3− (ArC-4), 133.5+ (d, 2JCP 19.6, PhC-2), 132.7+ (d, 2JCP 25.5, ArC-2), 129.3+, 129.0+ and 128.9+.4-(Benzyldiphenylphosphonio)benzoic acid bromide 1a
4-(Diphenylphosphanyl)benzoic acid 6 (1.37 g, 4.47 mmol) was suspended in dry toluene (10 cm3) under argon. Benzyl bromide (0.53 cm3, 4.47 mmol) in dry toluene (5 cm3) was added and the mixture refluxed for 24 h. The white precipitate was collected and washed with diethyl ether to give the phosphonium salt (2.07 g, 95%) as a powder, mp 308–310 °C (white plates from EtOH) (Found: C, 65.1; H, 4.6; P, 6.5. C26H22BrO2P requires C, 65.4; H, 4.65; P, 6.5%); νmax(Nujol)/cm−1 3500–2000 (COOH), 1710 (CO) and 1461 (P–Ph); δH(250 MHz; d6-DMSO) 13.7 (1 H, br s, COOH), 8.22 (2 H, dd, 4JHP 3.0 and J 8.4, ArH-3), 8.0–7.7 (12 H, m, ArH-2 and PPh), 7.29 (1 H, dt, 6JHP 1.6 and J 7.3, CH2PhH-4), 7.24 (2 H, br t, J 7.3, CH2PhH-3), 7.00 (2 H, dd, 4JHP 1.4 and J 7.3, CH2PhH-2) and 5.29 (2 H, d, 2JHP 15.8, PCH2); δC(101 MHz; d6-DMSO) 166.2− (COOH), 136.5− (ArC-4), 135.3+ (CH2PhC-4), 134.6+ (d, 4JCP 10.3, PPhC-4), 134.2+ (CH2PhC-2 or CH2PhC-3), 134.1+ (CH2PhC-2 or CH2PhC-3), 130.9+ (d, J 5.3, ArC-2 or ArC-3), 130.2+ (d, J 12.5, PPhC-2 or PPhC-3), 128.9+ (PPhC-2 or PPhC-3), 128.5+ (ArC-2 or ArC-3), 127.7− (d, 2JCP 8.6, CH2PhC-1), 122.6− (d, 1JCP 84.1, PPhC-1), 117.4− (d, 1JCP 85.6, ArC-1) and 28.1− (d, 1JCP 46.3, PCH2); m/z (+FAB) 397 (M+), 352 (M − COOH). M is defined here as the phosphonium cation.4-(Butyldiphenylphosphonio)benzoic acid bromide 1b
4-(Diphenylphosphanyl)benzoic acid 6 (1.886 g, 6.16 mmol) was stirred in n-bromobutane (10 cm3) at reflux overnight under argon. A white precipitate was collected and washed with diethyl ether to give the phosphonium salt (2.53 g, 92%) as a white powder, mp 233–238 °C (Found: C, 65.7; H, 5.3; P, 6.3. C23H24BrO2P requires C, 64.6; H, 5.3; P, 6.0%); νmax(Nujol)/cm−1 1709 (CO) and 1462 (P–Ph); δH(400 MHz; d6-DMSO) 13.7 (1 H, br s, COOH), 8.2 (2 H, dd, 4JHP 2.9 and J 8.4, ArH-3), 8.0–7.7 (12 H, m, ArH-2 and 2 × Ph), 3.6 (2 H, m, PCH2), 1.5 (4 H, m, PCH2CH2CH2Me) and 0.9 (3 H, t, J 6.8, Me); δC(101 MHz; d6-DMSO) 166.2− (COOH), 136.3− (ArC-4), 135.1+ (PhC-4), 134.2+ (d, 2JCP 10.4, ArC-2), 133.7+ (d, 2JCP 10.3, PhC-2), 130.5+ (d, 3JCP 12.6, ArC-3), 130.4+ (d, 3JCP 12.5, PhC-3), 123.4− (d, 1JCP 84.1, ArC-1), 118.1− (d, 1JCP 85.5, PhC-1), 23.8− (d, 2JCP 4.3, PCH2CH2), 23.2− (d, 3JCP 17.4, CH2Me), 19.9− (d, 1JCP 49.2, PCH2) and 13.3+ (Me); m/z (+FAB) 363.1533 (100%, M+. M requires 363.1544). M is defined here as the phosphonium cation.4-Methoxyphenyldiphenylphosphine 8 (4-OMe; n = 1)
By the method of McEwen et al.,16 the phosphine was obtained (86%) as needles. The melting point16 and 1H NMR data20 are consistent with previous data.3-Methoxyphenyldiphenylphosphine 8 (3-OMe; n = 1)
In the same way, the phosphine was obtained (55%) as needles. The melting point and 1H NMR data are consistent with previous data.16Bis(4-methoxyphenyl)phenylphosphine 8 (4-OMe; n = 2)
In the same way, the phosphine was obtained (76%) as plates, mp 87–88 °C (from MeOH) (lit.,21 89–90 °C); low-field 1H NMR data were recorded previously:22δH(400 MHz; CDCl3) 7.3 (3 H, m), 7.25 (4 H, dd, 4JHP 7.5 and J 8.4, ArH-3), 7.25 (2 H, m), 6.88 (4 H, br d, J 8.4, ArH-2) and 3.84 (6 H, s, Me); δC(101 MHz; CDCl3) 160.3− (ArC-4), 138.6− (d, 1JCP 10.5, ArC-1), 135.3+ (d, 2JCP 21.0, ArC-2), 134.0− (d, 1JCP 4.0, PhC-1), 133.1+ (d, 2JCP 18.7, PhC-2), 128.4+ (m, PhC-3 and PhC-4), 114.2+ (d, 3JCP 7.8, ArC-3) and 55.2+ (OMe).Bis(3-methoxyphenyl)phenylphosphine oxide 10
In the same way, dry magnesium turnings (6.00 g, 0.25 mol), 3-bromoanisole (34.0 cm3, 0.27 mol) and dichlorophenylphosphine (16.7 cm3, 0.13 mol) gave a yellow oil. 13C NMR analysis showed this to be a mixture of bis(3-methoxyphenyl)phenylphosphine and its oxide. The oil was dissolved in THF (50 cm3) and hydrogen peroxide (100 vol solution in water) was added dropwise until no further temperature rise was observed. The solution was evaporated under reduced pressure to give the phosphine oxide (38.5 g, 100%) as white needles, mp 68–70 °C (from cyclohexane); Rf(EtOAc) 0.23; νmax(Nujol)/cm−1 1595 and 1574 (Ph), 1463 (P–Ph) and 1285 (P=O); δH(400 MHz; CDCl3) 7.65–7.60 (2 H, m), 7.52–7.47 (1 H, br t, J 7.4, PhH-4), 7.43–7.40 (2 H, m), 7.32–7.23 (4 H, m), 7.13–7.08 (2 H, ddd, J 0.7, 7.4 and 11.7), 7.04–7.01 (2 H, m) and 3.74 (6 H, s, OMe); δC(101 MHz; CDCl3) 159.6− (d, 3JCP 15.0, ArC-3), 133.4− (d, 1JCP 103.9, ArC-1), 132.1+ (PhC-4), 132.1+ (d, 2JCP 10.3, PhC-2), 132.0− (d, 1JCP 104.9, PhC-1), 129.7+ (d, 3JCP 14.5, ArC-5), 128.6+ (d, 3JCP 12.3, PhC-3), 124.3+ (d, 2JCP 10.1, ArC-6), 118.3+ (d, 4JCP 2.3, ArC-4), 116.8+ (d, 2JCP 10.8, ArC-2) and 55.4+ (OMe); m/z (EI) 338.1076 (5%, M+. M requires 338.10723), 337.1 (12%, M − H), 188.0 (60), 84.0 (100) and 49.0 (85).Benzyl(4-methoxyphenyl)diphenylphosphonium bromide 9 (4-OMe; n = 1; R = Ph)
4-Methoxyphenyldiphenylphosphine 8 (4-OMe; n = 1) (12.00 g, 41.0 mmol) was suspended in dry toluene (150 cm3) under argon. Benzyl bromide (4.9 cm3, 41.0 mmol) was added and the mixture refluxed overnight. A white precipitate was collected and washed with diethyl ether to give the phosphonium salt (18.37 g, 97%) as a powder, mp 221–224 °C (white needles from EtOH–EtOAc) (lit.,16 225–228 °C). Only melting point and analytical data were previously published:16δH(500 MHz; d6-DMSO) 7.9 (2 H, br t, J 7.6, PPhH-4), 7.7 (4 H, dt, 4JHP 3.5 and J 7.6, PPhH-3), 7.6 (4 H, dd, J 7.6 and 3JHP 12.5, PPhH-2), 7.6 (2 H, dd, J 8.9 and 3JHP 12.0, ArH-2), 7.3 (3 H, m, ArH-3 and CH2PhH-4), 7.2 (2 H, t, J 7.1, CH2PhH-3), 7.0 (2 H, br d, J 7.1, CH2PhH-2), 5.1 (2 H, d, 2JHP 15.6, PCH2) and 3.9 (3 H, s, OMe); δC(101 MHz; CDCl3) 164.8− (ArC-4), 136.5+ (d, 2JCP 11.4, ArC-2), 134.8+ (d, 4JCP 2.6, PPhC-4), 134.2+ (d, 2JCP 9.8, PPhC-2), 131.5+ (d, 4JCP 5.3, CH2PhC-3), 130.1+ (d, 3JCP 12.5, PPhC-3), 128.8+ (d, 5JCP 3.0, CH2PhC-4), 128.3+ (d, 3JCP 3.6, CH2PhC-2), 127.3− (d, 2JCP 8.5, CH2PhC-1), 118.5− (d, 1JCP 86.3, PPhC-1), 116.0+ (d, 3JCP 13.9, ArC-3), 107.0− (d, 1JCP 93.6, ArC-1), 55.9+ (OMe) and 31.2− (d, 1JCP 48.0, PCH2).Butyl(4-methoxyphenyl)diphenylphosphonium bromide 9 (4-OMe; n = 1; R = Pr)
4-Methoxyphenyldiphenylphosphine 8 (4-OMe; n = 1) (5.96 g, 20.4 mmol) was stirred in n-bromobutane (30 cm3) at reflux overnight under argon. A white precipitate was collected and washed with diethyl ether to give the phosphonium salt (6.56 g, 75%) as a powder, mp 142–145 °C (Found: C, 64.1; H, 6.0; Br, 18.3; P, 7.4. C23H26BrOP requires C, 64.3; H, 6.1; Br, 18.6; P, 7.2%); νmax(Nujol)/cm−1 1587 (Ph) and 1462 and 1442 (P–Ph); δH(400 MHz; CDCl3) 7.70–7.55 (12 H, m), 7.11 (2 H, dd, 4JHP 2.6 and J 9.0, ArH-3), 3.80 (3 H, s, OMe), 3.47–3.38 (2 H, m, PCH2), 1.59–1.41 (4 H, m, PCH2CH2CH2Me) and 0.78 (3 H, t, J 6.9, CH2Me); δC(101 MHz; CDCl3) 164.8− (ArC-4), 135.7+ (d, 2JCP 11.5, ArC-2), 134.9+ (br s, PhC-4), 133.4+ (d, 2JCP 10.0, PhC-2), 130.4+ (d, 3JCP 12.5, PhC-3), 118.9− (d, 1JCP 86.4, PhC-1), 116.4+ (d, 3JCP 13.7, ArC-3), 107.3− (d, 1JCP 93.4, ArC-1), 56.1+ (OMe), 24.5− (d, 2JCP 4.3, PCH2CH2), 23.7− (d, 3JCP 16.3, CH2Me), 22.9− (d, 1JCP 51.5, PCH2) and 13.7+ (CH2Me); m/z (+FAB) 349.1702 (100%, M+. M requires 349.1721). M is defined here as the phosphonium cation.Benzyl(3-methoxyphenyl)diphenylphosphonium bromide 9 (3-OMe; n = 1; R = Ph)
3-Methoxyphenyldiphenylphosphine 8 (3-OMe; n = 1) (6.82 g, 23.3 mmol) was dissolved in dry toluene (100 cm3) under argon. Benzyl bromide (2.8 cm3, 23.5 mmol) was added and the mixture refluxed overnight. A white precipitate was collected and washed with diethyl ether to give the phosphonium salt (9.44 g, 87%) as a powder, mp 261–265 °C (from EtOH) [lit.,16 264–266 °C (from EtOH–Et2O)]. Only melting point and analytical data were previously published:16δH(500 MHz; CDCl3) 7.77–7.70 (6 H, m, PPhH-2 and PPhH-4), 7.61 (4 H, dt, 4JHP 3.5 and J 8.0, PPhH-3), 7.51 (1 H, dt, 4JHP 4.0 and 7.9, ArH-5), 7.25 (1 H, t, J 2.3), 7.23–7.17 (3 H, m), 7.12–7.11 (4 H, m), 5.43 (2 H, d, 2JHP 14.5, PCH2) and 3.84 (3 H, s, OMe); δC(101 MHz; CDCl3) 160.4− (d, 3JCP 15.6, ArC-3), 135.0+ (br s, PPhC-4), 134.4+ (d, 2JCP 9.8, PPhC-2), 131.5+ (d, 4JCP 5.5, CH2PhC-3), 131.4+ (d, 3JCP 15.0, ArC-5), 130.1+ (d, 3JCP 12.6, PPhC-3), 128.8+ (br s, CH2PhC-2), 128.4+ (br s, ArC-4), 127.3− (d, 2JCP 8.5, CH2PhC-1), 126.4+ (d, 2JCP 9.7, ArC-6), 121.3+ (br s, CH2PhC-4), 119.1− (d, 1JCP 85.4, ArC-1), 118.8+ (d, 2JCP 15.9, ArC-2), 117.9− (d, 1JCP 85.6, PPhC-1), 56.4+ (s, OMe) and 30.9− (d, 1JCP 46.5, PCH2).Butyl(3-methoxyphenyl)diphenylphosphonium bromide 9 (4-OMe; n = 1; R = Pr)
3-Methoxyphenyldiphenylphosphine 8 (3-OMe; n = 1) (4.02 g, 13.8 mmol) was stirred in n-bromobutane (50 cm3) at reflux temperature overnight under argon. The white precipitate was collected and washed with diethyl ether to give the phosphonium salt (4.78 g, 81%) as a powder, mp 154–155 °C (Found: C, 64.0; H, 6.0; Br, 18.2; P, 7.3. C23H26BrOP requires C, 64.3; H, 6.1; Br, 18.6; P, 7.2%); νmax(Nujol)/cm−1 1592 and 1574 (Ph) and 1463 (P–Ph); δH(400 MHz; CDCl3) 7.85–7.7 (6 H, m), 7.7–7.6 (4 H, m), 7.60–7.55 (1 H, m), 7.3–7.2 (3 H, m), 3.86 (3 H, s, OMe), 3.72 (2 H, m, PCH2), 1.7–1.5 (4 H, m, PCH2CH2CH2Me) and 0.88 (3 H, t, J 7.0, CH2Me); δC(101 MHz; CDCl3) 160.7− (d, 3JCP 15.8, ArC-3), 135.0+ (d, 4JCP 2.6, PhC-4), 133.7+ (d, 2JCP 9.9, PhC-2), 131.8+ (d, 3JCP 14.7, ArC-5), 130.5+ (d, 3JCP 12.5, PhC-3), 125.5+ (d, 2JCP 9.6, ArC-6), 120.7+ (d, 4JCP 2.4, ArC-4), 119.8− (d, 1JCP 85.3, ArC-1), 118.7+ (d, 2JCP 11.6, ArC-2), 118.3− (d, 1JCP 86.0, PhC-1), 56.4+ (OMe), 24.7− (d, 2JCP 4.4, PCH2CH2), 23.7− (d, 3JCP 16.4, CH2Me), 22.7− (d, 1JCP 50.0, PCH2) and 13.8+ (CH2Me); m/z (+FAB) 349.1704 (100%, M+. M requires 349.1721). M is defined here as the phosphonium cation.Benzyl[bis(4-methoxyphenyl)]phenylphosphonium bromide 9 (4-OMe; n = 2; R = Ph)
The phosphonium chloride has previously been reported.23 Bis(4-methoxyphenyl)phenylphosphine 8 (4-OMe; n = 2) (3.10 g, 9.6 mmol) was dissolved in dry toluene (150 cm3) under argon. Benzyl bromide (1.14 cm3, 9.6 mmol) was added and the mixture refluxed overnight. A white precipitate was collected and washed with diethyl ether to give the phosphonium salt (4.13 g, 87%) as a powder, mp 167–173 °C (white prisms from EtOH–EtOAc) (Found: C, 63.7; H, 5.5; P, 6.2. C27H26BrO2P requires C, 65.7; H, 5.3; P, 6.3.%); νmax(Nujol)/cm−1 1594 and 1566 (Ph) and 1455 (P-Ph); δH(500 MHz; CDCl3) 7.7 (1 H, m, PPhH-4), 7.6 (8 H, m), 7.2 (1 H, br t, J 7.3, CH2PhH-4), 7.1 (8 H, m), 5.1 (2 H, d, 2JHP 14.3, PCH2) and 3.8 (6 H, s, OMe); δC(101 MHz; CDCl3) 164.7− (d, 4JCP 2.7, ArC-4), 136.4+ (d, 2JCP 11.3, ArC-2), 134.7+ (d, 4JCP 2.5, PPhC-4), 134.1+ (d, 2JCP 9.7, PPhC-2), 131.4+ (d, 4JCP 5.3, CH2PhC-3), 130.0+ (d, 3JCP 12.4, PPhC-3), 128.8+ (d, 5JCP 3.1, CH2PhC-4), 128.3+ (d, 3JCP 3.6, CH2PhC-2), 127.6− (d, 2JCP 8.6, CH2PhC-1), 119.1− (d, 1JCP 86.7, PPhC-1), 115.9+ (d, 3JCP 13.6, ArC-3), 107.7− (d, 1JCP 93.9, ArC-1), 55.9+ (OMe) and 31.6− (d, 1JCP 49.0, PCH2); m/z 412.1580 (60%, M+ − H. C27H25O2P requires 412.1670) and 322.1 (100%, M+ − CH2Ph). M is defined here as the phosphonium cation.Benzyl[bis(3-methoxyphenyl)]phenylphosphonium bromide 9 (3-OMe; n = 2; R = Ph)
By the method of Coumbe et al.,17 titanium(IV) isopropoxide (17.3 cm3, 58.2 mmol) was added to a stirred solution of polymethylhydrosiloxane (34.7 cm3, 582 mmol, 10 eq.) and bis(3-methoxyphenyl)phenylphosphine oxide 10 (19.68 g, 58.2 mmol) in dry THF (100 cm3) under argon. The yellow solution was refluxed overnight. The solution was allowed to cool and benzyl bromide (7.0 cm3, 58.85 mmol) was added. The solution was refluxed for a further 4 h and, after cooling, was filtered and washed with diethyl ether to give the phosphonium salt (23.32 g, 81%) as a powder, mp 233–236 °C (yellow needles from EtOH–EtOAc); νmax(Nujol)/cm−1 1589 and 1571 (Ph) and 1462 (P–Ph); δH(500 MHz; CDCl3) 7.74–7.70 (3 H, m, PPhH-2 and PPhH-4), 7.60 (2 H, dt, 4JHP 3.4 and J 7.6, PPhH-3), 7.48 (2 H, dt, 4JHP 4.7 and 8.3, ArH-5), 7.26–7.19 (7 H, m), 7.13–7.11 (4 H, m), 5.44 (2 H, d, 2JHP 14.5, PCH2) and 3.83 (6 H, s, OMe); δC(101 MHz; CDCl3) 160.3− (d, 3JCP 15.9, ArC-3), 135.0+ (d, 4JCP 2.5, PPhC-4), 134.3+ (d, 2JCP 9.9, PPhC-2), 131.5+ (d, 3JCP 8.5, ArC-5), 131.4+ (s, CH2PhC-2), 130.1+ (d, 3JCP 12.5, PPhC-3), 128.8+ (d, 4JCP 2.8, CH2PhC-3), 128.4+ (d, 4JCP 3.6, ArC-4), 127.3− (d, 2JCP 8.5, CH2PhC-1), 126.3+ (d, 2JCP 9.6, ArC-6), 121.2+ (d, 5JCP 2.4, CH2PhC-4), 118.9− (d, 1JCP 85.5, ArC-1), 118.9+ (d, 2JCP 11.3, ArC-2), 117.7− (d, 1JCP 85.9, PPhC-1), 56.3+ (s, OMe) and 30.7− (d, 1JCP 46.7, PCH2); m/z (+FAB) 412.1598 (20%, M+ − H. C27H25O2P requires 412.1670) and 322.1 (100%, M+ − CH2Ph). M is defined here as the phosphonium cation.Benzyl(4-hydroxyphenyl)diphenylphosphonium bromide 2a
Benzyl(4-methoxyphenyl)diphenylphosphonium bromide 9 (4-OMe; n = 1; R = Ph) (36.60 g, 79 mmol) was stirred in hydrobromic acid (250 cm3 of a 48% solution in water) at reflux overnight under argon. A white precipitate was collected and washed with diethyl ether. It was then dried over two days under reduced pressure to give the phosphonium salt (35.21 g, 99%) as a white powder, mp 317–319 °C (from aqueous MeOH) (Found: C, 66.6; H, 4.9; Br, 17.6; P, 7.0. C25H22BrOP requires C, 66.8; H, 4.9; Br, 17.8; P, 6.9%); νmax(Nujol)/cm−1 1601 and 1578 (Ph) and 1461 (P–Ph); δH(500 MHz; d6-DMSO) 11.1 (1 H, s, OH), 7.9 (2 H, t, J 7.6, PPhH-4), 7.7 (4 H, dt, 4JHP 3.4 and J 7.6, PPhH-3), 7.6 (4 H, dd, J 7.6 and 3JHP 12.4, PPhH-2), 7.45 (2 H, dd, J 8.7 and 3JHP 11.9, ArH-2), 7.3 (1 H, br t, J 7.3, CH2PhH-4), 7.2 (2 H, t, J 7.3, CH2PhH-3), 7.1 (2 H, dd, 4JHP 2.5 and J 8.7, ArH-3), 7.0 (2 H, d, J 7.3, CH2PhH-2) and 5.1 (2 H, d, 2JHP 16.0, PCH2); δC(101 MHz; d6-DMSO) 163.6− (d, 4JCP 2.4, ArC-4), 136.3+ (d, 2JCP 12.0, ArC-2), 134.9+ (d, 4JCP 2.0, PPhC-4), 133.9+ (d, 2JCP 10.0, PPhC-2), 130.9+ (d, 4JCP 5.0, CH2PhC-3), 130.0+ (d, 3JCP 12.0, PPhC-3), 128.7+ (d, 5JCP 2.5, CH2PhC-4), 128.4− (d, 2JCP 9.0, CH2PhC-1), 128.3+ (d, 3JCP 2.5, CH2PhC-2), 118.9− (d, 1JCP 86.0, PPhC-1), 117.3+ (d, 3JCP 14.0, ArC-3), 104.6− (d, 1JCP 94.0, ArC-1) and 28.7− (d, 1JCP 48.0, PCH2); m/z (+FAB) 369.3 (100%, M+), 154.1 (70%) and 136.1 (55%). M is defined here as the phosphonium cation.Butyl(4-hydroxyphenyl)diphenylphosphonium bromide 2b
In the same way, butyl(4-methoxyphenyl)diphenylphosphonium bromide 9 (4-OMe; n = 1; R = Pr) (4.14 g, 9.7 mmol) gave the phosphonium salt (2.43 g, 61%) as needles, mp 177–180 °C (from EtOH–Et2O); νmax(Nujol)/cm−1 1598 and 1576 (Ph) and 1463 and 1436 (P–Ph); δH(400 MHz; d6-DMSO) 10.9 (1 H, br s, OH), 7.87 (2 H, dt, 5JHP 2.8 and J 5.9, PhH-4), 7.80–7.72 (8 H, m, PhH-2 and PhH-3), 7.60 (2 H, dd, J 8.8 and 3JHP 12.0, ArH-2), 7.13 (2 H, dd, 4JHP 2.6 and J 8.8, ArH-3), 3.52–3.44 (2 H, m, PCH2), 1.50–1.45 (4 H, m, PCH2CH2CH2Me) and 0.87 (3 H, t, J 6.8, Me); δC(101 MHz; d6-DMSO) 163.3− (ArC-4), 135.9+ (d, 2JCP 11.6, ArC-2), 134.6+ (PhC-4), 133.4+ (d, 2JCP 10.0, PhC-2), 130.2+ (d, 3JCP 12.3, PhC-3), 119.7− (d, 1JCP 86.1, PhC-1), 117.4+ (d, 3JCP 13.7, ArC-3), 105.6− (d, 1JCP 93.9, ArC-1), 23.9− (d, 2JCP 3.6, PCH2CH2), 23.2− (d, 3JCP 17.1, CH2Me), 20.6− (d, 1JCP 51.2, PCH2) and 13.4+ (Me); m/z (+FAB) 335.1554 (100%, M+. M requires 335.1565). M is defined here as the phosphonium cation.Benzyl(3-hydroxyphenyl)diphenylphosphonium bromide 3a
In the same way, benzyl(3-methoxyphenyl)diphenylphosphonium bromide 9 (3-OMe; n = 1; R = Ph) (3.50 g, 7.6 mmol) gave the phosphonium salt (3.22 g, 95%) as a powder, mp 279–281 °C (white prisms from aqueous MeOH) (Found: C, 66.5; H, 4.9; Br, 17.6; P, 6.9. C25H22BrOP requires C, 66.8; H, 4.9; Br, 17.8; P, 6.9%); νmax(Nujol)/cm−1 1602 and 1574 (Ph) and 1460 and 1436 (P–Ph); δH(500 MHz; d6-DMSO) 7.86 (2 H, br t, J 7.6, PPhH-4), 7.70 (4 H, dt, 4JHP 3.3 and J 7.6, PPhH-3), 7.64 (4 H, dd, J 7.6 and 3JHP 12.3, PPhH-2), 7.51 (1 H, dt, 4JHP 4.7 and 7.7, ArH-5), 7.28–7.23 (2 H, m, CH2PhH-4 and ArH-4), 7.19 (2 H, t, J 7.4, CH2PhH-3), 7.07 (1 H, dd, J 7.8 and 3JHP 12.1, ArH-6), 6.98 (2 H, br d, J 7.4, CH2PhH-2), 6.92 (1 H, br d, 3JHP 14.1, ArH-2) and 5.15 (2 H, d, 2JHP 15.65, PCH2); δC(101 MHz; d6-DMSO) 158.4− (d, 3JCP 16.0, ArC-3), 140.3+ (br s, PPhC-4), 139.2+ (d, 2JCP 9.8, PPhC-2), 136.7+ (d, 3JCP 15.0, ArC-5), 136.1+ (d, 4JCP 5.4, CH2PhC-3), 135.3+ (d, 3JCP 12.4, PPhC-3), 133.95+ (d, 3JCP 2.5, CH2PhC-2), 133.5+ (d, 4JCP 3.2, ArC-4), 133.3− (d, 2JCP 8.6, CH2PhC-1), 129.65+ (d, 2JCP 9.5, ArC-6), 127.3+ (br s, CH2PhC-4), 125.4+ (d, 2JCP 10.9, ArC-2), 123.9− (d, 1JCP 84.8, ArC-1), 123.1− (d, 1JCP 85.5, PPhC-1) and 28.3− (d, 1JCP 46.8, PCH2); m/z (+FAB) 369.1404 (100%, M+. M requires 369.1408). M is defined here as the phosphonium cation.Butyl(3-hydroxyphenyl)diphenylphosphonium bromide 3b
In the same way, butyl(3-methoxyphenyl)diphenylphosphonium bromide 9 (3-OMe; n = 1; R = Pr) (2.40 g, 5.6 mmol) gave the phosphonium salt (2.12 g, 91%) as needles, mp 164–167 °C (from EtOH–Et2O); νmax(Nujol)/cm−1 1597 and 1567 (Ph) and 1463 (P–Ph); δH(400 MHz; d6-DMSO) 7.89 (2 H, dt, 5JHP 1.3 and J 6.8, PhH-4), 7.84–7.74 (8 H, m, PhH-2 and PhH-3), 7.58 (1H, dt, 4JHP 4.7 and J 8.2, ArH-5), 7.27 (1 H, dd, 5JHP 1.0 and J 8.2, ArH-4), 7.22 (1 H, dd, J 8.2 and 3JHP 12.4, ArH-6), 7.07 (1 H, d, 3JHP 14.1, ArH-2), 3.60–3.51 (2 H, m, PCH2), 1.52–1.45 (4 H, m, PCH2CH2CH2Me) and 0.88 (3 H, t, J 6.9, Me); δC(101 MHz; d6-DMSO) 158.4− (d, 3JCP 15.7, ArC-3), 134.8+ (PhC-4), 133.5+ (d, 2JCP 10.1, PhC-2), 131.7+ (d, 3JCP 14.9, ArC-5), 130.2+ (d, 3JCP 12.4, PhC-3), 123.9+ (d, 2JCP 9.6, ArC-6), 121.9+ (ArC-4), 119.6+ (d, 2JCP 11.1, ArC-2), 119.3− (d, 1JCP 85.0, ArC-1), 118.6− (d, 1JCP 85.5, PhC-1), 23.9− (d, 2JCP 4.2, PCH2CH2), 23.1− (d, 3JCP 17.1, CH2Me), 20.1− (d, 1JCP 49.9, PCH2) and 13.3+ (Me); m/z (+FAB) 335.1540 (100%, M+. M requires 335.1565). M is defined here as the phosphonium cation.Benzyl[bis(4-hydroxyphenyl)]phenylphosphonium bromide 4
In the same way, benzyl[bis(4-methoxyphenyl)]phenylphosphonium bromide 9 (4-OH; n = 2; R = Ph) (2.16 g, 4.4 mmol) gave the phosphonium salt (1.94 g, 95%) as a powder, mp 336–339 °C (white needles from aqueous MeOH) (Found: C, 64.0; H, 4.75; P, 6.9. C25H22BrO2P requires C, 64.5; H, 4.8; P, 6.7%); νmax(Nujol)/cm−1 1598 and 1575 (Ph) and 1462 (P–Ph); δH(500 MHz; d6-DMSO) 10.9 (2 H, s, OH), 7.8 (1 H, t, J 7.5, PPhH-4), 7.7 (2 H, dt, 4JHP 3.2 and J 7.5, PPhH-3), 7.6 (2 H, dd, J 7.5 and 3JHP 12.4, PPhH-2), 7.4 (4 H, dd, J 8.7 and 3JHP 11.9, ArH-2), 7.3 (1 H, br t, J 7.2, CH2PhH-4), 7.2 (2 H, t, J 7.2, CH2PhH-3), 7.05 (4 H, dd, 4JHP 2.6 and J 8.7, ArH-3), 6.95 (2 H, d, J 7.2, CH2PhH-2) and 4.9 (2 H, d, 2JHP 15.5, PCH2); δC(101 MHz; d6-DMSO) 163.3− (d, 4JCP 2.5, ArC-4), 136.2+ (d, 2JCP 11.4, ArC-2), 134.7+ (s, PPhC-4), 133.8+ (d, 2JCP 9.8, PPhC-2), 130.9+ (d, 4JCP 5.2, CH2PhC-3), 130.1+ (d, 3JCP 12.2, PPhC-3), 128.7+ (s, CH2PhC-4), 128.7− (s, CH2PhC-1), 128.2+ (s, CH2PhC-2), 119.9− (d, 1JCP 86.3, PPhC-1), 117.2+ (d, 3JCP 13.7, ArC-3), 105.9− (d, 1JCP 94.5, ArC-1) and 29.2− (d, 1JCP 49.2, PCH2); m/z (+FAB) 464.0577 (3%, M+. M requires 464.0541), 385.4 (100%, M − Br).Benzyl[bis(3-hydroxyphenyl)]phenylphosphonium bromide 5
In the same way, benzyl[bis(3-methoxyphenyl)]phenylphosphonium bromide 9 (3-OMe; n = 2; R = Ph) (22.3 g, 45.2 mmol) gave the phosphonium salt (8.41 g, 40%) as a powder, mp 268–272 °C (precipitated from water); νmax(Nujol)/cm−1 1596 and 1574 (Ph) and 1458 and 1436 (P–Ph); δH(400 MHz; d6-DMSO) 10.46 (2 H, br s, OH), 7.89 (1 H, br t, J 7.6, PPhH-4), 7.73 (2 H, dt, 4JHP 3.6 and J 7.6, PPhH-3), 7.66 (2 H, dd, J 7.6 and 3JHP 12.3, PPhH-2), 7.54 (2 H, dt, 4JHP 4.6 and 8.0, ArH-5), 7.31–7.25 (2 H, m, CH2PhH-4 and ArH-4), 7.23 (2 H, t, J 7.3, CH2PhH-3), 7.10 (2 H, dd, J 8.0 and 3JHP 12.2, ArH-6), 7.01 (2 H, br d, J 7.3, CH2PhH-2), 6.94 (2 H, br d, 3JHP 14.1, ArH-2) and 5.10 (2 H, d, 2JHP 15.7, PCH2); δC(101 MHz; d6-DMSO) 158.3− (d, 3JCP 15.9, ArC-3), 135.1+ (br s, PPhC-4), 134.0+ (d, 2JCP 9.7, PPhC-2), 131.5+ (d, 3JCP 14.9, ArC-5), 130.9+ (d, 4JCP 5.2, CH2PhC-3), 130.1+ (d, 3JCP 12.3, PPhC-3), 128.8+ (s, CH2PhC-2), 128.4+ (s, ArC-4), 128.1− (d, 2JCP 8.5, CH2PhC-1), 124.4+ (d, 2JCP 9.4, ArC-6), 122.1+ (s, CH2PhC-4), 120.2+ (d, 2JCP 10.8, ArC-2), 118.8− (d, 1JCP 84.8, ArC-1), 118.1− (d, 1JCP 85.4, PPhC-1) and 28.3− (d, 1JCP 46.8, PCH2); m/z (+FAB) 385.1345 (100%, M+. M requires 385.1357). M is defined here as the phosphonium cation.Typical Wittig reaction in water
Benzyl(3-hydroxyphenyl)diphenylphosphonium bromide 3a (0.203 g, 0.45 mmol) and 4-nitrobenzaldehyde (83.1 mg, 0.55 mmol) were suspended in 1 cm3 water. NaOH (1 cm3 of a 10% solution in water) was added dropwise and the phosphonium salt dissolved. The solution was left to stir overnight at room temperature. It was then filtered and washed with base to give the stilbene 14 (100 mg, 98%) as a mixture of stereoisomers (E∶Z, 60∶40). The aqueous solution was acidified by 3 M HCl to pH 1 and then filtered to give 3-hydroxyphenyldiphenylphosphine oxide 18 (117 mg, 87%).All alkenes synthesised are known compounds and the E∶Z ratios were all determined by integration of discrete peaks in the 1H NMR spectra of the crude isolated alkene. The alkenes include 1,2-diphenylethene 11,24 1-(4-methoxyphenyl)-2-phenylethene 12,25 1-(4-chlorophenyl)-2-phenylethylene 13,25 1-(4-nitrophenyl)-2-phenylethylene 14,25 2-(2-phenylethen-1-yl)furan26 and 2-(2-phenylethen-1-yl)pyridine.27
Attempted Wittig reaction between 4-(butyldiphenylphosphonio)benzoic acid bromide 1b and benzaldehyde
4-(Butyldiphenylphosphonio)benzoic acid bromide 1b (0.208 g, 0.47 mmol) was suspended in water (1 cm3). Benzaldehyde (50 μm3, 0.47 mmol) was added and the suspension stirred. NaOH (1 cm3 of a 10% solution in water) was added dropwise to the mixture. The phosphonium salt dissolved upon addition of the aqueous base. The solution was stirred overnight at room temperature and extracted with diethyl ether (3 × 2 cm3). The combined organic extracts were dried (MgSO4) and evaporated under reduced pressure to give the phosphine oxide2817 (37 mg, 31%) as yellow crystals. The aqueous fraction was acidified to pH 1 with 3 M HCl and extracted with dichloromethane (3 × 5 cm3). The combined organic extracts were dried (MgSO4) and evaporated under reduced pressure to give the phosphine oxide18 (96 mg, 67%) as a white foam; δH(400 MHz; d6-DMSO) 7.98–7.72 (9 H, m) 2.45–2.40 (2 H, m, PCH2), 1.39–1.34 (4 H, m, PCH2CH2CH2Me) and 0.8 (3 H, t, J 6.9, Me).Attempted Wittig reaction between butyl(3-hydroxyphenyl)diphenylphosphonium bromide 3b and benzaldehyde
In the same way, butyl(3-hydroxyphenyl)diphenylphosphonium bromide 3b (0.174 g, 0.42 mmol) and benzaldehyde (50 μm3, 0.47 mmol) in water (1 cm3) were reacted, except that NaOH (1 cm3 of a 30% solution in water) was used and the mixture was stirred overnight at reflux. The dichloromethane extract was dried (MgSO4) and evaporated under reduced pressure to give butyl(3-hydroxyphenyl)phenylphosphine oxide20 (66 mg, 58%); δH(400 MHz; d6-DMSO) 9.9 (1 H, br s, OH), 7.8–7.7 (2 H, m, PhH-3), 7.53–7.47 (3 H, m, PhH-2 and PhH-4), 7.3 (1 H, dt, 4JHP 3.8 and J 7.8, ArH-5), 7.2 (1 H, d, J 7.8, ArH-4), 7.1 (1 H, d, 3JHP 2.3, ArH-2), 6.9 (1 H, dd, 3JHP 2.3 and J 7.8, ArH-6), 2.33–2.26 (2 H, m, PCH2), 1.43–1.29 (4 H, m, PCH2CH2CH2Me) and 0.8 (3 H, t, J 7.0, Me); δC(101 MHz; d6-DMSO) 157.5− (d, 3JCP 14.1, ArC-3), 135.0− (d, 1JCP 95.5, ArC-1 or PhC-1), 133.8− (d, 1JCP 96.0, ArC-1 or PhC-1), 131.5+ (PhC-4), 130.4+ (d, 2JCP 9.3, PhC-2), 130.0+ (d, 3JCP 13.5, ArC-5), 128.7+ (d, 3JCP 11.1, PhC-3), 120.9+ (d, 2JCP 9.2, ArC-2 or ArC-6), 118.6+ (ArC-4), 117.0+ (d, 2JCP 9.7, ArC-2 or ArC-6), 28.1− (d, 1JCP 71.8, PCH2), (23.25–22.38)− (m, PCH2CH2CH2Me) and 13.6+ (Me).Acknowledgements
We thank the EPSRC Clean Technology Unit and Zeneca Specialties for grants (to M. G. R.). We also wish to thank Dr A. Collins of Zeneca Specialties for his support throughout the project.References
- M. G. Russell and S. Warren, Tetrahedron Lett., 1998, 39, 7995 CrossRef CAS
.
- G. Märkl and A. Merz, Synthesis, 1973, 295 CrossRef
- W. Tagaki, I. Inoue, Y. Yano and T. Okonogi, Tetrahedron Lett., 1974, 2587 CrossRef CAS
- C. Piechucki, Synthesis, 1976, 187 CrossRef CAS
- E. D’Incan and J. Seyden-Penne, Synthesis, 1975, 516 CrossRef CAS
- M. Mikolajczyk, S. Grzejszczak, W. Midura and A. Zatorski, Synthesis, 1975, 278 CrossRef CAS
- M. Delmas, Y. Le Bigot and A. Gaset, Tetrahedron Lett., 1980, 4831 CrossRef CAS
- M. Delmas, Y. Le Bigot, A. Gaset and J. P. Garrichon, Synth. Commun., 1981, 125 CAS
- M. Delmas, Y. Le Bigot, A. Gaset and J. P. Garrichon, Synth. Commun., 1982, 107
- M. Butcher, R. J. Mathews and S. Middleton, Aust. J. Chem., 1973, 26, 2067 CAS
- R. Broos, D. Tavernier and M. Anteunis, J. Chem. Educ., 1978, 55, 813 Search PubMed
- K. Schleich
, C. H. Zollikerberg
, H. Stoller
and C. H. Reinach
, 1977, GP 2733231 C2
(Chem. Abstr., 1977, 88, P 191170e).
Search PubMed
- X. M. Zhang and F. G. Bordwell, J. Am. Chem. Soc., 1994, 116, 968 CrossRef CAS
- M. Grayson and P. T. Keough, J. Am. Chem. Soc., 1960, 82, 3919 CrossRef CAS
- V. Ravindar, H. Hemling, H. Schumann and J. Blum, Synth. Commun., 1992, 22, 841 CAS
- W. E. McEwen, W.-I. Shiau, Y.-I. Yeh, D. N. Schulz, R. U. Pagilagan, J. B. Levy, C. Symmes Jr., G. O. Nelson and I. Granoth, J. Am. Chem. Soc., 1975, 97, 1787 CrossRef CAS
- T. Coumbe, N. J. Lawrence and F. Muhammad, Tetrahedron Lett., 1994, 35, 625 CrossRef CAS
- C. Larpent and H. Patin, Tetrahedron, 1988, 44, 6107 CrossRef CAS
- R. A. Y. Jones
, Physical and Mechanistic Organic Chemistry, Cambridge University Press, Cambridge, 1984.
Search PubMed
- S. E. Tunney and J. K. Stille, J. Org. Chem., 1987, 52, 748 CrossRef CAS
- A. E. Senear, W. Valient and J. Wirth, J. Org. Chem., 1960, 25, 2001 CrossRef CAS
- C. M. Whitaker, K. L. Kott and R. J. McMahon, J. Org. Chem., 1995, 60, 3499 CrossRef CAS
- G. L. Kelden and W. E. McEwen, J. Am. Chem. Soc., 1978, 100, 7312 CrossRef
- Comparison with authentic samples obtained from Aldrich Chemical Co..
- W. J. Ward Jr. and W. E. McEwen, J. Org. Chem., 1990, 55, 493 CrossRef
- G. Karminski-Zamola and K. Jakopcic, J. Heterocycl. Chem., 1981, 18, 193 Search PubMed
- T. Sakamoto, H. Arakida, K. Edo and H. Yamanaka, Chem. Pharm. Bull., 1982, 30, 3647 Search PubMed
- A. D. Buss and S. Warren, J. Chem. Soc., Perkin Trans. 1, 1985, 2307 RSC
| This journal is © The Royal Society of Chemistry 2000 |