Alkoxy/siloxy group exchange in the system vinyltrialkoxysilane–iridium(I) siloxide complex
Ireneusz
Kownacki
,
Bogdan
Marciniec
* and
Maciej
Kubicki
Faculty of Chemistry, Adam Mickiewicz University, 60-780 Poznan, Poland. E-mail: marcinb@main.amu.edu.pl
First published on 25th November 2002
Abstract
A study of reactions of dimeric siloxide iridium complex, [{(cod)Ir(μ-OSiMe3)}2] (1) with vinyltriethoxysilane and vinyltrimethoxysilane has revealed a new type of the reaction—alkoxy group transfer from silicon to iridium with a simultaneous transfer of a siloxy group from iridium to silicon—as a result of which vinyldialkoxytrimethyldisiloxane and dimeric alkoxide iridium complex [{(cod)Ir(μ-OR)}2] (3) are formed. The structure of [{(cod)Ir(μ-OEt)}2] (3a) has been solved by X-ray diffraction.
According to a general idea presented by Wolczanski, alkoxide and siloxide ligands are alternatives to cyclopentadienyl complexes of TMS. They can be bound preferably through a σ-type orbital (sp hybrid) and via π-donation of two pπ-orbitals.1 The strength of both σ- and π-interactions depends strongly on the electrophilicity of the metal center. Thanks to the more positive character of the silicon atom, when compared to carbon, siloxide ligands are generally less basic than alkoxides.2 Although dπ–pπ bonding (or σ*–pπ) as an interaction of fairly low-lying empty 3d/σ* molecular orbitals with oxygen pπ orbitals explains the π-accepting capability of silicon in the Si–O bonding, the inductive effects of substituents at silicon can additionally influence the electronic properties of the TM–O–SiR3 system.1
The composite (σ + π) electron donor ability of the group X in, for example, the complex of formula [RuH(X)(CO)(PBu2Me)2], determined by Caulton et al. using spectroscopic data, increased in the sequence OSiPh3 < OSiMe3 < OEt.3,4
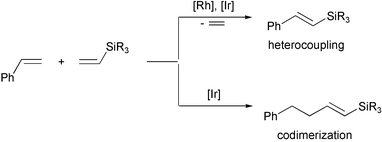
The stereoelectronic properties of the siloxy groups, particularly in metal siloxides, can be effectively exploited in molecular catalysis, either by using the above groups as ancillary ligands of early-TM complexes (see comprehensive studies by Wolczanski et al.1,4–6 and Feher et al.7,8) or by exploiting their ability to form bridging structures in a low oxidation state of the late-TM complexes (e.g. Rh(I)9–11 and Ir(I)12). The latter possibility opens a new catalytic route to obtaining precursors for catalysis involving coordination of olefins (or other π-acceptors) and oxidative addition steps.2
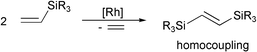
The dimeric siloxy-rhodium(I) complex of the general formula [{(cod)Rh(μ-OSiMe3)}2] appeared to be a very effective catalyst for homocoupling of vinyltrisubstituted silanes10 and their stereoselective heterocoupling with styrenes.11
 | (1) |
 | (2) |
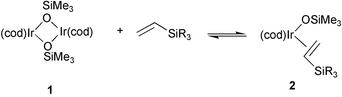
The reactions proceed via Rh–H and Rh–Si 16e intermediates generated in situ as a result of preliminary coordination of vinylsilane (or styrene) to a rhodium atom followed by their oxidative addition (activation of ![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–H bond). The analogous complex of iridium [{(cod)Ir(μ-OSiMe3)}2] (1), which was recently synthesized and its X-ray structure solved, was found to be similar to the rhodium complex. It appeared to be a stereoselective catalyst for the coupling of styrene with vinyltrisubstituted silanes R3SiCHCH2 (where R3 = Me2Ph and (OEt)3), whereas in the reaction of styrene with vinylsilanes containing three bulky substituents at silicon, e.g. (OSiMe3)3, (OtBu)3, selective co-dimerization was observed.12 However, 1H NMR study has confirmed that in all cases a preliminary dissociation of iridium dimer with coordination of vinylsilane occurs according to the following equation:
C–H bond). The analogous complex of iridium [{(cod)Ir(μ-OSiMe3)}2] (1), which was recently synthesized and its X-ray structure solved, was found to be similar to the rhodium complex. It appeared to be a stereoselective catalyst for the coupling of styrene with vinyltrisubstituted silanes R3SiCHCH2 (where R3 = Me2Ph and (OEt)3), whereas in the reaction of styrene with vinylsilanes containing three bulky substituents at silicon, e.g. (OSiMe3)3, (OtBu)3, selective co-dimerization was observed.12 However, 1H NMR study has confirmed that in all cases a preliminary dissociation of iridium dimer with coordination of vinylsilane occurs according to the following equation:
 | (3) |
The 1H NMR spectrum of the complex [(cod)Ir(CH2CHSi(OEt)3)(OSiMe3)], recorded after 30 min, revealed no signal at 0.312 ppm assigned to bridged ligands of OSiMe3 in the initial dimeric complex. Two singlets at 0.252 and 0.258 ppm, corresponding to the protons from OSiMe3 ligands of monomeric complex, have appeared instead. The spectrum also revealed a shift of signals from cyclooctadiene ligand to the higher magnetic field (by about 0.054 ppm). However, after 24 h the 1H NMR spectrum of this complex showed the appearance of a new signal at 0.166 ppm, assigned to the protons of the trimethylsilyl group of CH2CHSi(OEt)2OSiMe3, and new groups of signals coming from the new iridium(I) complex 3a. GC-MS analysis of organosilicon compounds has confirmed the formation of vinyldiethoxytrimethylsiloxane according to the following eqn. (4)
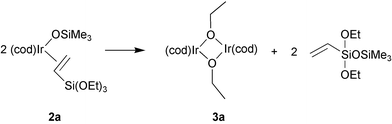 | (4) |
Vinyltrimethoxysilane undergoes the same transformation yielding analogous dimeric complex [{(cod)Ir(μ-OMe)}2] 3b and eliminating CH2CHSi(OMe)2OSiMe3. Contrary to vinyltrimethoxy- and vinyltriethoxysilane, CH2CHSi(OEt)Me2 and CH2CHSi(OtBu)3 yield monomeric complexes of the type 2, while cleaving siloxide bridge in 1, but no siloxy–alkoxy-transfer was observed.
Additional experiments using higher concentrations of the substrates and 10-fold excess of vinyltriethoxysilane allowed us to isolate iridium-ethoxide dimeric complex 3a. Here we present the synthesis and the structure of 3a, determined by X-ray diffraction.†
A solution of 0.15 g (0.193 mmol) [{(cod)Ir(μ-OSiMe3)}2] in 2 ml of dried and deoxidised pentone were placed in a Schlenk’s flask under argon. Then 0.37 g (1.95 mmol) of CH2CHSi(OEt)3 was added to the solution. The reaction was conducted for 2 hours at room temperature. After that time, the liquid was decanted and the orange crystals were three times washed with pentane by decantation at minus 15 °C. The yield was 62%.
1H NMR δ(ppm, C6D6) = 1.1 (t, 6H, –CH3); 1.39 (m, 8H, –CH2–); 2.22 (m, 8H, –CH2–); 3.31 (q, 4H, –OCH2–); 3.46 (m, 8H, CH–)
The molecular structure of this complex, determined by X-ray diffraction, with the atom numbering scheme is depicted in Fig. 1. The complex 3a lies on the twofold axis (the asymmetric part of the unit cell contains half of the molecule). The geometry of the complex is A-frame bis-square planar with the roof angle of 122.7°, slightly larger than that in similar trimethylsiloxy compounds (119.7°). The crystal structure of the isostructural complex of rhodium has been published previously.13
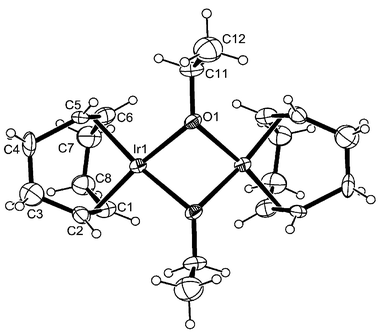 | ||
| Fig. 1 ORTEP view of the complex 3a, showing the labelling scheme (33% probability ellipsoids). The symmetry-related atoms are not labelled for clarity. Relevant distances and angles (Å, deg) Ir1⋯Ir1a 2.8958(11), Ir1–O1 2.075(10), Ir1–O1A 2.080(9), Ir1–X1 1.976(15), Ir1–X2 1.978(16); X1–Ir1–X2 87.8(8), X1–Ir1–O1 173.5(5), X2–Ir1-O1 98.4(6), X1–Ir1–O1a 98.8(6), X2–Ir1–O1a 173.0(7), O1–Ir1–O1a 74.9(4), Ir1–O1–Ir1a 88.4(4). Index a relates to the symmetry operation ½ − x, y, ½ − z. X1, X2 denote the middle points of the C1–C2 and C5–C6 bonds, respectively. | ||
In order to explain a mechanism for the siloxy ligand migration from iridium to silicon of vinyltrialkoxysilane π-bonded iridium complexes, a simple geometrical model of (1) interaction with vinyltrimethoxysilane has been considered. Typical bond lengths and bond angles were used for geometrical calculations.
We assumed the square-planar coordination of Ir and the obvious fact that both OSiMe3 and vinyltrimethoxysilane groups occupy the neighbouring coordination sites. There are conformations of vinyltrimethoxysilane (with double bond approximately perpendicular to the coordination plane) in which one of the oxygen atoms is close to the axial position with respect to the Ir atom, and in quite a typical Ir⋯O distance of ca. 2.1 Å (Fig. 2). In such conformations another oxygen atom from the vinyltrimethoxysilane group can come close to the Si atom from OSiMe3 group. Interestingly, there are no other obvious steric hindrances for this geometry.
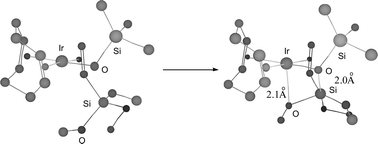 | ||
| Fig. 2 The model of 1 interaction with vinyltrimethoxysilane. | ||
The driving force for this migration is the cleavage of the siloxide dimer 1 by vinylsilane to generate the 16e complex (2) of square geometry recorded by 1H and 13C NMR spectroscopy [see eqn. (4)].12 There is no migration observed when methyltrialkoxysilane is used instead of vinyltrialkoxysilane.
This model is interpreted mainly in terms of the interaction of n-electrons of one of the methoxy groups of vinylsilane with the iridium atom and simultaneous interaction of n-electrons of the siloxy ligand with the silicon atom of vinyltrimethoxysilane. It results in the formation of a tetra-centered transition state with possible cleavage of the π-bonding of the vinyl group to iridium(I) (see Fig. 3).
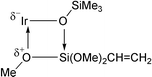 | ||
| Fig. 3 Proposed transition state in the reaction of 1 with vinyltrimethoxysilane. | ||
It is hard to distinguish whether initially a nucleophilic attack of the oxygen of the methoxy group on the iridium atom reduces dπ–pπ (or σ*–pπ ) back interaction and hence enables, if spatially feasible, a nucleophilic attack of the siloxy oxygen on the silicon atom of vinylsilane, or alternatively the driving force is a nucleophilic attack of the oxygen of iridium(I) siloxide on the silicon atom of the coordinated molecule of vinylsilane containing three electron-withdrawing substituents at silicon inducing a nucleophilic attack of oxygen of one of the methoxy groups in close to axial position of the iridium complex.
The above interactions lead to elimination of vinyldi(methoxy)trimethyldisiloxane and monomeric 14e iridium(I)(cod)methoxy complex which rapidly undergoes dimerization to yield finally 3b. Steric hindrance by the tert-butyl group in vinyltritert(butoxy)silane made the above mentioned interactions impossible, therefore no siloxy/alkoxy exchange was observed. The inactivity of vinylmethyldiethoxysilane in this rearrangement can be explained mainly by electronic effects. Apparently, at least two alkoxy (methoxy, ethoxy) substituents at silicon in the transition state (see Fig. 2 and 3) are necessary to ensure electron-acceptor character of the silicon atom and hence to induce nucleophilic attack of the oxygen of the siloxy ligand on this atom leading finally to the elimination of vinyldialkoxytrimethyldisiloxane. On the other hand, it is possible that three alkoxy groups in the parent vinylsilane are necessary to initiate a real interaction of one of them with the iridium atom, resulting in the transition state presented in Fig. 3.
The work was supported by the State Committee for Scientific Research, Project No.K026/T09/2001.
Notes and references
- P. T. Wolczanski, Polyhedron, 1995, 14, 3335 CrossRef CAS.
- B. Marciniec and H. Maciejewski, Coord. Chem. Rev., 2001, 223, 301 CrossRef CAS.
- J. T. Poulton, M. P. Sigalas, O. Eisenstein and K. G. Caulton, Inorg. Chem., 1993, 32, 5490 CrossRef CAS.
- J. T. Poulton, M. P. Sigalas, K. Folting, W. E. Streib, O. Eisenstein and K. G. Caulton, Inorg. Chem., 1994, 33, 1476 CrossRef CAS.
- K. J. Covert, D. R. Neithamer, M. C. Zonnevylle, R. E. LaPointe, C. P. Schaller and P. T. Wolczanski, Inorg. Chem., 1991, 30, 2494 CrossRef CAS.
- J. L. Bennett and P. T. Wolczanski, J. Am. Chem. Soc., 1994, 116, 2179 CrossRef CAS.
- F. J. Feher and T. A. Budzichowski, Polyhedron, 1995, 14, 3239 CrossRef CAS.
- F. J. Feher and T. L. Tajima, J. Am. Chem. Soc., 1994, 116, 2145 CrossRef CAS.
- B. Marciniec, P. Krzyzanowski, E. Walczuk-Gusciora and W. Duczmal, J. Mol. Catal., 1999, 144, 263 Search PubMed.
- B. Marciniec, E. Walczuk-Gusciora and P. Blazejewska-Chadyniak, J. Mol. Catal. A, 2000, 160, 165 CrossRef CAS.
- B. Marciniec, E. Walczuk-Gusciora and C. Pietraszuk, Organometallics, 2001, 20, 3423 CrossRef CAS.
- B. Marciniec, I. Kownacki and M. Kubicki, Organometallics, 2002, 21, 3263 CrossRef CAS.
- M. Ramm and D. Selent, Acta Cryst., 1996, C52, 2703 CAS.
Footnote |
| † Stoichiometric reactions of 1 with vinyltrisubstituted silanes. (1) (0.035 g; 4.5 × 10−5 mole) was dissolved under argon in 0.6 ml of C6D6 in a NMR tube and CH2CHSi(OEt)3 0.170 g (0.9 mmol) was added to the solution. The run of reaction was controlled by 1H NMR and GCMS. The corresponding reactions of [{Ir(cod)(μ-OSiMe3)}2] with CH2CHSi(OMe)3 0.067 g (0.45 mmol), with CH2CHSi(OEt)Me2 (0.117 g; 0.9 mmol) and with CH2CHSi(OtBu)3 (0.114 g; 0.45 mmol) were carried out using the same procedure as that for CH2CHSi(OEt)3.CCDC 192733. See http://www.rsc.org/suppdata/cc/b2/b208603b/ for crystallographic data in CIF or other electronic format. |
| This journal is © The Royal Society of Chemistry 2003 |