The elusive aldol reaction of enolates with aldolates—a highly stereoselective process using three different carbonyl components†
Michael
Schmittel
*a,
Andreas
Haeuseler
a,
Tom
Nilges
b and
Arno
Pfitzner
b
aFB 8 – OC1 (Chemie-Biologie), Universität Siegen, Adolf-Reichwein-Str, D-57068 Siegen, Germany. E-mail: schmittel@chemie.uni-siegen.de; Fax: (+49) 271 740 3270; Tel: (+49) 271 740 4356
bInstitut für Anorganische Chemie, Universität Regensburg, Universitätsstr. 31, D-93040 Regensburg, Germany
First published on 12th December 2002
Abstract
Three different carbonyl components are assembled to tetrahydropyran-2,4-diols by two successive diastereoselective aldol reactions.
Contrary to the ample usage of the aldol reaction in domino/tandem1 processes.2,3 its use in two consecutive aldol–aldol reactions is rare4 and often limited to trimerisation protocols.5 We have recently outlined the first examples of a highly diastereoselective and widely applicable one-pot domino–aldol–aldol–hemiacetal strategy using metal bisenolates (or polyenolates) 3 and various aldehydes 2 (Scheme 1, top route, R1 = R4; R2 = R5)6 yielding tetrahydropyran-2,4-diols 8 along the E1 + E1 + A route (using only one enol E1 and one aldehyde A). We now wish to report the first case of an E1 + E2 + A aldol–aldol protocol to yield structurally diversified tetrahydropyran-2,4-diols with up to 5 different groups R in a highly stereoselective manner.
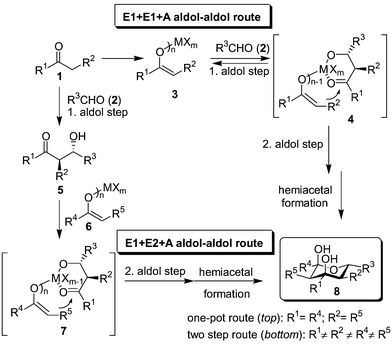 | ||
| Scheme 1 General concept for the synthesis of tetrahydropyran-2,4-diols by two successive aldol reaction steps (E1 and E2 denote the nucleophilic enolates, A the aldehyde component). | ||
As 4 is a plausible intermediate (the metal center coordinates both to the aldolate‡ and enolate) in the E1 + E1 + A reaction,6 we contemplated realising the elusive E1 + E2 + A aldol–aldol reaction via its structural analogue 7. In such an approach, however, one has to worry that rapid retro-aldol reaction, as observed in the E1 + E1 + A route (4 → 3 + 2), leads to a disastrous scrambling of the enol components, most likely the reason why any E1 + E2 + A reaction has been intangible so far.
Realistically, the E1 + E2 + A aldol–aldol reaction can only be orchestrated when (i) an adequate way to assemble the desired intermediate 7 is found, and (ii) a metal is met that renders the 2. aldol step (Scheme 1) more rapid than retro-aldol reaction. 7 may originate from the reaction of mono-aldolate 5·Li with metal enolate 6. (pathway 1, Scheme 2; X = leaving group) or alternatively from lithium enolate 10 and metal aldolate 9 (pathway 2). Independent of the pathway the aldolate must have the correct relative anti configuration as in the tetrahydropyran-2,4-diol.
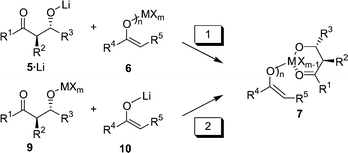 | ||
| Scheme 2 | ||
Following our earlier results,6 the influence of various metal fragments (MXm + n = TiCl4, TiCl4–Bu3N, Ti(OiPr)2Cl2, ZrCl4, SnCl4, InCl3, AlCl3, and ZnCl2) in the reaction of metal enolate 6a (R4 = Et, R5 = Me) with anti5a·Li7 (R1 = Ph, R2 = Me, R3 = Ph; d.e. = 75%) to afford 8a as the E1 + E2 + A product was explored (Scheme 3). From the metal fragments, only ZrCl4 (19%), SnCl4 (28%), InCl3 (7%) and ZnCl2 (14%) afforded 8a in some detectable yield.
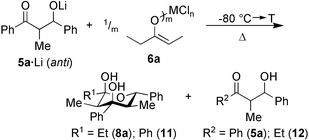 | ||
| Scheme 3 | ||
Most importantly, however, no retro-aldol cleavage of 5a was observed with SnCl4, whereas use of ZrCl4, InCl3, and ZnCl2 led additionally to tetrahydropyran-2,4-diol 11, propiophenone and β-hydroxyketone 12, in particular at higher temperatures. The formation of the latter compounds unequivocally indicates occurrence of the unwanted retro-aldol reaction. Thus, the reaction was optimized with SnCl4 varying the temperature, reaction time and stoichiometry. Finally, 8a was furnished in 63% at 40 °C, 4 h using SnCl4∶enolate∶monoaldolate = 1∶2∶2 attesting that two molecules of 8a form in the coordination sphere of one tin(IV) center. Further decrease of the SnCl4∶enolate ratio to 1∶5 failed to provide 8a, which precludes a catalytic route. Notably, all efforts to achieve the E1 + E2 + A reaction via pathway 2 (Scheme 2) proved far less successful.
The E1 + E2 + A product 8avia1H-NMR and X-ray structure analysis (Fig. 1) shows all alkyl and aryl substituents in the equatorial positions and both hydroxy groups axially. Typically, as already known from E1 + E1 + A products, the two methyl groups in 8a appear at high field (δ = 0.36 and 0.77 ppm).
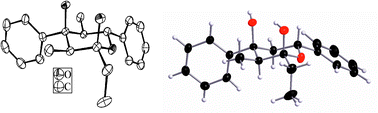 | ||
| Fig. 1 X-ray structure of 8a.ठEnantiomorphous crystals of 8a were received from EtOH (conglomerate). The ellipsoids (left) represent a probability factor of 50%; stick and ball representation (right). | ||
With a successful approach to 8a at hand, we now studied the reaction of 6a with aldolates 5a,b (for R1, R2 and R3, see Table 1) changing the syn∶anti ratio of the latter. Indeed, as predicted above, rather low yields of 8a, b, were received starting from syn enriched monoaldolates 5a,b while yields amounted to >50% with anti-aldolates as starting material (Table 2). Formation of 8b from syn-5b (entry d) is explained by partial syn → anti isomerisation of the β-hydroxyketone via a retro-aldol process, especially at elevated temperatures.7
| Entry | R1 | R2 | R3 | R4 | R5 | Product | Yielda (%) |
|---|---|---|---|---|---|---|---|
| a Isolated yields. b Reaction temperature = 0 °C. | |||||||
| 1 | Ph | Me | Ph | Et | Me | 8a | 63 |
| 2 | Ph | Me | iPr | Et | Me | 8b | 56 |
| 3 | Ph | Me | Ph | Ph | H | 8c | 53 |
| 4 | Ph | Me | iPr | Ph | H | 8d | 45 |
| 5 | Et | Me | Ph | Ph | Me | 8e | 43 |
| 6 | Et | Me | iPr | Ph | Me | 8f | 56 |
| 7 | Et | Me | Ph | Ph | H | 8g | 41 |
| 8 | Et | Me | iPr | Ph | H | 8h | 48 |
| 9 | Ph | H | Ph | Et | Me | 8i | 48b |
| 10 | Ph | H | iPr | Et | Me | 8k | 22b |
| 11 | Ph | H | Ph | Ph | Me | 8l | — |
| 12 | Ph | H | iPr | Ph | Me | 8m | — |
The general applicability of the concept was further explored by varying the enolates and aldehydes. Rewardingly, Table 1 documents that 10 out 12 desired E1 + E2 + A products could be prepared in a highly stereoselective manner. In no case were other diastereomeric tetrahydropyrandiols detected.
Some problems arise with β-hydroxyketones containing the acetophenone subunit as they easily dehydrate under the reacton conditions to afford α,β-unsaturated ketones. Dehydration could be minimized for entries 9 and 10 by reducing the reaction temperature to 0 °C. However, no formation of 8l,m was detected even at low temperatures (Table 1, entries 11 and 12).
A mechanistic rationale (Scheme 4) for these results has to acknowledge the anti configuration of the starting aldolate. Thus, to minimize steric interactions in the transition state for the 2. aldol step (TS1) a chair-twistboat conformation allows the bulky groups to assume a pseudo equatorial position. Similarly, 14 should be most stable in chair-boat conformation. Formation of the final hemiacetal viaTS2 should therefore be accompanied by a release of strain as all R1–R5 substituents move into equatorial positions.
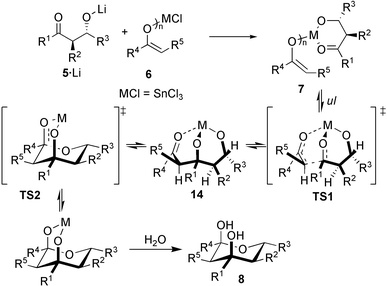 | ||
| Scheme 4 Mechanistic proposal for the formation of 8. | ||
In summary, a novel methodology is described for the highly stereoselective synthesis of tetrahydropyran-2,4-diols starting from simple carbonyl compounds in two sequential aldol reactions. The utility of the concept has been demonstrated preparing a variety of products from different alkyl and aryl ketones and aldehydes. Current investigations in our laboratories aim to use the diversified tetrahydropyran-2,4-diol structures as bisdentate ligands in metal catalysed reactions.
This work was supported by the Deutsche Forschungsgemeinschaft and by the Fonds der Chemischen Industrie.
Notes and references
- L. F. Tietze, Chem. Rev., 1996, 96, 115 CrossRef CAS.
- Aldol reaction as first step in a domino process: (a) P. Galatsis, S. D. Millan, P. Nechala and G. Ferguson, J. Org. Chem., 1994, 59, 6643 CrossRef CAS; (b) P. M. Bodnar, J. T. Shaw and K. A. Woerpel, J. Org. Chem., 1997, 62, 5674 CrossRef CAS; (c) H. W. Yang, C. X. Zhao and D. Romo, Tetrahedron, 1997, 53, 16471 CrossRef CAS; (d) G. W. Kabalka, D. Tejedor, N.-S. Li, R. R. Malladi and S. Trotman, J. Org. Chem., 1998, 63, 6438 CrossRef CAS; (e) L. Lu, H.-Y. Chang and J. -M. Fang, J. Org. Chem., 1999, 64, 843 CrossRef CAS; (f) C. Delas, O. Blacque and C. Moise, J. Chem. Soc., Perkin Trans. 1, 2000, 2265 RSC; (g) C. Schneider and M. Hansch, Chem. Commun., 2001, 1218 RSC.
- Aldol reaction as second step in a domino process: (a) T. Arai, H. Sasai, K. Aoe, K. Okamura, T. Date and M. Shibasaki, Angew. Chem., Int. Ed. Engl., 1996, 35, 104 CrossRef CAS; (b) B. L. Feringa, M. Pineschi, L. A. Arnold, R. Imbos and A. H. M. De Vries, Angew. Chem., Int. Ed. Engl., 1997, 36, 2620 CrossRef CAS; (c) M. Ono, K. Nishimura, Y. Nagaoka and K. Tomioka, Tetrahedron Lett., 1999, 40, 1509 CrossRef CAS; (d) X. Huang and M. H. Xie, Org. Lett., 2002, 4, 1331 CrossRef CAS; (e) P. Langer, N. N. R. Saleh and I. Freifeld, Chem. Commun., 2002, 168–169 RSC.
- F. Barba and J. L. de la Fuente, J. Org. Chem., 1996, 61, 8662 CrossRef CAS.
- (a) A. L. Henne and P. E. Hinkamp, J. Am. Chem. Soc., 1954, 76, 5147 CrossRef CAS; (b) R. A. Moore and R. Levine, J. Org. Chem., 1964, 29, 1439 CrossRef CAS; (c) F. G. Drakesmith, O. J. Stewart and P. Tarrant, J. Org. Chem., 1967, 33, 280; (d) P. Rollin, Bull. Soc. Chem. Fr., 1973, 1509 Search PubMed; (e) M. M. Dhingra and K. R. Tatta, Org. Magn. Reson., 1977, 9, 23 Search PubMed; (f) S.-S. Yun, I.-H. Suh, S.-S. Choi and S. Lee, Chem. Lett., 1998, 985 CrossRef.
- (a) M. Schmittel, M. K. Ghorai, A. Haeuseler, W. Henn, T. Koy and R. Söllner, Eur. J. Org. Chem., 1999, 2007 CrossRef CAS; (b) M. Schmittel and M. K. Ghorai, Synlett, 2001, 1992 CrossRef CAS.
- C. H. Heathcock, C. T. Buse, W. A. Kleschick, M. C. Pirrung, J. E. Sohn and J. Lampe, J. Org. Chem., 1980, 45, 1066 CrossRef CAS.
- (a) Y. Yoshida, R. Hayashi, H. Sumihara and Y. Tanabe, Tetrahedron Lett., 1997, 38, 8727 CrossRef CAS; (b) Y. Yoshida, N. Matsumoto, R. Hamasaki and Y. Tanabe, Tetrahedron Lett., 1999, 40, 4227 CrossRef CAS.
- G. M. Sheldrick, SHELX 97 Programs for the solution and refinement of crystal structures, University of Göttingen, Germany, 1997.
- H. D. Flack, Acta Crystallogr., Sect. A, 1983, 39, 876 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: synthesis and spectroscopic data for 8a. Crystallography for 8a. Fig. S1: crystal structure of 8a; Fig. S2: hydrogen bonding in 8a. See http://www.rsc.org/suppdata/cc/b2/b209536j/ |
| ‡ We use the expression aldolate also for a ketolate (= β-hydroxyketone). |
| § Crystal data for 8a: orthorhombic, space group Pna21 (No. 33), a = 10.9177(9), b = 17.2334(10), c = 9.4999(5) Å, V = 1787.4(2) Å3, Z = 4, ρcalc = 1.213 g cm−1, data collection: STOE IPDS, 27347 reflections, 4247 independent reflections, Rint = 0.0409, T = 173 K, Mo-Kα radiation (λ = 0.71069 Å), 2θmax = 56.22°, −14 ⩽ h ⩽ 14, −22 ⩽ k ⩽ 22, −12 ⩽ l ⩽ 12, crystal size 0.45 × 0.4 × 0.3 mm, no absorption correction, structure solution by direct methods, refinement against F2 (SHELX-979). The refinement of 322 parameters converged at R = 0.0292 and wR = 0.0732 (I > 2σ(I)) and R = 0.0324 and wR = 0.0746 (all reflections). Flack10 parameter 0.8(6). The absolute configuration could not be determined from X-ray. CCDC 163263. See http://www.rsc.org/suppdata/cc/b2/b209536j/ for crystallographic data in CIF or other electronic format. |
| This journal is © The Royal Society of Chemistry 2003 |