Asymmetric electrochemical lactonization of diols on a chiral 1-azaspiro[5.5]undecane N-oxyl radical mediator-modified graphite felt electrode
Yoshitomo
Kashiwagi
*a,
Futoshi
Kurashima
a,
Shinya
Chiba
a,
Jun-ichi
Anzai
a,
Tetsuo
Osa
a and
James M.
Bobbitt
b
aGraduate School of Pharmaceutical Sciences, Tohoku University, Aramaki, Aoba-ku, Sendai 980-8578, Japan. E-mail: kashi@mail.pharm.tohoku.ac.jp; Fax: +81-22-217-6840; Tel: +81-22-217-6842
bDepartment of Chemistry, University of Connecticut, Storrs, CT 06269-3060, USA
First published on 28th November 2002
Abstract
A graphite felt electrode modified with (6S,7R,10R)-4-amino-2,2,7-trimethyl-10-isopropyl-1-azaspiro[5.5]undecane N-oxyl was prepared for electrocatalytic oxidation of diols; electrolysis of diols on the modified electrode yielded optically active lactones (92.0–96.4%), with an enantiopurity of 82–99% ee.
The preparation of optically active lactones is of importance for the synthesis of chiral bioactive compounds and functional materials. 2,2,6,6-Tetramethylpiperidin-1-yloxyl (TEMPO) is known to be an effective redox mediator for the chemical or electrochemical oxidation of diols to lactones.1–10 Bobbitt et al. demonstrated the asymmetric oxidation of cis-1,2-cyclohexanedimethanol to the corresponding optically active lactones using 4-acetylamino-2,2,7-trimethyl-10-isopropyl-1-azaspiro[5.5]undecane N-oxyl (4-acetylamino-SPIROXYL) as a chiral nitroxyl radical via a non-electrochemical method.11 In order to construct a clean and simple reaction system, we report here the first efficient asymmetric electrocatalytic oxidation of diols to the corresponding optically active lactones on a SPIROXYL-modified graphite felt (GF) electrode.
Four isomers of optically active 4-amino-SPIROXYL were prepared by the reaction of 2,2,4,4,6-pentamethyl-2,3,4,5-tetrahydropyrimidine with (+)-dihydrocarvone as the starting material.11 The SPIROXYL-modified GF electrode was prepared in a similar manner as in the preparation of a TEMPO-modified GF electrode12 by attaching 4-amino-SPIROXYL to the carboxyl groups of a thin poly(acrylic acid) (PAA) layer coated on GF.13Fig. 1 shows the cyclic voltammogram (CV) of a (6S,7R,10R)-SPIROXYL-modified GF electrode, in which a reversible redox couple was observed. This redox couple corresponds to the one-electron oxidation of nitroxyl radical to oxoammonium ion. The immobilized (6S,7R,10R)-SPIROXYL on the electrode surface was quite stable and no deactivation was observed in CV after repeated potential scanning. The oxidation potential was found at +0.58 V vs. Ag/AgCl and the peak separation between the positive and negative peak potentials was 65 mV. The amount of electroactive (6S,7R,10R)-SPIROXYL on the electrode surface as determined by integrating the oxidation current peak of the CV and applying Faraday’s law was ca. 8.4 × 10−6 mol cm−3. This means that ca. 20% of the carboxyl groups of the PAA layer on the GF electrode were modified with (6S,7R,10R)-SPIROXYL. When 3-methyl-1,5-pentanediol was added to the electrolytic solution bathing the (6S,7R,10R)-SPIROXYL-modified GF electrode, an increase of the nitroxyl radical oxidation current was observed (Fig. 1). The oxoammonium ion reduction current disappeared, and the new oxidation peak current was proportional to the concentration of 3-methyl-1,5-pentanediol. The ratio of this new oxidation peak’s height to the reversible nitroxyl radical oxidation peak height decreased as the scan rate increased. 3-Methyl-1,5-pentanediol is not oxidized directly on a bare GF electrode below +1.0 V vs. Ag/AgCl. All these results are characteristic of electrochemical catalysis14,15 of the oxidation of 3-methyl-1,5-pentanediol. Similar results were seen with the other diols used.
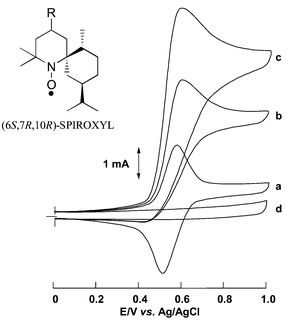 | ||
| Fig. 1 Cyclic voltammograms of a (6S,7R,10R)-SPIROXYL-modified GF electrode (1.0 × 1.0 × 0.5 cm) at 50 mV s−1 in 0.1 M NaClO4/CH3CN: with (a) 0 M 3-methyl-1,5-pentanediol; (b) 0.1 M 3-methyl-1,5-pentanediol and 0.2 M 2,6-lutidine; (c) 0.2 M 3-methyl-1,5-pentanediol and 0.2 M 2,6-lutidine; (d) 0.1 M 3-methyl-1,5-pentanediol on a bare GF electrode. | ||
Preparative potential-controlled electrolysis was performed on the (6S,7R,10R)-SPIROXYL-modified GF electrode (1.0 × 1.0 × 0.5 cm) in MeCN solution, using an ‘H’ type divided cell separated by a cationic exchange membrane (Nafion 117). The anolyte contained 0.5 mmol of substrate, 2 mmol of 2,6-lutidine as a deprotonating agent, 0.5 mmol of tetralin as a chromatographic standard and 0.5 mmol of NaClO4 as a supporting electrolyte in a total volume of 5 ml. The catholyte was 5 ml of MeCN solution containing 0.5 mmol of NaClO4. The electrolysis was carried out at +0.8 V vs. Ag/AgCl. During electrolysis, aliquots of anolyte were analyzed occasionally by GC† and HPLC.‡ The consumption of 3-methyl-1,5-pentanediol and formation of 3-methyl-δ-valerolactone are plotted against electrolysis time in Fig. 2. After 10 h of electrolysis, 3-methyl-1,5-pentanediol was oxidized to the (R)- and (S)-forms of 3-methyl-δ-valerolactone in 1.0% and 95.4% yield, respectively. Thus, the ee of the formed lactone was 98%. The current efficiency and turnover number (given by ratio of mole of product × 4/mol of (6S,7R,10R)-SPIROXYL) were 97.0% and 459.0, respectively, at 10 h of electrolysis. The catalytic activity of the modified electrode remained high after several runs.
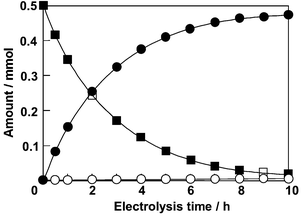 | ||
| Fig. 2 Macroelectrolysis of 3-methyl-1,5-pentanediol using the (6S,7R,10R)-SPIROXYL-modified GF electrode in the presence of 2,6-lutidine: (■) 3-methyl-1,5-pentanediol, (○) (R)-3-methyl-δ-valerolactone and (●) (S)-3-methyl-δ-valerolactone. | ||
When we carried out the oxidation reaction of 3-methyl-1,5-pentanediol on a bare GF with (6S,7R,10R)-4-acetylamino-SPIROXYL in solution or in a homogeneous chemical system under similar conditions, the stereoselectivity was rather poor (Table 1). Thus, this asymmetric oxidation reaction was achieved only on the (6S,7R,10R)-SPIROXYL-modified GF electrode.
| Method | Current efficiency (%) | Ee (%) | Isolated yield (%) | Turnover number |
|---|---|---|---|---|
| a In the presence of 0.5 mmol 3-methyl-1,5-pentanediol and 2 mmol 2,6-lutidine in each reaction. b 0.05 mmol (6S,7R,10R)-4-acetylamino-SPIROXYL. c Ref. 11. | ||||
| Electrocatalysis on (6S,7R,10R)-SPIROXYL-modified GF | 97.0 | 98 | 96.4 | 459.0 |
| Electrocatalysis on bare GFb | 78.5 | 38 | 86.5 | 34.7 |
| Reagent oxidationc | — | 16 | 85.0 | 34.0 |
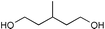
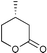
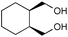
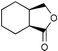
The results from the oxidation reactions of a variety of diols are shown in Table 2. (S)-(+)-1,4-Pentanediol was converted to (S)-(−)-2-methyl-γ-butyrolactone in high enantiomeric excess of 99%. cis-1,2-Cyclohexanedimethanol was also oxidized to the corresponding cis-(1R,6S)-(+)-8-oxabicyclo[4.3.0]nonan-7-one in an enantioselectivity of 82%. They were oxidized to the corresponding lactones in 94.9–97.0% current efficiency and 92.0–96.4% yield. The turnover numbers based on (6S,7R,10R)-SPIROXYL are greater than 430. (S)-(+)-1,3-Butanediol did not lead directly to the corresponding β-lactone; however, it was oxidized to a optically active hydroxyaldehyde without loss of optical purity (99% ee). The current efficiency and turnover number (given by ratio of mole of product × 2/mol of (6S,7R,10R)-SPIROXYL) were 84.6% and 191.4, respectively. This hydroxyaldehyde was oxidized directly with sodium chlorite to the corresponding hydroxy acid in 80.4% yield to give an unstable compound, and the closure of the hydroxy acid to β-lactone was carried out using PhSO2Cl (Scheme 1).16
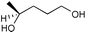
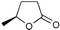
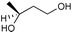
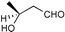
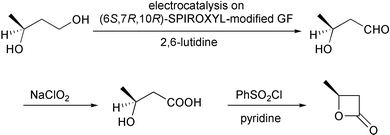 | ||
| Scheme 1 | ||
We have described the first efficient, asymmetric oxidation of a number of diols using a chemically modified electrode. Electrolysis on the (6S,7R,10R)-SPIROXYL-modified GF electrode gave selectively (S)-isomers of lactones. We are now investigating the other isomer-modified GF electrodes for the asymmetric oxidation of diols.
This work was supported in part by the Foundation for Earth Environment, and by a Grant-in-Aid for Scientific Research on Encouragement Research (No. 13750792) from the Ministry of Education, Culture, Sports, Science and Technology of Japan.
Notes and references
- T. Miyazawa and T. Endo, J. Org. Chem., 1985, 50, 3930 CrossRef CAS.
- T. Miyazawa and T. Endo, J. Mol. Catal., 1985, 32, 357 CrossRef CAS.
- P. L. Anelli, S. Banfi, F. Montanari and S. Quici, J. Org. Chem., 1989, 54, 2970 CrossRef CAS.
- T. Inokuchi, S. Matsumoto, T. Nishiyama and S. Torii, J. Org. Chem., 1990, 55, 462 CrossRef CAS.
- R. Siedlecka, J. Skarzewski and J. Mlochowski, Tetrahedron Lett., 1990, 31, 2177 CrossRef CAS.
- Z. Ma and J. M. Bobbitt, J. Org. Chem., 1991, 56, 6110 CrossRef CAS.
- M. F. Semmelhack, C. S. Chou and D. A. Cortes, J. Am. Chem. Soc., 1983, 105, 4492 CrossRef CAS.
- T. Inokuchi, S. Matsumoto and S. Torii, J. Org. Chem., 1991, 56, 2416 CrossRef CAS.
- T. Osa, Y. Kashiwagi, K. Mukai, A. Ohsawa and J. M. Bobbitt, Chem. Lett., 1990, 75 CAS.
- H. Tanaka, Y. Kawakami, K. Goto and M. Kuroboshi, Tetrahedron Lett., 2001, 42, 445 CrossRef CAS.
- Z. Ma, Q. Huang and J. M. Bobbitt, J. Org. Chem., 1993, 58, 4837 CrossRef CAS.
- Y. Kashiwagi, H. Ono and T. Osa, Chem. Lett., 1993, 257.
- T. Osa, Y. Kashiwagi, M. Nakamura and M. Ohba, JP Pat., 12 776, 1999.
- R. W. Murray, in Electroanalytical Chemistry, vol. 13, ed. A. J. Bard, Marcel Dekker, New York, 1984, p. 191 Search PubMed.
- R. W. Murray, Molecular Design of Electrode Surfaces, Techniques of Chemistry Series 22, ed. R. W. Murray, Wiley-Interscience, New York, 1992 Search PubMed.
- P. M. Wovkulich, K. Shankaran, J. Kiegiel and M. R. Uskoković, J. Org. Chem., 1993, 58, 832 CrossRef CAS.
Footnotes |
| † The GC analysis was carried out using a CP-Cyclodextrin-B-2,3,6-M-19 capillary column (0.25 mm ϕ × 25 m). The column temperature increased at 3 °C min−1 from 80 to 150 °C. The injection and detector temperatures were constant at 200 and 240 °C, respectively. |
| ‡ The HPLC analysis was carried out using a Daisel CHIRALCEL® OD column (4.6 mm ϕ × 250 mm). The column temperature was constant at 40 °C. The analytes were eluted by PriOH–n-hexane (2∶33) at 0.7 ml min−1 flow rate, and detected by UV absorption at 254 nm. |
| This journal is © The Royal Society of Chemistry 2003 |