Enantioselective molecularly imprinted polymers via ring-opening metathesis polymerisation
Alpesh Patel, Sandra Fouace† and Joachim H. G. Steinke*
Department of Chemistry, Imperial College of Science, Technology and Medicine, South Kensington, London, UK SW7 2AY
First published on 29th November 2002
Abstract
Enantioselective molecularly imprinted polymers (MIPs) have been synthesised via ROMP for the first time.
The considerable interest in molecularly imprinted polymers (MIPs) derives from their potential to perform as artificial enzymes and catalysts.1,2 Indeed for certain cases molecularly imprinted polymers (MIPs) have been shown to successfully mimic the recognition and catalysis behaviour associated with enzyme and antibody activity.1 Ever increasing effort is being devoted to investigate MIPs for a wide variety of applications involving highly selective molecular recognition events (such as affinity chromatography (enantiopolishing) and sensing layers (ELISA assays)). However, the way in which these polymers are being synthesised has remained essentially unchanged since the introduction of the free radically crosslinked network approach by Takagishi and Klotz3 and more ingeniously by Wulff et al. in the early 1970s.3
The process of forming a MIP is in many ways the nanoscale version of producing a plaster cast of a three-dimensional object (the template) (Fig. 1). MIPs are made via free radical polymerization in the presence of a template molecule. Removal of the template leaves behind cavities that are complementary to it in size, shape and electron density distributions. The functional monomers act as binding sites and self-assemble around the template prior to polymerisation. Once the template molecule has been removed these binding sites are arranged in a specific three-dimensional relationship very much in analogy to the specific arrangements of amino acid sidechains within the active site of an enzyme (Fig. 1).1 It is clear from these considerations that MIPs possess the potential to be developed into synthetic enzymes and antibodies. To date a serious downside to the application of MIPs has been lack of synthetic control. The statistical, kinetically driven nature of the network forming process makes it impossible to achieve monoclonality. Instead polyclonal cavities are being formed exhibiting a wide range of sizes and shapes and as a consequence a wide range of selectivities and/or catalytic activities thereby limiting the potential of MIPs drastically.1,2,4
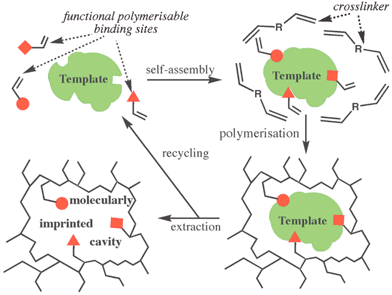 | ||
| Fig. 1 Molecularly imprinted polymers (MIPs)—The concept. | ||
A conceptual approach of addressing the issue of polyclonality in MIPs is to investigate the possibility of utilising thermodynamically controlled bond forming (polymerisation) reactions. The ability of silica (surfaces), which was used in early MIP syntheses as network forming material, to dynamically rearrange and reorganise itself in the presence of traces of water makes it eligible for such an approach.1 Implicitly Morihara et al., by allowing the surface of a silica network to form slowly around the template, elaborated successfully on this methodology some twenty years ago in a series of intriguing publications on ‘footprint’ catalysis.5 It seems that the brittle nature of silica and its susceptibility to surface rearrangements might have been reasons why other groups have not pursued this direction any further.1,3,6
Recent advances in the reactivity and functional group tolerance of ruthenium alkylidene catalysts7 and the concurrent development of the formation of highly crosslinked networks via ring-opening metathesis polymerisation with high conversion by Mühlebach et al.8 and Buchmeiser et al.,9 together with the incorporation of functional norbornene monomers by Feast et al.10 made us decide to evaluate ROMP (incl. cross metathesis) as a thermodynamically controlled network forming strategy. A complementary ‘small molecule’ strategy has been developed by Sanders et al., who recently applied ring-opening/ring-closing metathesis catalysis in the synthesis of templated supramolecular assemblies.11
In our case the template was covalently attached to a ROMP-polymerisable monomer, thereby ensuring maximum compatibility with the catalyst.7 Upon hydrolysis the covalent interaction is replaced by reversible non-covalent ones, an approach pioneered by Whitcombe et al.12 Thus monomer 2-L was synthesized from the chiral template L-menthol, 1-L (Scheme 1). Homopolymerisation, and copolymerisation of 2-L with norbornene, both to high conversions using Grubbs’ catalyst, provided us with the necessary evidence of monomer/catalyst compatibility. We also established that L-menthol could be hydrolysed from the copolymer almost quantitatively by using an excess of potassium trimethylsilanoate in THF.
![Synthesis of ROMP monomer 2-L. (i) Triphosgene, py, DCM, −70 °C → rt, 12 h. (not isolated) (ii) exo-N-hydroxy-7-oxabicyclo[2.2.1]hept-5-ene-2,3-dicarboximide, NEt3, DMF, −30 °C → rt, 12 h 76%.](/image/article/2003/CC/b208922j/b208922j-s1.gif) | ||
| Scheme 1 Synthesis of ROMP monomer 2-L. (i) Triphosgene, py, DCM, −70 °C → rt, 12 h. (not isolated) (ii) exo-N-hydroxy-7-oxabicyclo[2.2.1]hept-5-ene-2,3-dicarboximide, NEt3, DMF, −30 °C → rt, 12 h 76%. | ||
Finally we embarked on the synthesis of the MIPs, substituting norbornene with the crosslinker dicyclopentadiene essentially following reaction conditions established by Mühlebach et al.8 (Scheme 2). The polymer was obtained as a lightweight, brittle solid after 36 h at 80 °C in a sealed ampoule. The crosslinked network was crushed into small particles, extracted in a Soxhlet extractor and then dried (yield 99%). After lengthy optimisation a 10-fold excess of n-hexylamine and triethylamine allowed us to remove more than 90% (92–94%) of L-menthol (Scheme 2).
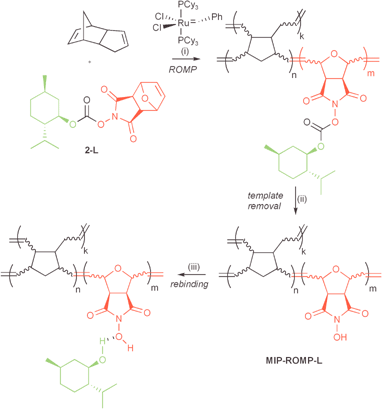 | ||
| Scheme 2 Synthesis of MIP-ROMP-L. (i) 5 mol% 2-L, 95 mol% dicyclopentadiene, 0.1 mol% Grubbs’ catalyst, DCM∶2-propanol, 80 °C, 36 h, 99%, (ii) 10 mol eq. NEt3, 10 mol eq. n-hexylamine, dioxane, rt, 12 h, 93%, (iii) for details see Table 1. | ||
| Supernatant | MIP | D,L-Methanol | Sep. factor | ||||
|---|---|---|---|---|---|---|---|
| Menthol | Menthol | Supernat. | MIP | ||||
| Polymer | L (%) | D (%) | L (%) | D (%) | [mmol] | [mmol] | α-Value |
| a 5.1 × 10−2 mmol of a 1∶1 mixture of L- and D-menthol in 10 ml of hexane∶chloroform (1∶1 (v/v)). | |||||||
| MIP-ROMP-L | 40.1 | 59.9 | 58.7 | 41.3 | 0.013 | 0.038 | 2.1 |
| MIP-ROMP-D | 58.7 | 41.3 | 40.8 | 59.2 | 0.012 | 0.039 | 2.1 |
| MIP-ROMP-DL | 49.6 | 50.4 | 49.4 | 50.6 | 0.013 | 0.038 | 1.0 |
| Pre-equilibration | 49.6 | 50.4 | n/a | n/a | 0.051 | n/a | n/a |
Molecular recognition studies were carried out as batch mode equilibrations. Preference was given to this analytical method as it measures the thermodynamic preference of the polymer for its template in line with the thrust of our investigation.2 The MIP was suspended in a 1∶1 mixture of hexane and chloroform containing equimolar amounts of L- and D-menthol. We established that 24 h were sufficient for the polymer to equilibrate fully. The supernatant was collected and the filtered polymer extracted exhaustively with chloroform to recover all menthol. The supernatant and the collected polymer extract were analysed by chiral GC (see Table 1).
It was satisfying to see that the template L-menthol was bound to its polymer preferentially. In fact the final composition on the polymer was 60% of L-menthol and 40% of D-menthol, which is equivalent to an enantiomeric excess (ee) of almost 20%. With about three quarters of the total amount of menthol absorbed by the MIP it is also possible to calculate the separation factor, which is a useful quantity when considering chromatographic investigations.13 A value of 2.1 indicates that the MIP outperforms many chiral stationary phases.13 Interestingly, one has to infer that recognition takes place as a result of a single accurately positioned alcohol functionality within a chirally imprinted cavity, since a three point binding model is usually invoked to rationalise chiral discrimination, though in this MIP only a single hydrogen bonding interaction can be present within each cavity.13
In earlier work imprinting with a single menthol enantiomer via the sol-gel process resulted in non-stereoselective MIPs.14 Percival et al. on the other hand prepared non-covalently L-menthol imprinted MIPs as sensing layer for a quartz crystal microbalance. A three-fold preference for the template molecule was established through separate binding assays so that a direct comparison with our data at this stage is not possible.15
To add further proof that the observed chiral discrimination is the result of cavities chirally imprinted by the template, we synthesised two related MIPs, MIP-ROMP-D and MIP-ROMP-DL. In MIP-ROMP-D the template monomer 2-L was replaced by its enantiomer, 2-D, and subsequently polymerised in identical fashion to MIP-ROMP-D. The other MIP (MIP-ROMP-DL) was synthesised in the presence of an equimolar mixture of 2-L and 2-D. MIP-ROMP-D exhibited the same level of chiral discrimination for its template (D-menthol) had been found for MIP-ROMP-L. MIP-ROMP-DL on the other hand showed no preference for either L- or D-menthol (Table 1). Both findings are fully consistent with our expectations and demonstrate that chiral recognition is controlled entirely through the presence of chirally imprinted cavities.
We are now investigating reaction conditions and catalysts to influence the selectivity distribution of the cavities found in these MIPs with the thrust of our investigation aimed at reducing polyclonality.
We would like to thank Fred Goldberg and Dr Alan Armstrong for access to and their assistance in the GC analyses, and Dr Guy Clarkson for helpful discussions. Funding by the Engineering and Physical Sciences Research Council (EPSRC), UK, is gratefully acknowledged.
Notes and references
- G. Wulff, Chem. Rev., 2002, 102, 1 CrossRef CAS.
- G. Wulff, Angew. Chem., Int. Ed. Engl., 1995, 34, 1812 CrossRef CAS.
- J. H. G. Steinke, D. C. Sherrington and I. R. Dunkin, Adv. Polym. Sci., 1995, 123, 81 Search PubMed.
- B. Wandelt, P. Turkewitsch, S. Wysocki and G. D. Darling, Polymer, 2002, 43, 2777 CrossRef CAS.
- K. Morihara, S. Doi, M. Takiguchi and T. Shimada, Bull. Chem. Soc. Jpn., 1993, 66, 2977 CAS.
- T. Shimada, R. Hirose and K. Morihara, Bull. Chem. Soc. Jpn., 1994, 67, 227 CAS.
- T. M. Trnka and R. H. Grubbs, Acc. Chem. Res., 2001, 34, 18 CrossRef CAS.
- A. Della Martina, J. G. Hilborn and A. Mühlebach, Macromolecules, 2000, 33, 2916 CrossRef CAS.
- F. Sinner and M. R. Buchmeiser, Macromolecules, 2000, 33, 5777 CrossRef CAS.
- P. J. Hine, T. Leejarkpai, E. Khosravi, R. A. Duckett and W. J. Feast, Polymer, 2001, 42, 9413 CrossRef CAS.
- S. J. Rowan, S. J. Cantrill, G. R. L. Cousins, J. K. M. Sanders and J. F. Stoddart, Angew. Chem., Int. Ed., 2002, 41, 898 CrossRef.
- M. J. Whitcombe, M. E. Rodriguez, P. Villar and E. N. Vulfson, J. Am. Chem. Soc., 1995, 117, 7105 CrossRef CAS.
- B. Sellergren, J. Chromatogr. A, 2001, 906, 227 CrossRef CAS.
- C. Pinel, P. Loisil and P. Gallezot, Adv. Mater., 1997, 9, 582 CAS.
- C. J. Percival, S. Stanley, M. Galle, A. Braithwaite, M. I. Newton, G. McHale and W. Hayes, Anal. Chem., 2001, 73, 4225 CrossRef CAS.
Footnote |
| † Current address: Université de Perpignan 52, Avenue de Villeneuve, 66 860 Perpignan, France. |
| This journal is © The Royal Society of Chemistry 2003 |