Synthesis of new types of polysiloxane based surfactants
Josef Bauer, Nicola Hüsing and Guido Kickelbick*
Institut für Anorganische Chemie, Technische Universität Wien, Getreidemarkt 9, A-1060, Wien, Austria.. E-mail: kickelgu@mail.zserv.tuwien.ac.at
First published on UnassignedUnassigned3rd January 2001
Abstract
New types of amphiphilic polysiloxane diblock copolymers containing a pure polysiloxane backbone were prepared by the functionalization of poly(dimethylsiloxane)-block-poly(methylvinylsiloxane) copolymers, synthesized by ‘living’ anionic ring opening polymerisation of 1,3,5,7-tetramethyl-1,3,5,7-tetravinylcyclotetrasiloxane (D4v) and hexamethylcyclotrisiloxane (D3).
Amphiphilic block copolymers find widespread technological applications especially as non-ionic polymeric surfactants. The coexistence of the dissimilar segments, which show a different solubility in a specific solvent, produces unique properties because of their ability to self-organize at interfaces and in solution and thus modify interfacial properties and enhance compatibility or separation.1 Typically, purely organic backbones are found in this type of surfactant, nevertheless inorganic polymer segments, especially polysiloxanes show properties that are advantageous, such as biocompatibility, robustness, flexibility etc.2 Although polysiloxanes have superior properties compared to common organic polymers their controlled formation as block copolymers is nearly unexplored.
In recent years the interest in amphiphilic block copolymers has grown especially due to their surface activity and their lyotropic behaviour.3 Amphiphilic block copolymers with a hydrophobic polysiloxane part are predominantly known in combination with organic hydrophilic blocks such as poly(ethylene oxide) or polymethacrylates.4 As far as we know there is only one example in literature citing a block copolymer with a pure polysiloxane backbone bearing carboxyl groups pendant to the polysiloxane chain.5
In this paper we present the formation and post-functionalization of block copolymers of poly(dimethylsiloxane) (PDMS) and poly(methylvinylsiloxane) by ‘living’ anionic ring opening polymerisation.
Valuable precursors for a post-functionalization are silane-bonded vinyl groups. For the preparation of a vinyl-functionalized block via anionic ring opening polymerisation a cyclic monomer with a sufficient reactivity is needed. The only commercially available monomer that fulfils this demand is 1,3,5,7-tetramethyl-1,3,5,7-tetravinylcyclotetrasiloxane (D4v). D4v did not show satisfactorily polymerisation initiated by butyllithium with the usual donor solvents (THF, DMF, DMSO, HMPA). Therefore another initiator with a higher basicity, i.e. trimethylsilylmethyllithium, (CH3)3SiCH2Li, was used to transform the carbanionic acids into the silanolate species.6 Usually polymerisations of unstrained cyclic siloxanes, such as cyclotetrasiloxanes or larger rings, are more difficult due to the presence of intra- and intermolecular reactions of the active ends leading to an equilibrium reaction between the ring, an oligomeric, and a polymeric species. It was shown that the equilibrium is shifted to the polymeric species if additives able to separate the ion pair between the oxidic end group and the lithium ion are added to the reaction solution. Typical additives are ethylenediamine, diglyme, cryptands and crown ethers like 12-crown-4.7
Block copolymers from ‘living’ ring opening polymerisations are obtained by using a polymer as macroinitiator and extending it with a second monomer. For the PDMS-block-poly(methylvinylsiloxane) we observed that the extension of a PDMS chain with poly(methylvinylsiloxane) only leads to the addition of up to five monomeric units until the final equilibrium between polymeric and ring species is reached, while the polymerisation of D4v first allows for a much better control of the equilibrium. Therefore, the polymerisation of vinylmethylcyclotetrasiloxane was initiated with trimethylsilylmethyllithium in a first step and the chain was extended by PDMS in a second step8 (Scheme 1). With this procedure block copolymers with polydispersities around 1.40 and block lengths up to 10.000 with varying poly(methylvinylsiloxane) content were obtained (Table 1).
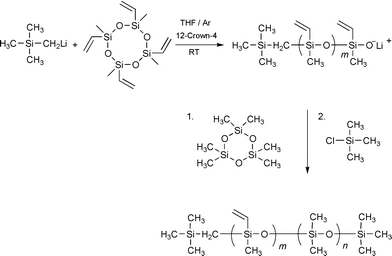 | ||
| Scheme 1 | ||
| Sample | Mn (Mw/Mn) (PD4v-block) | m | n | Mn (Mw/Mn) (PD4v-PD3- diblock) | Ratio by 1H NMR (PD4v/PD3) | Ratio by SEC (PD4v/PD3) |
|---|---|---|---|---|---|---|
| 1 | 2840 (1.36) | 33 | 96 | 9940 (1.46) | 0.38 | 0.40 |
| 2 | 1900 (1.36) | 22 | 22 | 3520 (1.38) | 0.86 | 0.85 |
| 3 | 1730 (1.21) | 20 | 82 | 7840 (1.36) | 0.25 | 0.28 |
| 4 | 1480 (1.14) | 17 | 50 | 5190 (1.31) | 0.35 | 0.40 |
The resulting block copolymers have purely hydrophobic properties but the vinyl groups can easily be transformed into a variety of other functional groups. The double bonds of the methylvinylsiloxane blocks were modified either by hydrosilation9 or by epoxidation.10 In the latter case further modification reactions of the polyepoxide block were carried out11 (Scheme 2).
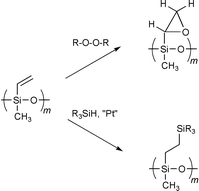 | ||
| Scheme 2 | ||
The hydrosilation of the double bonds with Karstedt’s catalyst at 70 °C worked quantitatively as confirmed by the disappearance of the vinylic protons in 1H NMR. HSi(OEt)3, HSi(OMe)3 and HSi(CH3)2Cl were used for this reaction to obtain a functionalized polysiloxane block. While the trialkoxysilane groups can be hydrolysed and used for cross-linking reactions, the chlorosilanes are able to react with a variety of nucleophiles, like alcohols, carboxylic acids, etc.
Another possibility of the double bond functionalization is the
epoxidation12 followed by the subsequent
ring opening of the oxirane ring. The epoxide was quantitatively formed by
a selective oxidation of the double bonds with
m-chloroperoxybenzoic acid (MCPBA) in methylene chloride or
toluene proved by 1H NMR. SEC measurements of the epoxidised
samples showed that only a small amount (<4%) of high molecular weight
(cross-linked) polymer was formed. The cross-linking was almost
quantitatively suppressed by working in high dilutions.13 The obtained epoxide was opened with different
mono- or difunctional nucleophiles, such as hydroxide ions, diamines,
diols, dicarboxylic acids, hydroxy-functionalized ethers and carboxylic
acid chlorides (Scheme 3). These
reactions resulted in the formation of a hydroxide group at one carbon atom
and the coupling product with the nucleophile at the other. Modification of
the epoxide block by reaction with amines or alcohols was catalysed by
LiClO4. Nucleophilic attack of the epoxide by the nucleophile on
the more hindered carbon atom (SiCH![[double bond, length half m-dash]](https://www.rsc.org/images/entities/char_e006.gif) CH2) was not
observed in 1H NMR spectra which indicates an almost
quantitative attack on the terminal methylene carbon atom. In the case of
modification with oxalic acid the reaction was performed in
N,N′-dimethylformamide to dissolve the reactant and
additionally catalysed by pyridine. Hydrolysis of the epoxidised block
copolymers leads to the corresponding 1,2-diol side groups and may either
be acid- or base-catalysed. Epoxide opening worked quantitatively, which
was proved by 1H NMR (disappearance of the signals of the
epoxide protons at 2.2, 2.7 and 2.9 ppm). The processing parameters of the
ring opening reactions allow for the preparation of different materials.
High dilution of the block copolymers in the solvent during the ring
opening leads to linear polymers while cross-linked hydrogels are formed in
low dilution. To completely suppress cross-linking reactions using
multifunctional nucleophiles a large excess of reactants is necessary and
harsh purification conditions should be avoided. For a total prevention of
cross-linking reactions it is advisable to use monofunctional protected
ring opening agents and remove the protecting groups afterwards. Additional
advantages of this procedure are simpler purification and analyses. In
those cases in which hydroxy groups are formed during the ring opening or
other functionalities are present, no elution in the SEC experiments was
obtained due to interaction with the column material. This was avoided by
protection of the free functional groups.
CH2) was not
observed in 1H NMR spectra which indicates an almost
quantitative attack on the terminal methylene carbon atom. In the case of
modification with oxalic acid the reaction was performed in
N,N′-dimethylformamide to dissolve the reactant and
additionally catalysed by pyridine. Hydrolysis of the epoxidised block
copolymers leads to the corresponding 1,2-diol side groups and may either
be acid- or base-catalysed. Epoxide opening worked quantitatively, which
was proved by 1H NMR (disappearance of the signals of the
epoxide protons at 2.2, 2.7 and 2.9 ppm). The processing parameters of the
ring opening reactions allow for the preparation of different materials.
High dilution of the block copolymers in the solvent during the ring
opening leads to linear polymers while cross-linked hydrogels are formed in
low dilution. To completely suppress cross-linking reactions using
multifunctional nucleophiles a large excess of reactants is necessary and
harsh purification conditions should be avoided. For a total prevention of
cross-linking reactions it is advisable to use monofunctional protected
ring opening agents and remove the protecting groups afterwards. Additional
advantages of this procedure are simpler purification and analyses. In
those cases in which hydroxy groups are formed during the ring opening or
other functionalities are present, no elution in the SEC experiments was
obtained due to interaction with the column material. This was avoided by
protection of the free functional groups.
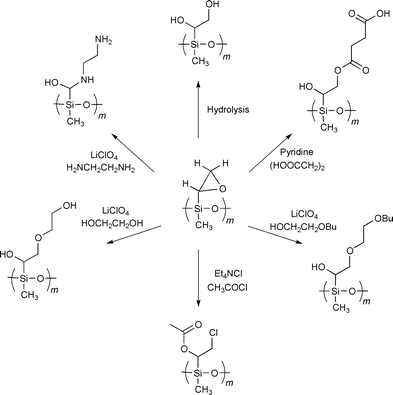 | ||
| Scheme 3 | ||
In summary new methylvinyl-substituted polysiloxane diblock copolymers were prepared by ‘living’ anionic ring opening polymerisation. The vinyl group was used for a further functionalization to obtain silicon alkoxide substituted or amphiphilic block copolymers with a variety of functional groups.
Acknowledgements
We gratefully acknowledge the financial support by the Fonds zur Förderung der wissenschaftlichen Forschung Austria and Wacker-Chemie for their kind donation of chemicals.Notes and references
- P. Alexandridis, U. Olsson and B. Lindeman, Langmuir, 1998, 14, 2627 CrossRef CAS.
- P. R. Dvornic and R. W. Lenz, High temperature siloxane elastomers, ed. P. Dvornic and R. W. Lenz, Hüthig & Wepf, Verlag, 1990. Search PubMed.
- S. Förster and M. Antonietti, Adv. Mater., 1998, 3, 195 CrossRef.
- H.-W. Haesslin, Makromol. Chem., 1985, 186, 357 Search PubMed.
- M. Scibiorek, N. K. Gladkova and J. Chojnowski, Polymer Bulletin, 2000, 44, 377 CrossRef CAS.
- R. P. Quirk and D. E. Kester, J. Organomet. Chem., 1977, 127, 111 CrossRef CAS.
- J. Chojnowki, K. Ròzga, W. Foruniak and A. Kowalewska, Makromol. Chem., Makromol. Symp., 1993, 67, 183 Search PubMed.
- Typical procedure: to a solution of D4v in
THF (CH3)3SiCH2Li was added. The amount of
the lithium compound was chosen depending on the target molecular weight of
the poly(methylvinylsiloxane)
([I] = initiator concentration; [M]0 = monomer concentration; DP = degree of polymerisation). After stirring the solution at rt for 1 h 12-crown-4 was added. Propagation progress was determined during the polymerisation by taking samples, quenching the reaction with chlorotrimethylsilane at various reaction times and determining the molecular weights by size exclusion chromatography (SEC) analyses. By adding D3 to the ‘living’ polymer the PDMS-blocks were formed. The diblock copolymers were quenched by chlorotrimethylsilane and analysed by SEC and NMR measurements. SEC was performed using a Waters 717 autosampler, 515 HPLC pump, 2410 RI detector and Styragel columns in THF at 40 °C at a rate of 1 ml min−1 applying linear polystyrene standards. The DP of each block was calculated by the SEC results of homopolymer and diblock copolymer and compared with the results of the composition determined by 1H NMR. 29Si NMR analyses allowed the determination of the sequence distribution of repeat units, which showed no random copolymerization of D4v and D3.. - Y. Chang, Y. C. Kwon, S. C. Lee and C. Kim, Polym. Prepr. (Am. Chem. Soc. Div. Polym. Chem.), 1999, 40, 269 Search PubMed.
- F. Macchia, P. Crotti and M. Chini, Tetrahedron Lett., 1990, 31, 4661 CrossRef.
- A. Hassner, in Small Ring Heterocycles, ed. A. Weissberger and E. C. Taylor, John Wiley & Sons, New York, 1976. Search PubMed.
- General procedure: MCPBA was dissolved in toluene and dried over Na2SO4. Depending on the block length of the poly D4v block, a calculated amount of the solution was added to the block copolymer. The resulting mixture was stirred overnight at rt. The organic layer was extracted with an aqueous sodium bicarbonate solution and deionised water several times. The organic layers were collected and dried over Na2SO4. Afterwards the solution was filtered and evaporated..
- K. Udipi, J. Appl. Polym. Sci., 1979, 23, 3301 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2001 |