Anion-sealed single-molecule capsules†
Jerry L.
Atwood
* and
Agnieszka
Szumna
Department of Chemistry, University of Missouri-Columbia, Columbia MO 65211, USA. Fax: +1 573 882 2754; Tel: +1 573 882 2730E-mail: atwoodj@missouri.edu
First published on 31st March 2003
Abstract
A new approach to anion recognition utilizing electrostatic and hydrogen bonding interactions has been demonstrated by placement of the whole ion-pair in a molecular capsule.
Introduction
Non-covalent interactions have played a dominant role in the development of molecular capsules.1 Dimers,2 trimers,3 tetramers,4 hexamers,5 or larger supramolecular assemblies6 have proved useful in confining cationic,6 neutral,7 and anionic guests.8 For the construction of effective anion receptors many kinds of non-covalent interactions have been applied.9 Among them, the most commonly used are ion–ion, ion–dipole (hydrogen bonding), and Lewis acid–base interactions (coordination). All of these have advantages and disadvantages for anion complexation. Electrostatic interactions are strong, but not directional; hydrogen bonding is directional, but usually not strong enough to compete with multiple solvation effects in polar protic media. The straightforward solution for anion complexation seems to be a combination of these two interactions. Some examples of such a combination have been reported. For example, Beer and coworkers constructed many transition metal complex–amide based receptors.10 Steed and coworkers synthesized receptors containing positively charged pyridinium groups and hydrogen bonding amino groups.11 Both of these approaches may be viewed as in Fig. 1a.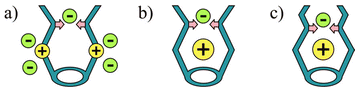 | ||
| Fig. 1 Receptors based on electrostatic (yellow) and hydrogen bonding interactions (pink): 1a shows possible positions for anions. | ||
In our approach, we also combine electrostatic interactions and hydrogen bonding. However, the positively charged site, which is responsible for the electrostatic driving of an anion into the binding site, is not covalently bound to the receptor shell, but resides inside the pocket (Fig. 1b,c). This makes it easier to separate the charged site from the environment and thus restrict possible pathways of anion approach, i.e.
“directionalize” the electrostatic interaction. This method of anion binding may also be considered in terms of ion-pair recognition.12,13 A recent example13 of such ion-pair recognition utilized a metallocrown rim capable of selective binding of Li+
(inside) and F−. The selectivity gate was considered to operate on the basis of steric hindrance. In this contribution, we present a new system, based on more fragile interactions (cation⋯π and hydrogen bonding), which is effective and selective for ion complexation.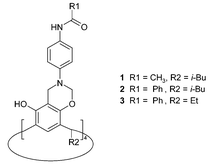
Previously, we reported that 1 forms very stable complexes with tetramethylammonium salts (TMAX).14 The Me4N+ cation resides deeply inside the cavity, held by multiple C–H⋯π interactions with eight surrounding aromatic rings. The cavity appears to be completely selective for Me4N+, since we were not able to obtain complexes with any other ammonium or metal cations. The position of the anion is solvent-dependent for 1: in chloroform Cl− resides at the top and is hydrogen bonded; in the presence of methanol, the anion is no longer bound. The crystal structure, 1·TMACl, provides an explanation of these solution observations, since it is clear that the Cl− is exposed to interaction with the solvent (Fig. 2).
 | ||
| Fig. 2 X-Ray structure of 1·TMACl. Chloride anion (green) interacts with Me4N+ cation, amide hydrogen atoms and with a CHCl3 molecule.14 | ||
Based on the information provided by the 1·TMACl structure, we have now designed and synthesized compound 2, as a second-generation anion receptor. In order to restrict solvent access to the anion, in 2 we have introduced bulky groups at the upper rim of the macrocycle.
Compound 2 can be synthesized in high yield using the Mannich reaction in a 1∶1 mixture of methanol and chloroform. When powdered 2 is stirred overnight in methanol in the presence of Me4NX salts (X = Cl, Br, I), the solid 1∶1 complexes are formed (2·TMAX). The spectral characteristics of the complexes in chloroform are similar to those reported earlier for 1·TMAX, i.e., the spectra indicate binding of the ion pair by the receptor. However, in this case, contrary to that for 1, the addition of methanol (10–50% v/v) does not remove the anion from its binding site. Further verification that the anion remains bound was obtained from a comparison experiment. Two complexes, 2·TMACl and 2·TMABr, were used and the spectrum of the mixture was recorded. In pure chloroform two sets of signals corresponding to two different complexes are visible (Fig. 3a). This means that the complexes are thermodynamically and kinetically stable on the NMR time scale (kexchange << 60 s−1 at 300 K). After addition of methanol (10% v/v, Fig. 3b) only one set of signals can be observed. This means that either the anions were released and now all complexed species are identical (2·TMA+) or that there is a fast anion exchange between complexes. Subsequent experiments (Fig. 3c) showed that signals could be separated at lower temperatures. The more methanol added, the lower the temperature required for separation of the peaks. Thus, methanol accelerates exchange of anions, but does not destroy the anion complex.
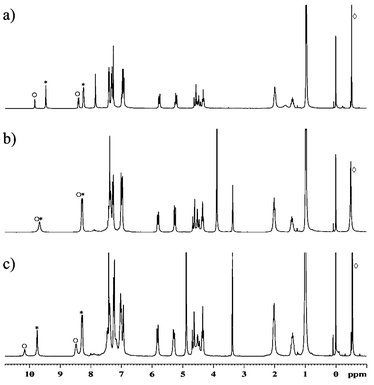 | ||
| Fig. 3 NMR spectra of the mixture of 2·TMACl(○) and 2·TMABr(*) (300 MHz, ⋄ TMA+). a) in CDCl3, 300 K; b) CDCl3–MeOH 9∶1, 300 K; c) CDCl3–MeOH 9∶1, 253 K. | ||
In order to gain insight into the anion selectivity of 2·TMA+ we have measured the relative association constants in chloroform against 2·TMAI as the reference complex. We expected this complex to have a moderate anion association constant and reasonable solubility (ClO4− and PF6− containing complexes displayed poor solubility). The results show the remarkable preference for spherical halide anions. After addition of one equiv. of (n-Bu)4NCl to the reference complex, it is quantitatively transformed into 2·TMACl, which means that the selectivity factor must be higher than 103. For bromide the selectivity factor was calculated as 25. No change was detected after addition of 5 equiv. of (n-Bu)4NClO4.
To estimate the extraction potential of the system, we have checked the ability of 2·TMA+ to extract various anions from water to an organic phase (chloroform). A 20 mM solution of 2 in CDCl3 was stirred together with a water solution containing a mixture of salts (NaCl, NaBr, TMAI, NaAcO, NaH2PO4, NaNO3, NaClO4 — 0.1 M each, at least one salt is used as TMAX in order to supply the appropriate cation). Since in chloroform different complexes are easily recognizable (slow exchange) the competition extraction can be followed by NMR spectroscopy. After 24 h of equilibration no free 2 can be detected. Instead, integration of the spectra indicates quantitative formation of 2·TMAX, X = Cl− (5% mol/mol), Br− (30%) and I− (65%). Thus the extraction experiment confirms the selectivity of 2·TMA+ towards halides. However, the amounts of extracted halides do not follow the selectivity trend in chloroform. Instead, the order agrees with the Hofmeister series15 — larger and more charge-diffuse anions are extracted better to the organic phase.
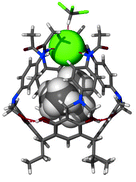
Anion confinement by the receptor has been verified by an X-ray structural analysis.‡ In the structure of 3·TMACl the anion remains in proximity to the Me4N+ cation, and is symmetrically bound by all four NHamide hydrogen bonds (Fig. 4, N–H⋯Cl distances are 2.5–2.6 Å). Additionally, from the top, four aromatic hydrogens (from ortho- positions) are in close proximity to the chloride (C–H⋯Cl distances are 2.7–3.1 Å). This weak interaction also adds to the overall binding efficiency and selectivity. Indeed, it is seen that the anion effectively seals the molecular capsule. The view from the top (Fig. 4b) also reveals the depth to which the anion is embedded and isolated from the environment by the bulky phenyl groups.
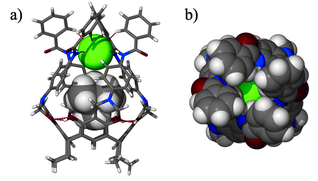 | ||
| Fig. 4 Crystal structure of 3·TMACl (obtained from the mixture of CHCl3–nitrobenzene as 3·TMACl·4.5PhNO2). a) Side view with receptor molecule in stick representation, guest as van der Waals spheres; b) top view — space filling (Cl− green). | ||
In conclusion, we have proved that ion pair recognition can be achieved based on the sum of weak interactions. Further, the cationic site inside the cavity, even though not covalently bound, remains remarkably stable over a variety of conditions. Of the solvents studied, cation release is found only in DMSO.
We are grateful to the National Science Foundation for support of this research.
Notes and references
- For reviews see: L. R. MacGillivray and J. L. Atwood, Angew. Chem., Int. Ed. Engl., 1999, 38, 1018 Search PubMed; J. Rebek Jr., Chem. Commun., 2000, 637 CrossRef CAS; V. Bohmer and M. O. Vysotsky, Aust. J. Chem., 2001, 54, 671 RSC; D. A. Rudkevich, Bull. Chem. Soc. Jpn., 2002, 75, 393 CrossRef CAS.
- R. S. Meissner, J. Rebek Jr. and J. de Mendoza, Science, 1995, 270, 1485 CAS; J. de Mendoza, Chem. Eur. J., 1998, 4, 1373 CrossRef CAS; K. N. Rose, L. J. Barbour, G. W. Orr and J. L. Atwood, Chem. Commun., 1998, 407 RSC; J. L. Atwood, L. J. Barbour, P. J. Nichols, C. L. Raston and C. A. Sandoval, Chem. Eur. J., 1999, 5, 990 CrossRef CAS; A. Shivanyuk and J. Rebek Jr., Chem. Commun., 2001, 2374 RSC.
- C. L. Raston, J. L. Atwood, P. J. Nichols and I. B. N. Sudria, Chem. Commun., 1996, 2615 RSC.
- D. W. Johnson, F. Hof, P. M. Iovine, C. Nuckolls and J. Rebek Jr., Angew. Chem., Int. Ed. Engl., 2002, 41, 3793 CrossRef CAS.
- L. R. MacGillivray and J. L. Atwood, Nature, 1997, 389, 469 CrossRef CAS; T. Gerkensmeier, W. Iwanek, C. Agena, R. Frolich, S. Kotila, C. Naher and J. Mattay, Eur. J. Org. Chem., 1999, 64, 2257 CrossRef; J. L. Atwood, L. J. Barbour and A. Jerga, Chem. Commun., 2001, 2376 RSC; J. L. Atwood, L. J. Barbour and A. Jerga, J. Supramol. Chem., 2001, 1, 131 Search PubMed.
- G. W. Orr, L. J. Barbour and J. L. Atwood, Science, 1999, 285, 1049 CrossRef.
- J. L. Atwood, L. J. Barbour and A. Jerga, Science, 2002, 296, 2367 CrossRef CAS; J. L. Atwood, L. J. Barbour, A. Jerga and B. L. Schottel, Science, 2002, 298, 1000 CrossRef CAS.
- O. Hayashida, A. Shivanyuk and J. Rebek Jr., Angew. Chem., Int. Ed. Engl., 2002, 41, 3423 CrossRef CAS.
- A. Bianchi, K. Bowman-James and E. Garcia-Espana, Supramolecular Chemistry of Anions, Wiley-VCH, New York, 1997 Search PubMed; J. W. Steed and J. L. Atwood, Supramolecular Chemistry, Wiley, Chichester, 2000 Search PubMed.
- P. D. Beer and P. A. Gale, Angew. Chem., Int. Ed., 2001, 40, 486 CrossRef.
- L. O. Abouderbala, W. J. Belcher, M. G. Boutelle, P. J. Cragg, J. Dhaliwal, M. Fabre, J. W. Steed, D. R. Turner and K. J. Wallace, Chem. Commun., 2002, 358 RSC.
- J. M. Mahoney, A. M. Beatty and B. D. Smith, J. Am. Chem. Soc., 2001, 123, 5847 CrossRef CAS; H. Piotrowski, G. Hilt, A. Schulz, P. Mayer, K. Polborn and K. Severin, Chem. Eur. J., 2001, 7, 3196 CrossRef CAS; T. Tuntulani, S. Poompradub, P. Thavornyutikarn, N. Jaiboon, V. Ruangpornvisuti, N. Chaichit, Z. Asfari and J. Vicens, Tetrahedron Lett., 2001, 42, 5541 CrossRef CAS; A. Arduini, E. Brindani, G. Giorgi, A. Pochini and A. Secchi, J. Org. Chem., 2002, 123, 5847.
- M. L. Lehaire, R. Scopelliti, H. Piotrowski and K. Severin, Angew. Chem., Int Ed. Engl., 2002, 41, 1419 CrossRef CAS.
- J. L. Atwood and A. Szumna, J. Am. Chem. Soc., 2002, 124, 10646 CrossRef CAS.
- F. Hofmeister, Arch. Exp. Pathol. Pharmakol., 1888, 24, 247 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details. See http://www.rsc.org/suppdata/cc/b3/b301511d/ |
| ‡ Crystal data for 3·TMACl: C126.50H118.50ClN13.50O21 (3 × TMACl × 4.5PhNO2), M = 2199.30, monoclinic, P21/c (No. 14), a = 18.891(7), b = 15.913(6), c = 37.075(14) Å, β = 99.032(7)°, V = 11007(7) Å3, Z = 4, Dc = 1.327 g cm−3, µ = 0.115 mm−1, F000 = 4628, SMART CCD, λ = 0.71073 Å, T = 173(2) K, 2θmax = 45.2°, 28678 reflections collected, 14428 unique (Rint = 0.167). Final GOF = 1.199, R1 = 0.1307, wR2 = 0.2865, R indices based on 5261 reflections with I >2σ(I) (refinement on F2), 1385 parameters, 10 restraints. The Rint value given includes all data measured up to an angle 2θ = 45°. This large value is caused by a high proportion of reflections of very low intensity in the high-angle range. If, for example, only reflections up to an angle of 2θ = 30° are taken into consideration, then Rint value decreases to a value of 0.086. CCDC 203670. See http://www.rsc.org/suppdata/cc/b3/b301511d/ for crystallographic data in .cif or other electronic format. |
| This journal is © The Royal Society of Chemistry 2003 |