Variable temperature electrochemistry as a powerful method for conformational investigations on the fluxional organometallic complex Mo(His-Nε-C2H4CO2 Me)(η-allyl)(CO)2 (His = Nδ, N,O-L-histidinate)†
Dave R. van Staveren, Eberhard Bothe, Thomas Weyhermüller and Nils Metzler-Nolte‡*
Max-Planck-Institut für Strahlenchemie, Stiftstraße 34-36, D-45470, Mülheim/Ruhr, Germany
First published on 19th December 2000
Abstract
Square-wave voltammograms of the complex Mo(His-Nε-C2H4CO 2Me)(η-allyl)(CO)2 (His = Nδ,N,O-L-histidinate) 2 recorded at variable temperatures establish the existence of two isomers in solution in both the reduced and the oxidized paramagnetic form of 2; a complete analysis yields thermodynamic parameters such as equilibrium constants, ΔG and ΔH for all species.
Fluxionality is frequently observed in organometallic complexes in solution.1,2 In the course of our studies on the labelling of biomolecules with organometallic complexes we have reinvestigated the compound Mo(His)(η-allyl)(CO)21 (His = Nδ,N,O-L-h istidinate). The synthesis of this complex was first reported by Beck and coworkers twenty years ago.3,4 Since that time, fluxionality in diamagnetic complexes of the general formula M(η-allyl)(CO)2L2X (M = Cr, Mo, W) has been studied in some detail by NMR spectroscopy in solution.5–7 For some derivatives, a good stability of the one-electron oxidized species was demonstrated,8,9 but related fluxionality was never mentioned. There seems to be no obvious method for investigations of this kind on the paramagnetic species [M(η-allyl)(CO)2L2X]+.
We were intrigued by the excellent stability of complex 1 and its potential as a covalent marker for biomolecules, in particular by IR or electrochemical detection. In complex 1, three different orientations of the His ligand with respect to the (allyl)(CO)2 plane are possible. The stereochemical implications of fluxional processes in M(η-allyl)(CO)2L2X have been discussed, but all complexes investigated so far were either achiral or racemic mixtures.7 Here, we report the synthesis of the enantiomerically pure methyl propionate derivative Mo(His-Nε-C2H4 CO2Me)(η-allyl)(CO)22 and its complete characterization. The presence of two isomers of 2 is shown by NMR and electrochemical investigations. An unusual case of coalescence in temperature-dependent square-wave voltammograms is reported and we demonstrate how a complete analysis of the electrochemical data gives detailed quantitative information on both isomers of the paramagnetic, one-electron oxidized 2+. This information is difficult to obtain otherwise with such accuracy.
Compound 2 was synthesised in 65% yield by reacting Mo(His)(allyl)(CO)23 with 3-bromomethylpropionate in the presence of equimolar amounts of Cs2CO3 in DMF, under an atmosphere of Ar. Compound 2 is completely stable under Ar and can be stored in air on the benchtop for several days without decomposition.
X-Ray quality single crystals of 2·2MeOH were grown by slow pentane diffusion into MeOH at +4 °C. An ORTEP representation of 2·2MeOH§ is depicted in Fig. 1. The chirality around the metal atom is clearly determined by the known stereochemistry of the amino acid L-His. The Mo(allyl)(CO)2 moiety is in a facial arrangement, with the terminal carbon atoms of the allyl ligand oriented towards the carbonyl ligands. Such a conformation has been shown to be energetically favourable10 and is observed in all solid-state structures of this type reported thus far.6,7,11–13 The allyl ligand is located trans relative to the histidine-Nδ atom [N(8)] and bound symmetrically to the Mo atom, with Mo–C distances of 2.328(3) Å [Mo(1)–C(17) and Mo(1)–C(19)] and 2.209(2) Å [Mo(1)-C(18)]. The two MeOH molecules form hydrogen bonds with the carboxylato group, with O⋯O contacts of 2.750(10) Å [O(14)⋯O(20)] and 2.800(10) Å [O(13)⋯O(30)].
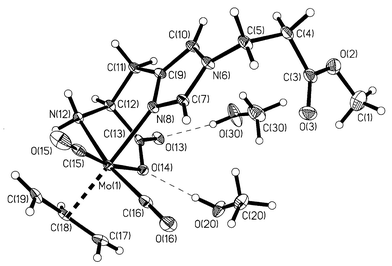 | ||
Fig. 1 ORTEP plot for 2·2MeOH, with thermal ellipsoids at 50%
probability level. Selected bond distances (Å): Mo(1)–N(8)
2.216(2), Mo(1)–N(12) 2.256(2), Mo(1)–O(14) 2.214(2),
Mo(1)–C(15) 1.945(3), Mo(1)–C(16) 1.944(3),
Mo(1)–C(17)![[double bond, length half m-dash]](https://www.rsc.org/images/entities/char_e006.gif) Mo(1)–C(19) 2.328(3), Mo(1)–C(18)
2.209(2). Mo(1)–C(19) 2.328(3), Mo(1)–C(18)
2.209(2). | ||
In solution, the situation is more complex. Two sets of signals are observed in the 1H NMR as well as in the 13C NMR spectrum of 2, indicating the presence of two isomers (2a and 2b, see Scheme 1) in solution. The ratio between both isomers is solvent dependent [DMSO 53∶47, MeCN (293 K) 57∶43, MeOH 77∶23]. The presence of two isomers in the special case of Mo(allyl)(CO)2L2X with L2X = cyclopentadienyl (Cp) has been explained by rotation of the allyl group. In the more general case where L2X comprise heterodonor atoms (N, O, P etc.) fluxionality is brought about by a trigonal twist of the L2X plane with respect to the Mo(allyl)(CO)2 core.12 Recently, we reported the presence of two different allyl orientations in the same crystal structure of a CpMo(allyl)(CO)2 derivative.14 For 2, if one of the isomers has the conformation observed in the solid-state structure, the other detectable isomer could either have the NH2 or the carboxylato group trans to the allyl ligand. In accordance with NMR data (vide infra), preliminary results from DFT calculations show the latter is the energetically favoured conformer of the two,15 with the third isomer being too high in energy to be observed by NMR spectroscopy (<0.1% at 500 MHz 1H NMR). The chemical shift of the imidazole proton signals are at δ 8.62 and 7.12 in isomer 2a and δ 8.02 and 6.92 in isomer 2b. We assume that the imidazole nitrogen atom is trans to the allyl ligand in isomer 2a and trans to a CO in 2b. The chemical shift difference may then be rationalized by the different trans influences of the allyl and CO ligands. Variable temperature measurements in MeCN yielded ΔG‡ = 66.7 ± 0.5 kJ mol−1, consistent with a trigonal twist mechanism of interconversion.11,12
 | ||
| Scheme 1 Molecular structure of the two isomers of 2. | ||
The cyclic voltammogram (CV) of 2 in MeCN (scan rate of 0.1 V s−1, 25 °C) shows a wave with reversible appearance at E1/2 = +86 mV vs. Fc/Fc+. This value is in the range reported for several other analogous pseudo-octahedral Mo complexes.8,9 Controlled potential coulometry (at +0.5 V vs. Fc/Fc+) reveals that the wave arises from a one-electron oxidation of 2. However, the oxidation of 2 does not proceed via an uncomplicated reversible single electron process. At slow scan rates, the peak separation increases with increasing scan rates from 85 mV at 0.025 V s−1 to 140 mV at 0.8 V s−1, but at even faster scan rates (up to the highest value measured of 25 V s−1) it remains almost constant (±10 mV). This behaviour can be accommodated in a ‘square scheme’ shown in Scheme 2, assuming the presence of two isomers in both the oxidized and reduced form of 2.
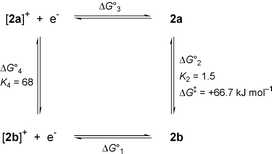 | ||
| Scheme 2 Square scheme showing the redox reactions and rerrangements of 2. At −60 °C: ΔG01 = −40.1 kJ mol−1, ΔG02 = −0.72 kJ mol−1, ΔG03 = +48.3 kJ mol−1, ΔG04 = −7.5 kJ mol−1. From electrochemical experiments at variable temperatures: ΔH02 = −2.5 kJ mol−1, ΔH04 = −8.3 kJ mol−1. | ||
The presence of two species was readily detected by square-wave voltammetry (SWV). Square-wave voltammograms at four different temperatures are shown in Fig. 2. At −60 °C, the anodic and cathodic scans are fully symmetric [curve (d)]. This shows that the equilibrium composition of the reduced form (the starting material 2) is maintained after the oxidation for the time period of equilibration at +0.6 V and during the time of the scan, i.e. for at least a few seconds. Because the equilibrium constants for the oxidized and reduced forms are quite different (see below), the isomerisation reaction must be correspondingly slow at −60 °C. At −20 °C anodic and cathodic scans are different [curve (b)] and it is observed that oxidized 2+ exhibits a preference for the state with the lower redox potential. At even higher temperatures two components are no longer discernible by SWV and a situation analogous to coalescence in NMR experiments is reached [curve (a)]. Curve fitting (COOL kinetic software) to the model of reversible oxidation of two independent species with equal diffusion coefficients was performed. At −60 °C the values E1(1/2) = +15 mV and E2(1/2) = +100 mV vs. Fc/Fc+ were obtained, with relative contributions of 40 and 60% for the species with E1(1/2) and E2(1/2), respectively in accordance with 1H NMR data. From the equilibrium data at −60 °C the following ΔG0 and K values of Scheme 2 are calculated [counter-clockwise direction, starting with the reduction reaction and assuming E°(Fc/Fc+) = 0.400 V vs. NHE]: ΔG10 = −40.1 kJ mol−1, ΔG20 = −0.72 kJ mol−1, ΔG30 = +48.3 kJ mol−1, ΔG40 = −7.5 kJ mol−1, K2 = 1.5 and K4 = 68. It should be noted that in reduced 2 the form with the higher redox potential is slightly favoured, whereas the oxidized form exhibits a strong preference for the species with the lower redox potential. Analysis in terms of the van’t Hoff isochore yields standard reaction enthalpies ΔH20 = −2.5 kJ mol−1 and ΔH40 = −8.3 kJ mol−1 for the reduced and the oxidized forms of 2, respectively. Small values are expected because the same types of bonds and presumably similar geometric parameters are present in all species. The difference of ca. 6 kJ mol−1 may well be attributed to increased solvation of the oxidized species as a consequence of their positive charge.
![Square-wave voltammograms of 2 at 25 °C [(a), scan rate 60
Hz], −20 °C [(b), scan rate 20 Hz], −40 °C [(c), 20 Hz]
and −60 °C [(d), 20 Hz]. Concentration of 2 1 mM, 0.1 M
NBu4PF6 in EtCN, vs. Fc/Fc+ as
reference.](/image/article/2001/CC/b007822k/b007822k-f2.gif) | ||
| Fig. 2 Square-wave voltammograms of 2 at 25 °C [(a), scan rate 60 Hz], −20 °C [(b), scan rate 20 Hz], −40 °C [(c), 20 Hz] and −60 °C [(d), 20 Hz]. Concentration of 2 1 mM, 0.1 M NBu4PF6 in EtCN, vs. Fc/Fc+ as reference. | ||
Poli and coworkers have investigated stable paramagnetic organometallic Mo compounds in some detail.16,17 As shown herein, electrochemical measurements provide valuable information about equilibrium constants and thermodynamic parameters for fluxional paramagnetic species. This information cannot easily be derived by other common analytical techniques in organometallic chemistry. Currently, detailed calculations are performed to reveal the structure of all isomers and the mechanism of interconversion along with applications of compound 2 for the labelling of biomolecules.
Acknowledgements
We are grateful to Professor K. Wieghardt for his support of this work.Notes and references
- A. Haaland, Acc. Chem. Res., 1979, 12, 415 CrossRef CAS.
- N. J. Long, Metallocenes, Blackwell Science, Oxford, 1998..
- W. Beck, W. Petri and J. Meder, J. Organomet. Chem., 1980, 191, 73 CrossRef CAS.
- H.-J. Meder and W. Beck, Z. Naturforsch., Teil B, 1986, 41, 1247 Search PubMed.
- J. W. Faller and B. C. Whitmore, Organometallics, 1986, 5, 752 CrossRef CAS.
- K.-B. Shiu, K.-S. Liou, C. P. Cheng, B.-R. Fang, Y. Wang, G.-H. Lee and W.-J. Vong, Organometallics, 1989, 8, 1219 CrossRef CAS.
- P. Espinet, R. Hernando, G. Iturbe, F. Villafañe, A. G. Orpen and I. Pascual, Eur. J. Inorg. Chem., 2000, 1031 CrossRef CAS.
- B. J. Brisdon, K. A. Conner and R. A. Walton, Organometallics, 1983, 2, 1159 CrossRef CAS.
- B. Brisdon, S. K. Enger, M. J. Weaver and R. A. Walton, Inorg. Chem., 1987, 26, 3340 CrossRef CAS.
- M. D. Curtis and O. Eisenstein, Organometallics, 1984, 3, 887 CrossRef CAS.
- B. J. Brisdon and A. A. Woolf, J. Chem. Soc., Dalton Trans., 1978, 291 RSC.
- J. W. Faller, D. A. Haitko, R. D. Adams and D. F. Chodosh, J. Am. Chem. Soc., 1979, 101, 865 CrossRef CAS.
- K.-B. Shiu, C.-J. Chang, S.-L. Wang and F.-L. Liao, J. Organomet. Chem., 1991, 407, 225 CrossRef CAS.
- D. R. van Staveren, T. Weyhermüller and N. Metzler-Nolte, Organometallics, 2000, 19, 3730 CrossRef CAS.
- D. R. van Staveren, E. Bothe, T. Weyhermüller, M. Bühl and N. Metzler-Nolte, 2000, manuscript in preparation..
- R. Poli and L.-S. Wang, Coord. Chem. Rev., 1998, 180, 169 CrossRef.
- L.-S. Wang, J. C. Fettinger and R. Poli, J. Am. Chem. Soc., 1997, 119, 4453 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: analytical data for 2. See http://www.rsc.org/suppdata/cc/b0/b007822k |
| ‡ Present address: Institute for Pharmaceutical Chemistry, University of Heidelberg, Im Neuenheimer Feld 364, D-69120 Heidelberg, Germany. E-mail: Nils.Metzler-Nolte@urz.uni-heidelberg.de |
| § Crystal data for 2: SMART-CCD diffractometer,
Mo-Kα radiation (λ = 0.71073 Å).
C17H27MoN3O8, M =
497.36, monoclinic, space group P21, a =
12.0928(14), b = 7.5347(8), c = 12.1177(14) Å,
β = 101.59(2)° (unit cell parameters determined from 5092
reflections), V = 1081.6(2) Å3, T =
100(2) K, Z = 2, μ(Mo-Kα) = 0.653
mm−1, 11346 reflections measured, 4668 unique
(Rint = 0.0509) which were used in all calculations.
R1 = 0.0281 (all data) and
wR2(F2) = 0.0692 (all data). 7
restraints were used. Allyl C–H bond lengths were restrained to be
equal within certain errors (0.005 Å). Flack parameter is
−0.03(3), in agreement with the absolute configuration (L)
of the histidine. CCDC 182/1846. See http://www.rsc.org/suppdata/cc/b0/b007822k/ for crystallographic files in .cif format. |
| This journal is © The Royal Society of Chemistry 2001 |