Silica gel catalysed generation of episulfonium ions: a mild method for the synthesis of heterocycles
Lorenzo
Caggiano
,
David J.
Fox
and
Stuart
Warren
*
University Chemistry Laboratory, Lensfield Road, Cambridge, UK CB2 1EW
First published on 30th September 2002
Abstract
β-Carbonyloxy sulfides fragment when heated with silica or alumina to produce reactive episulfonium ion intermediates. Intramolecular activation of the leaving group and concurrent protection of a nitrogen or oxygen nucleophile via a cyclic carbamate or carbonate leads to the formation of pyrrolidines or cyclic ethers.
Many allylic sulfides and oxygen heterocycles have been synthesised from β-hydroxy sulfides by treatment with catalytic strong acids.1 These transformations proceed via reactive episulfonium ions trapped by by nucleophilic opening by a remote hydroxy group to produce a cyclic ether (Scheme 1). Similar ether syntheses can be initiated by electrophilic iodine2 and selenium3,4 reagents forming corresponding reactive three-membered ring intermediates from olefins.
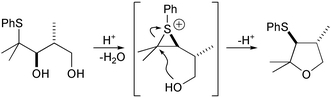 | ||
| Scheme 1 | ||
In order to make nitrogen heterocycles via episulfonium ions it has been necessary to reduce the basicity of the nitrogen by attaching a tosyl group.1 This method has also been used in similar iodonium ion5 and bromonium ion6 mediated reactions. While this strategy gave high yields of heterocycle the removal of the sulfonyl group to produce the free amine required strongly reducing conditions incompatible with many functional groups. Methods are available for halogen7 and sulfur8 mediated cyclisation of free amines but they involve the addition of an electrophilic reagent to an olefin. We wished to develop a simple method of cyclising free amines onto stereospecifically generated episulfonium ions without strong acid catalysis. Recently we have reported the use of the weaker acid pyridinium p-toluene sulfonate as a catalyst in cyclo-etherifications,1 but these conditions would still not be effective in the cyclisation of a free amine due to the protonation of the nucleophile. A method for generating episulfonium ions under essentially neutral conditions was therefore needed.
It was hoped that, if the hydroxy group could be activated sufficiently, the episulfonium ion could be formed with the aid of a very mild non-acidic catalyst. The activated compounds should however be stable enough for easy synthesis and isolation in stereochemically pure fashion. Conversion of the alcohol into an ester or related group should meet these criteria. In order to test this proposal, alcohol11 was converted into benzoate ester 2, a stable compound at ambient temperature that could be purified by silica chromatography or heated in chloroform at reflux without chemical change. The proven stability of the episulfonium ion precursor to both column chromatography and heating is vital if it is to be a useful synthetic intermediate.
The formation of the episulfonium ion from benzoate 2 therefore requires a Lewis acid to coordinate to the carbonyl group of the ester and increase its leaving group ability. Silica gel is a mild acid and although it does not cause degradation of ester 2 during purification at ambient temperature, when benzoate 2 was heated in chloroform in the presence of ten times its mass of silica gel, it reacted to form allylic sulfide 7, the isomeric ester 5 and benzoic acid. Prolonged reaction times (4 and 8 h) increased the conversion to rearranged ester 5. After 16 h both esters were converted into allylic sulfide 7 and benzoic acid (Table 1, entries 1–4). The [1,2]-migration of the PhS group during the reaction suggests an episulfonium ion intermediate, like those formed in the strong acid catalysed rearrangements.1 The transient appearance of ester 5 is consistent with the reversible formation of the episulfonium ion which can be trapped by the benzoate anion bound to the silica surface. This mechanism is consistent with the treatment of independently synthesised ester 5 in the same reaction conditions to give a similar mixture including the primary benzoate 2 approaching an equilibrium from the other direction (Table 1, entry 5, Scheme 2). A similar silica catalysed rearrangement occurs for a phenylselenyl acetate in which the selenium migrated from a secondary to a primary carbon while the acetate moved in the other direction.9
| Sulfides in reaction mixtureb (%) | ||||||
|---|---|---|---|---|---|---|
| Entry | Starting material | C/Sa | Time/h | 2 or 3 | 5 or 6 | 7 |
| a Silica catalyst-to-substrate ratio by mass. b Measured by NMR spectroscopy. | ||||||
| 1 | 2 | 10 | 1 | 86 | 5 | 9 |
| 2 | 2 | 10 | 4 | 34 | 24 | 41 |
| 3 | 2 | 10 | 8 | 17 | 26 | 56 |
| 4 | 2 | 10 | 16 | <1 | <1 | >98 |
| 5 | 5 | 10 | 16 | 3 | 63 | 34 |
| 6 | 3 | 5 | 8 | 85 | 1 | 14 |
| 7 | 3 | 5 | 16 | 80 | 1 | 19 |
| 8 | 3 | 10 | 8 | 75 | 2 | 23 |
| 9 | 3 | 10 | 16 | 45 | 10 | 45 |
| 10 | 3 | 30 | 8 | 21 | 23 | 56 |
| 11 | 3 | 30 | 16 | 13 | 20 | 67 |
| 12 | 6 | 10 | 16 | 6 | 68 | 26 |
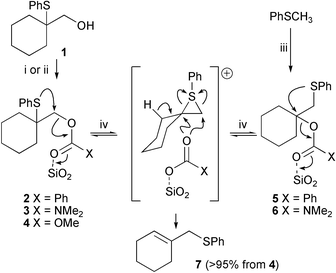 | ||
| Scheme 2 Reagents and conditions: i, PhCOCl, pyridine, 88%; ii, NaH, DMF; Me2NCOCl or MeOCOCl, (3) 91%; (4) 78%; iii, n-BuLi, DABCO, THF; cyclohexanone; then PhCOCl or Me2NCOCl, (5) 5%; (6) 4%; iv, SiO2, CHCl3, Δ, see Table 1. | ||
Diethyl carbamate 3 and methyl carbonate 4 were also synthesised from alcohol 1. These compounds are also stable to silica gel chromatography and heating, but on heating with silica gel in chloroform they too formed allylic sulfide 7, showing that this mild method of forming episulfonium ions is applicable to a range of carbonyl containing leaving groups (Scheme 2). A proportion of the carbamate also rearranged to the tertiary carbamate 6 by PhS migration before elimination to the olefin (Table 1, 6–11). The intermolecular recapture of the episulfonium ion by the carbamate anion may seem surprising given that the reaction is occurring in refluxing chloroform and in the presence of mildly acidic silica gel, given that the decarboxylation of carbamate ions is acid catalysed.10 Once again the reversible nature of the isomerisation was confirmed by reaction of the primary sulfide 6 (Table 1, entry 12). Only elimination was observed for carbonate 4.
Alumina also catalyses the reaction of the carbonyl derivatives on heating, but does not do so at ambient temperature. Other solids (TiO2, MgSO4) do not catalyse the transformation. The choice of solvent is also important: the reaction does not occur in solvents that reduce the activity of silica gel such as methanol and THF, but does so in less Lewis basic solvents (PhMe and EtOAc). Addition of water inhibits the reactions, whereas drying the silica gel at elevated temperature in vacuo increases the activity of the catalyst. The residual ethanol present in commercial chloroform also inhibits the reaction. The reactions were performed in chloroform that had been purified by passage through a column of silica gel.
A range of cyclisation reactions in which the episulfonium ion could be captured by an intramolecular nucleophile was then attempted. Activation of the leaving group while temporarily protecting the nucleophile could be achieved with a cyclic carbamate or carbonate (Scheme 3). The generation of the episulfonium ion would then be followed by a ring closure with loss of carbon dioxide. Treatment of the cyclic carbamate 8 with silica gel in refluxing chloroform produced the pyrrolidine 9 in excellent yield. Filtration, washing of the silica gel and removal of solvent produces essentially pure cyclic amine. In identical conditions the cyclic carbonate reacted quantitatively to give a roughly equal mixture of THF 11 and allylic sulfide 12 in chloroform, but more cyclisation occurred in CCl4. As it is known that the equivalent free hydroxyl group readily cyclises to give a quantitative yield of THF,1 the silica gel reactions indicate that the carbonate group must have an appreciable life-time after episulfonium ion generation. For both the ether and amine cyclisations, slow decarboxylation may occur before the cyclisation, but this seems unlikely given the isolations of the rearranged products 5 and 6. Alternatively the nucleophile could be the less effective alkylated-carbonate-oxygen, or carbamate nitrogen, and decarboxylation is concurrent with, or occurs after, the ring closure (Scheme 4). Cyclisation of alkyl carbamates to form nitrogen heterocycles can be initiated by selenium reagents, but in these cases nitrogen-decarboxylation does not occur in situ.4,10–12
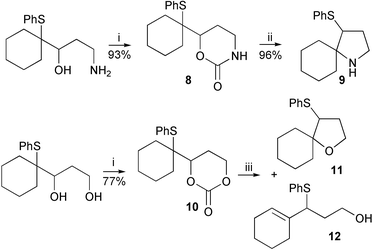 | ||
| Scheme 3 Reagents and conditions: i, CDI, CH3CN; ii, SiO2, CHCl3, Δ; iii, SiO2, CHCl3 (11∶12 =50∶50) or CCl4 (11∶12 = 86∶14), Δ. | ||
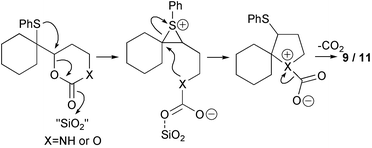 | ||
| Scheme 4 | ||
The seven-membered carbamate 14 was synthesised from ketone113 (Scheme 5). Treatment of this cyclic compound with silica gel in refluxing chloroform produced a mixture of unrearranged pyrrolidine 15 and rearranged piperidine 16 (Scheme 5). The nitrogen nucleophile attacks the proximal end of the episulfonium ion more readily to form a five-membered ring, than the distal end to produce a six-membered ring. In the equivalent strong-acid-catalysed ether-cyclisation reaction, the six-membered-ring compound is the only product, and the reaction is under thermodynamic control. In this silica gel catalysed process however it seems that the loss of CO2 prevents the regeneration of an episulfonium ion and that the product ratio is controlled by the relative rates of cyclisation rather than the ultimate stability of the products. Similar product ratios have been observed in other non-equilibrating cyclisation reactions.1,9
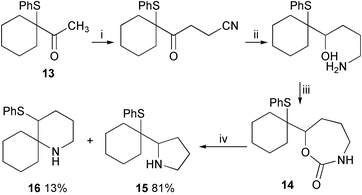 | ||
| Scheme 5 Reagents and conditions: i, LDA, THF, −78 °C then BrCH2CN, HMPA, 43%; ii, LiAlH4, Et2O, 99%; iii, CDI, DMF, 65%; iv, SiO2, CHCl3, Δ. | ||
We thank the EPSRC for a grant to L. C.
Notes and references
- D. J. Fox, D. House and S. Warren, Angew. Chem., Int. Ed., 2002, 41, 2462 CrossRef CAS.
- J. M. Barks, G. G. Weingarten and D. W. Knight, J. Chem. Soc., Perkin Trans. 1, 2000, 3496 Search PubMed.
- T. Wirth, Angew. Chem., Int. Ed., 2000, 39, 3740 CrossRef CAS.
- R. Deziel and E. Malenfant, J. Org. Chem., 1995, 60, 4660 CrossRef CAS.
- A. D. Jones, D. W. Knight and D. E. Hibbs, J. Chem. Soc., Perkin Trans. 1, 2001, 1182 RSC.
- Y. Tamaru, S. Kawamura, T. Bando, K. Tanaka, M. Hojo and Z. Yoshida, J. Org. Chem., 1988, 53, 5491 CrossRef.
- W. S. Lee, K. C. Jang, J. H. Kim and K. H. Park, Chem. Commun., 1999, 251 RSC.
- T. Ohsawa, M. Ihara, K. Fukumoto and T. Kametani, J. Org. Chem., 1983, 48, 3644 CrossRef CAS.
- D. Brugier, F. Outurquin and C. Paulmier, J. Chem. Soc., Perkin Trans. 1, 2001, 37 RSC.
- M. Caplow, J. Am. Chem. Soc., 1968, 90, 6795 CrossRef CAS.
- D. L. J. Clive, C. K. Wong, W. A. Kiel and S. M. Menchen, J. Chem. Soc., Chem. Commun., 1978, 379 RSC.
- K. C. Nicolaou, D. A. Claremon, W. E. Barnette and S. P. Seitz, J. Am. Chem. Soc., 1979, 101, 3704 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2002 |