Thermodynamically controlled cyclisation reactions with double phenylsulfanyl migration
Julie
Carlisle
,
David J.
Fox
and
Stuart
Warren
*
University Chemical Laboratory, Lensfield Road, Cambridge, UK CB2 1EW. E-mail: sw134@cam.ac.uk
First published on 30th September 2003
Abstract
Enantiomerically enriched C2-symmetric tetrols were synthesised by a route involving a ‘self-metathesis’ reaction with Grubbs' second-generation ruthenium catalyst; these tetrols produced interesting bicyclic products when rearranged under acidic conditions.
The acid-catalysed generation of episulfonium ions from β-phenylsulfanyl alcohols has been extensively studied by our research group as a useful method for the synthesis of saturated oxygen heterocycles.1 In particular, competitive cyclisation reactions of compounds in which there is more than one possible internal nucleophile have been investigated; the thermodynamic product is always formed under acidic conditions and the structure of this product is dependent on both stereochemistry and the degree of substitution around the ring.
Most recently, triols such as 1 were synthesised and cyclisation of these triols under thermodynamic control resulted exclusively in tetrahydrofuran (THF) formation; the alternative tetrahydropyran (THP) product was not formed.2 This was attributed to the presence of the gem-dimethyl migration origin, which destabilises the THP relative to the THF due to the unfavourable diaxial interactions which would be present in the six-membered ring.
We subsequently wished to investigate further levels of competition in molecules such as tetrols 2 and 3 (Fig. 1). In the cyclisation of these molecules, there is potentially competition between two internal nucleophiles and between two episulfonium ions.
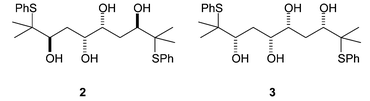 | ||
| Fig. 1 Structures of 2 and 3. | ||
The required tetrols were synthesised in enantiomerically enriched form by a short synthetic route starting from aldehyde 5 and involving a ‘self-metathesis’ reaction using Grubbs' second-generation ruthenium catalyst 4 (Fig. 2) as a key step (Scheme 2).3 The second-generation catalyst is tolerant of the sulfide functionality in these molecules, unlike the first-generation ruthenium catalysts which are completely deactivated by the sulfur. This result was also recently reported by Nolan et al.4 The resulting disubstituted alkene 6, resulting from PCC oxidation, could be recrystallised to give pure E isomer, which was dihydroxylated using Sharpless' (DHQD)2PHAL ligand to give diol 7 in excellent enantiomeric excess.5 This diol was then converted to tetrol 2, via a 1,3-anti stereoselective reduction,6 or to tetrol 3, via a 1,3-syn stereoselective reduction.7
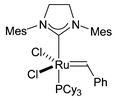 | ||
| Fig. 2 Grubbs' second generation metathesis catalyst 4. | ||
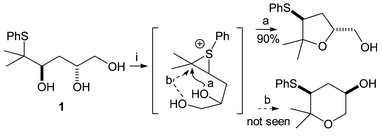 | ||
| Scheme 1 Reagents: i. TsOH, CH2Cl2, reflux. | ||
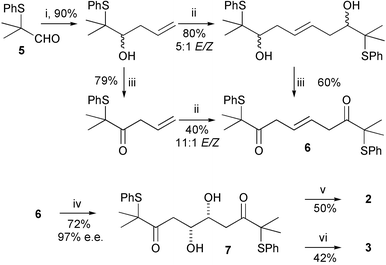 | ||
Scheme 2
Reagents: i. CH2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHCH2MgCl, THF, rt; ii. Ru catalyst 4, 65 °C; iii. PCC, CH2Cl2, rt. iv. K2OsO4.2H2O, (DHQD)2PHAL, K2CO3, K3Fe(CN)6, MeSO2NH2, tBuOH–H2O, 0 °C; v. Me4NBH(OAc)3, AcOH–MeCN, −20 °C, 7 days; vi. Et2BOMe, NaBH4, THF–MeOH, −78 °C. CHCH2MgCl, THF, rt; ii. Ru catalyst 4, 65 °C; iii. PCC, CH2Cl2, rt. iv. K2OsO4.2H2O, (DHQD)2PHAL, K2CO3, K3Fe(CN)6, MeSO2NH2, tBuOH–H2O, 0 °C; v. Me4NBH(OAc)3, AcOH–MeCN, −20 °C, 7 days; vi. Et2BOMe, NaBH4, THF–MeOH, −78 °C. | ||
Based on the previous results with the analogous triols we expected that a bis-THF would be formed in each case, by a mechanism like that in Scheme 1, involving reversible episulfonium formation and capture by the remaining hydroxyl groups.
Anti,syn,anti-tetrol 2 behaved as expected (Scheme 3). After prolonged reflux in DCM in the presence of acidic Amberlyst-15 resin, a single product had been formed in quantitative yield and this was the bis-THF 9. This product was also exclusively formed from the cyclisation of tetrol 8†, which produces the same episulfonium ion intermediate. This kind of bis-THF motif is common among biologically active natural products, most notably the annonaceous acetogenins.8
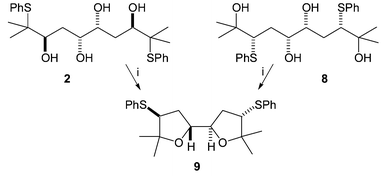 | ||
| Scheme 3 Reagents: i. Amberlyst, CH2Cl2, reflux, 72 h. | ||
On one occasion some of the Z isomer of alkene 6 was carried through the AD/anti reduction sequence as an impurity. In this case a small amount of another cyclic product crystallised out from the cyclisation reaction mixture. This was shown by X-ray crystallography to be the centrosymmetric trans-fused bis-THP 10 (Fig. 3), which results from cyclisation of the corresponding anti meso-tetrol. A similar bis-THP was desymmetrised by Nelson in a formal synthesis of hemibrevetoxin B.9 Resubmitting this bis-THP to the cyclisation conditions resulted in its conversion to an equilibrium mixture with mesobis-THF 11 (Scheme 4).
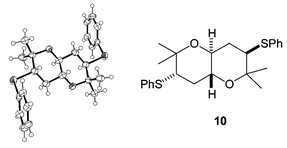 | ||
| Fig. 3 Centrosymmetric by-product. | ||
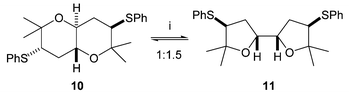 | ||
| Scheme 4 Reagents: i. Amberlyst, CDCl3, 60 °C. | ||
Cyclisation of the all syn tetrol 3 produced a mixture of products (Scheme 5). Under the same conditions as those used initially for tetrol 2 (Amberlyst, CH2Cl2, reflux) a 1 ∶ 1 equilibrium mixture of bis-THF 12 and fused bis-THP 13 was formed. The two products could be easily separated by chromatography, and re-submitting either product to the original reaction conditions resulted in the same 1 ∶ 1 ratio. The structure of the cis-fused bis-THP 13 was confirmed by X-ray crystallography (Fig. 4).
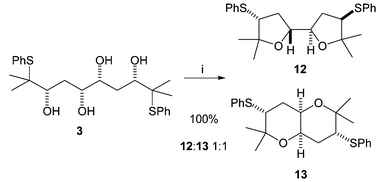 | ||
| Scheme 5 Reagents: Amberlyst, CH2Cl2, reflux, 72 h. | ||
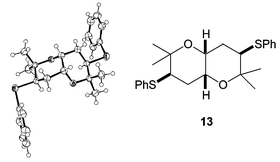 | ||
| Fig. 4 Crystal structure of THP 13. | ||
We were very interested to observe that the position of the equilibrium was affected by both temperature and solvent, as summarised in Table 1. In a less polar solvent (toluene), the fused bis-THP 12 becomes the major product (in marked contrast with the cyclisation of diastereomeric tetrol 2 and with our earlier work on triols such as 1), whereas increasing the temperature favours the bis-THF 13. This is consistent with the bis-THF being more polar and higher in entropy than the fused bis-THP. From this data estimates of ΔH and ΔS for the reaction could be calculated (ΔH −10.5 kJ mol−1, ΔS −24.3 J mol−1 K−1).
This property can be exploited to synthesise either ring system in useful synthetic yields from the same tetrol starting material, by choosing appropriate reaction conditions. The unwanted isomer can be recycled to further increase the overall yield.
In conclusion, the acid-catalysed phenylsulfanyl migration reaction can be used to create two rings simultaneously and interesting bicyclic systems can be synthesised by this method. The selectivity of the cyclisation depends on both stereochemistry and reaction conditions.
Notes and references
- D. J. Fox, D. House and S. Warren, Angew. Chem., Int. Ed. Engl., 2002, 41, 2462 CrossRef CAS.
- L. Caggiano, D. J. Fox, D. House, Z. A. Jones, F. Kerr and S. Warren, J. Chem. Soc., Perkin Trans. 1, 2002, 2634 RSC.
- M. Scholl, S. Ding, C. W. Lee and R. H. Grubbs, Org. Lett., 1999, 1, 953 CrossRef CAS; T. M. Trnka and R. H. Grubbs, Acc. Chem. Res., 2001, 34, 18 CrossRef CAS.
- G. Spagnol, M. P. Heck, S. P. Nolan and C. Mioskowski, Org. Lett., 2002, 4, 1767 CrossRef CAS.
- H. C. Kolb, M. S. VanNieuwenhze and K. B. Sharpless, Chem. Rev., 1994, 94, 2483 CrossRef CAS.
- D. A. Evans, K. T. Chapman and E. M. Carreira, J. Am. Chem. Soc., 1988, 110, 3560 CrossRef CAS.
- K. M. Chen, G. E. Hardtmann, K. Prasad, O. Repic and M. J. Shapiro, Tetrahedron Lett., 1987, 28, 155 CrossRef CAS.
- L. Zeng, Q. Ye, N. H. Oberlies, G. Shi, Z. M. Gu, K. He and J. L. McLaughlin, Nat. Prod. Rep., 1996, 13, 275 RSC.
- J. M. Holland, M. Lewis and A. Nelson, Angew. Chem., Int. Ed. Engl., 2001, 40, 4082 CrossRef CAS; J. M. Holland, M. Lewis and A. Nelson, J. Org. Chem., 2003, 68, 747 CrossRef CAS.
- G. M. Sheldrick, (1997). SHELXS-97/SHELXL-97. University of Göttingen, Germany.
Footnote |
| † Synthesised in low yield from trans-1,4-dibromobut-2-ene.Crystal data for 10: C24H30O2S2, M = 414.60, monoclinic, space group P2(1)/c (no. 14), a = 7.2728(4), b = 5.6573(3), c = 26.5782(16) Å, β = 96.130(2)°, U = 1087.29(11) Å3, Z = 2, μ(Mo-Kα) = 0.262 mm−1, 5145 reflections measured at 180(2) K using an Oxford Cryosystems Cryostream cooling apparatus, 1861 unique (Rint = 0.044); R1 = 0.050, wR2 = 0.114 [I>2σ(I)]. The structure was solved with SHELXS-97,10 and refined with SHELXL-97.10 CCDC 216120.Crystal data for 13: C24H30O2S2, M = 414.60, monoclinic, space group P2(1) (no. 4), a = 9.7835(3), b = 11.2223(4), c = 10.6280(3) Å, β = 109.981(2)°, U = 1096.64(6) Å3, Z = 2, μ(Mo-Kα) = 0.260 mm−1, 6935 reflections measured at 180(2) K using an Oxford Cryosystems Cryostream cooling apparatus, 4285 unique (Rint = 0.041); R1 = 0.038, wR2 = 0.072 [I>2σ(I)]. Absolute structure parameter −0.04(6). The structure was solved with SHELXS-97,10 and refined with SHELXL-97.10 CCDC 216121. See http://www.rsc.org/suppdata/cc/b3/b308609g/ for crystallographic data in .cif or other electronic format. |
| This journal is © The Royal Society of Chemistry 2003 |