A persistent C–H⋯C(π) T-stacked cation†
Colin D. Abernethya, Charles L. B. Macdonalda, Jason A. C. Clyburneb and Alan H. Cowley*a
aDepartment of Chemistry and Biochemistry, The University of Texas at Austin, Austin, Texas 78712, USA.. E-mail: cowley@mail.utexas.edu
bDepartment of Chemistry, Acadia University, Wolfville, Nova Scotia B0P 1X0, Canada
First published on 15th December 2000
Abstract
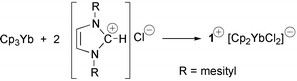
The reaction of 2 equivalents of 1,3-dimesitylimidazolium chloride with Cp3Yb affords the salt [bis(1,3-dimesitylimidazolium)cyclopentadienide] [bis(cyclopentadienyl)dichloroytterbate]; the T-stacked cation is composed of two imidazolium fragments that are C–H⋯C(π) hydrogen bonded to the bridging cyclopentadienide anion.
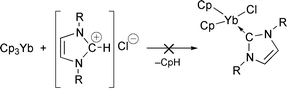
N-Heterocylic carbene transition-metal complexes have been the subject of intense study in recent years.1 Öfele,2 and more recently Tilset and coworkers,3 and our group4 have demonstrated the utility of imidazolium salts for the preparation of transition-metal (TM) carbene complexes via protonolysis of Cp–TM and other suitable ligand-TM bonds. Encouraged by the fact that N-heterocyclic carbene complexes of the type Cp′2M(carbene) (M = Sm, Yb; Cp′ = EtMe4C5) have been prepared by traditional methods,5 we were prompted to seek a new route to such compounds by treatment of lanthanocenes with imidazolium salts. However, the outcome of one of these experiments was unexpected and resulted in the formation of a remarkably persistent T-stacked cation. There is considerable current interest in such X–H⋯π systems, not only from the structural and theoretical standpoints, but also because of the recognition of this type of interaction in peptide and protein structures6 and supramolecular architectures.7 Furthermore, the reactivity patterns of imidazolium salts are of interest because of their use as ionic liquids.8
The reaction of 2 equivalents of 1,3-dimesitylimidazolium chloride (ImidCl) with Cp3Yb in refluxing tetrahydrofuran afforded the golden-orange salt [bis(1,3-dimesitylimidazolium)(μ-cyclopentadienide)][bis(cyclopentadien yl)dichloroytterbate], [Imid2Cp][Cp2YbCl2], upon recrystallisation from toluene.9 Although the spectroscopic data were consistent with the proposed formulation, unambiguous identification of the structure of the salt required an X-ray crystallographic experiment.‡ The most remarkable structural feature is the unprecedented geometry of the complex cation, [Imid2Cp]+ (1+) which can be regarded as two “interlocked” cationic 1,3-dimesitylimidazolium fragments encapsulating a cyclopentadienide anion (Fig. 1). Interestingly, the “glue” that binds this cation together appears to be two C–H⋯C(π Cp) hydrogen bonds complemented by interionic attractions. The confined cyclopentadienide anion, which is slightly disordered, has an average C–C bond distance of 1.341(10) Å and an average C–C–C bond angle of 108.0(6)°. Both values are close to those reported for a typical “free” [C5H5]− anion.10 Likewise, the metrical parameters for the 1,3-dimesitylimidazolium moieties are virtually identical to those reported for 1,3-dimesitylimidazolium chloride.11 The only significant difference is the slight shortening of the C(1)–H bond in 1+ [0.819(9) vs. 0.944(9) Å for ImidCl] which is an expected consequence of the weaker C–H⋯Cp hydrogen bond (vs. C–H⋯Cl). The C–X (X is the Cp ring centroid) distance is 3.086(8) Å which is significantly shorter than the 3.85 Å distance in e.g. the neutral T-stacked 4-methylpyridine hexamer.7 Likewise, the H⋯X distance is 2.295(9) Å and lies at the short end of the range of C–H⋯Cp contacts reported recently by Harder for ammonium and phosphonium cyclopentadienide salts (2.30–2.63 Å).12 The C–H–X and H–X–H angles are 162.4(5) and 167.9(5)°, respectively. The average H–C(Cp) distance (<d> = 2.561 Å) and the average C(1)–H–C(Cp) angle (<ψ> = 150.9°) yield a relatively high value of 341 for each of the hydrogen bonds using Harder’s C–H⋯Cp hydrogen bonding scale.13 Somewhat surprisingly, the two 1,3-dimesitylimidazolium moieties are not mutually orthogonal but are arranged at an angle of 70° with respect to each other. This structural feature is most likely a consequence of crystal packing effects. For example, there is a somewhat close interaction of H(3) with Cl(1) on an adjacent [Cp2YbCl2]− anion (2.662 Å) as shown in Fig. 2. Although the structure of [Cp2YbCl2]− has not been reported previously, the metrical parameters are unexceptional and are not commented on here.
![Thermal ellipsoid plot (30% probability surface) of
1[Cp2YbCl2] (most hydrogen atoms omitted for
clarity). Selected metrical parameters including bond lengths (Å) and
angles (°); note that X is the Cp ring centroid: C(1)–H(1)
0.819(9), C(1)–N(1) 1.330(4), C(1)–N(2) 1.334(4),
N(1)–C(3) 1.381(4), N(2)–C(2) 1.378(4), C(2)–C(3)
1.335(5), N(1)–C(21) 1.451(4), N(2)–C(31) 1.457(4),
C(11)–C(12) 1.311(6), C(12)–C(13) 1.352(7), C(13)–C(13A)
1.378(10), H(1)–X(1A) 2.295(9), av H(1)–C(Cp) 2.561(9),
Yb(1)–Cl(1) 2.5530(9), Yb(1)–X(1B) 2.329(5), av
Yb(1)–C(Cp) 2.614(5); H(1)–X(1A)–H(1A) 167.9(5),
X(1A)–H(1)–C(1) 162.4(5), H(1)–C(1)–N(1) 124.1(1),
H(1)–C(1)–N(2) 127.9(1), N(1)–C(1)–N(2) 108.0(3),
C(1)–N(1)–C(3) 108.8(3), C(1)–N(2)–C(2) 108.7(3),
N(1)–C(3)–C(2) 107.2(3), N(2)–C(2)–C(3) 107.4(3),
(X1B)–Yb(1)–Cl(1) 107.8(1), (X1B)–Yb(1)–X(1C)
127.3(1), Cl(1)–Yb(1)–Cl(1A) 95.03(4).](/image/article/2001/CC/b005450j/b005450j-f1.gif) | ||
| Fig. 1 Thermal ellipsoid plot (30% probability surface) of 1[Cp2YbCl2] (most hydrogen atoms omitted for clarity). Selected metrical parameters including bond lengths (Å) and angles (°); note that X is the Cp ring centroid: C(1)–H(1) 0.819(9), C(1)–N(1) 1.330(4), C(1)–N(2) 1.334(4), N(1)–C(3) 1.381(4), N(2)–C(2) 1.378(4), C(2)–C(3) 1.335(5), N(1)–C(21) 1.451(4), N(2)–C(31) 1.457(4), C(11)–C(12) 1.311(6), C(12)–C(13) 1.352(7), C(13)–C(13A) 1.378(10), H(1)–X(1A) 2.295(9), av H(1)–C(Cp) 2.561(9), Yb(1)–Cl(1) 2.5530(9), Yb(1)–X(1B) 2.329(5), av Yb(1)–C(Cp) 2.614(5); H(1)–X(1A)–H(1A) 167.9(5), X(1A)–H(1)–C(1) 162.4(5), H(1)–C(1)–N(1) 124.1(1), H(1)–C(1)–N(2) 127.9(1), N(1)–C(1)–N(2) 108.0(3), C(1)–N(1)–C(3) 108.8(3), C(1)–N(2)–C(2) 108.7(3), N(1)–C(3)–C(2) 107.2(3), N(2)–C(2)–C(3) 107.4(3), (X1B)–Yb(1)–Cl(1) 107.8(1), (X1B)–Yb(1)–X(1C) 127.3(1), Cl(1)–Yb(1)–Cl(1A) 95.03(4). | ||
![Partial crystal packing diagram for
1[Cp2YbCl2] showing the cation–anion
C–H⋯Cl interaction (most hydrogen atoms omitted for
clarity).](/image/article/2001/CC/b005450j/b005450j-f2.gif) | ||
| Fig. 2 Partial crystal packing diagram for 1[Cp2YbCl2] showing the cation–anion C–H⋯Cl interaction (most hydrogen atoms omitted for clarity). | ||
Interestingly, the complex cation persists in toluene solution. Unfortunately, 1[Cp2YbCl2] is virtually insoluble in aromatic solvents after initial crystallisation and decomposes rapidly in coordinating solvents hence 13C NMR experiments were not informative. However, it was possible to demonstrate the persistence of 1+ by means of an electrospray ionization (ESI) mass spectrum of a saturated toluene solution of 1[Cp2YbCl2] which exhibited an envelope with the anticipated isotopic intensity distribution in the region of m/z 674–678. This experiment confirmed the presence of 1+ in solution and revealed that the only other major peak in the positive mode corresponds to the 1,3-dimesitylimidazolium cation. As expected, electron impact (EI) and chemical ionization (CI) mass spectra of solid samples of 1[Cp2YbCl2] did not evidence significant peaks attributable to 1+, presumably because the high energy required to volatilize the solid exceeds the weak binding interactions of the complex cation.
The reaction clearly proceeds differently than had been expected and, although the mechanism remains unclear, some insight into the process can be gleaned from the nature of the products. Instead of protolytic cleavage of a Cp ligand and formation of CpH [eqn. (1)], Cp3Yb can be regarded as
 | (1) |
 | (2) |
Interestingly, density functional theory (DFT) calculations (see ESI for details†) indicate that the reaction of Cp− with a model imidazolium cation (R = Me) is highly exothermic (91.4 kcal mol−1). However, we have not yet calculated the activation energy for such a process; moreover, no account has been taken of solvent effects. Single-point DFT calculations on a model system (R = Ph) predict the bond energy of the imidazolium–Cp interaction to be 31.4 kcal mol−1. As such, this interaction is of a similar magnitude to H⋯O–R hydrogen bonding and represents the strongest C–H⋯C(π) bond yet reported.
In conclusion, the formation and persistence of the T-stacked cation 1+ stems both from electrostatic attractions and also from the appreciable acidity of the imidazolium protons which facilitates rather strong C–H⋯Cp (π) hydrogen bonding. In a sense, 1+ can be regarded as an organic analogue of an “inverse sandwich” complex.
Acknowledgements
We are grateful to the National Science Foundation, the Robert A. Welch Foundation, the Natural Sciences and Engineering Research Council (Canada) and Acadia University for financial support. We would also like to thank the Atlantic Region Magnetic Resonance Center (Dalhousie University) for the acquisition of some NMR spectra.Notes and references
- See, for example: D. Bourissou, O. Guerret, F. Gabbaï and G. Bertrand, Chem. Rev., 2000, 100, 39 CrossRef; W. A. Herrmann and C. Köcher, Angew. Chem., Int. Ed. Engl., 1997, 36, 2162 CrossRef CAS.
- K. Öfele, J. Organomet. Chem., 1968, 12, P42 CrossRef.
- M. H. Voges, C. Rømming and M. Tilset, Organometallics, 1999, 18, 529 CrossRef CAS.
- C. D. Abernethy, J. A. C. Clyburne, A. H. Cowley and R. A. Jones, J. Organomet. Chem., 2000, 596, 3 CrossRef CAS.
- H. Schumann, M. Glanz, J. Winterfeld, H. Hemling, N. Kuhn and T. Kratz, Chem. Ber., 1994, 127, 2369 CrossRef CAS; H. Schumann, M. Glanz, J. Winterfeld, H. Hemling, N. Kuhn and T. Kratz, Angew. Chem., Int. Ed. Engl., 1994, 33, 1733 CrossRef.
- G. R. Desiraju and T. Steiner, The Weak Hydrogen Bond in Structural Chemistry and Biology, Oxford University Press, Oxford, 1999; Search PubMed; N. Motohiro, M. Hirota and Y. Umezawa, The CH/π Interaction. Evidence, Nature, and Consequences, Wiley-VCH, Inc., New York, 1998. Search PubMed.
- See, for example: K. Biradha and M. J. Zaworotko, J. Am. Chem. Soc., 1998, 120, 6431 CrossRef CAS.
- T. Welton, Chem. Rev., 1999, 99, 1071 CrossRef.
- A mixture of Cp3Yb (1.0 g, 2.7 mmol) and
dimesitylimidazolium chloride11 (1.8 g, 5.4
mmol) was suspended in 150 mL of tetrahydrofuran then refluxed for 3 h. The
volatiles were removed in vacuo and the residue was extracted with
150 mL of hot toluene. The resulting orange solution was filtered while hot
and the volume was reduced to 100 mL. A crop of golden-orange crystals of
1[Cp2YbCl2] (0.85 g, 30% yield)formed upon storage of the filtrate at 25 °C for 5 days. Mp
158–160 °C (decomp.). Anal. Calc. for
C57H65Cl2N4Yb: C, 65.20; H,
6.24; N, 5.34; Cl, 6.75. Found: C, 64.34; H, 6.32; N, 5.22; Cl, 6.65. Mass
spectra: EI, m/z 305 (imid+, 100%); CI,
m/z 305 (imid+, 100%), 675
(1+, <1%); ESI, av. m/z
305 (imid+, 100%), 675 (1+, 15%).
NMR (note that the salt is virtually insoluble in benzene and toluene after
initial precipitation and decomposes rapidly in THF and dichloromethane,
thus only the 1H spectra in the latter solvents, acquired
immediately after dissolution, are reported):
δH(THF-d8): 1.51 (s, 12 H,
p-Me), 2.34 (s, 24 H, o-Me), 6.35 (s, 5 H, Cp-H), 6.41
(br s, 8H, Mes-H), 6.52 (br s, 4 H, imid C
![[double bond, length half m-dash]](https://www.rsc.org/images/entities/char_e006.gif) CH), 8.51 (broad s, 2 H,
NCHN). δH(CD2Cl2): 1.67 (s,
24 H, o-Me), 1.97 (s, 12 H, p-Me), 6.41 (br s, 8H,
Mes-H), 6.49 (br s, 4 H, imid CCH), 6.55 (s, 5 H, Cp-H), 7.03
(sharp s, 2 H, NCHN). δH(toluene-d8):
2.09 (s, 24 H, o-Me), 2.12 (s, 12 H, p-Me), 2.68 (br s, 5
H, Cp-H), 6.28 (br s, 4 H, imid CCH), 6.47 (br s, 8H, Mes-H), 7.08
(sharp s, 2 H, NCHN)..
CH), 8.51 (broad s, 2 H,
NCHN). δH(CD2Cl2): 1.67 (s,
24 H, o-Me), 1.97 (s, 12 H, p-Me), 6.41 (br s, 8H,
Mes-H), 6.49 (br s, 4 H, imid CCH), 6.55 (s, 5 H, Cp-H), 7.03
(sharp s, 2 H, NCHN). δH(toluene-d8):
2.09 (s, 24 H, o-Me), 2.12 (s, 12 H, p-Me), 2.68 (br s, 5
H, Cp-H), 6.28 (br s, 4 H, imid CCH), 6.47 (br s, 8H, Mes-H), 7.08
(sharp s, 2 H, NCHN).. - See, for example: C. D. Abernethy, J. A. C. Clyburne, A. H. Cowley and R. A. Jones, J. Am. Chem. Soc., 1999, 121, 2329 CrossRef CAS.
- A. J. Arduengo, III, S. F. Gamper, M. Tamm, J. C. Calabrese, F. Davidson and H. A. Craig, J. Am. Chem. Soc., 1995, 117, 572 CrossRef Refer to CSD #YOFKOT for metrical parameters..
- S. Harder, Chem. Eur. J., 1999, 5, 1852 CrossRef CAS.
- The value is defined as 1000(−cos<ψ>)/<d>. See ref. 12 for details..
Footnotes |
| † Electronic supplementary information (ESI) available: DFT calculations. See http://www.rsc.org/suppdata/cc/b0/b005450j/ |
| ‡ Crystal data for 1[Cp2YbCl2]. Formula (half of each cation and anion in the asymmetric unit): C28.5H32.5Cl1N2Yb0.5 , Mw = 525.04, orange blocks, orthorhombic, space group Pnma, a = 16.785(4), b = 19.727(6), c = 15.847(3) Å, V = 5247(2) Å3, Z = 8, Dc = 1.329 g cm−3, μ(Mo-Kα) = 1.923 mm−1 . A single crystal of 1[Cp2YbCl2]was covered with perfluoro(poly)ether and mounted on a Nonius KAPPA CCD diffractometer at 153(2) K. A total of 29722 reflections were collected in the range of 2.39 < θ < 29.12° using Mo-Kα radiation (λ = 0.71073 Å). Of these, 7037 independent reflections (Rint = 0.0859) were used to solve (direct methods, SHELXS) and refine (full matrix, least squares on F2, SHELXL) the structure of 1[Cp2YbCl2]; wR2 = 0.0664, R = 0.0403. Note: all hydrogen atoms except those on the encapsulated Cp ring were found in the Fourier difference maps; those on the said Cp ring were placed in calculated positions. CCDC 182/1848. See http://www.rsc.org/suppdata/cc/b0/b005450j/ for crystallographic data in .cif format. |
| This journal is © The Royal Society of Chemistry 2001 |