Preparation and electrochemical behaviour of hydrophobic vitamin B12 covalently immobilized onto platinum electrode
Hisashi
Shimakoshi
,
Mami
Tokunaga
,
Keita
Kuroiwa
,
Nobuo
Kimizuka
and
Yoshio
Hisaeda
*
Department of Chemistry and Biochemistry, Graduate School of Engineering, Kyushu University, Hakozaki, Higashi-ku, Fukuoka 812-8581, Japan. E-mail: yhisatcm@mbox.nc.kyushu-u.ac.jp; Fax: 81-92-632-4718; Tel: 81-92-642-3592
First published on 2nd December 2003
Abstract
Hydrophobic vitamin B12 was covalently immobilized onto a platinum electrode surface, and the immobilized complex exhibits Co(II)/Co(I) redox couple and in situ the Co(I) species reacts with phenethyl bromide to form styrene under irradiation with visible light with a turnover number of over 6000 for 1 h.
The functionalization of an electrode surface has stimulated continuing interest in a wide range of chemical areas because of its significance and utility for catalyst, sensor, and microelectronic devices.1 In particular, modification with a metal complex provided a good catalyst for electroorganic syntheses.2 Such modified electrodes are generally considered to offer many advantages, such as utilization of small amounts of catalyst species, ready separation of products, and the achievement of various electroorganic synthesis. Previously, we reported some methods for the fixation of vitamin B12 model complexes onto an electrode surface.3–5 Unfortunately, in these methods the stability of the modified electrode is not so high because of the noncovalent weak interactions between the complex and the electrode. Such problems prompted us to prepare a covalently immobilized B12 modified electrode for practical use.6–8 We have been dealing with heptamethyl cobyrinate perchlorate, hydrophobic vitamin B12, which performs various electroorganic reactions.9 In this paper, we report the synthesis of a new hydrophobic vitamin B12 derivative which has a trimethoxysilyl group at a peripheral position, and the complex was covalently immobilized onto a platinum electrode surface.
A hydrophobic vitamin B12 derivative (1) was synthesized as shown in Scheme 1. Monocyanocobyrinic acid hexamethyl ester, [(CN)(H2O)Cob(III)6C1ester]ClO4, was used as a synthetic intermediate.10 In 10 mL of dry DMF, 50 mg (0.047 mmol) of [(CN)(H2O)Cob(III)6C1ester]ClO4 was dissolved and the brown solution was cooled to 0 °C using an ice-bath under nitrogen atmosphere. To the solution was added 15 mg (0.092 mmol) of diethylphosphoryl cyanide (DEPC).11 After stirring for 30 min at 0 °C, 18.1 mg (0.08 mmol) of 3-(trimethoxysilyl)propylamine and 16 µL (0.096 mmol) of triethylamine were added and further stirred for 6 h at 0 °C, then 15 h at room temperature under nitrogen atmosphere. To the resulting purple solution was added 60 mL of dichloromethane and washed with water (60 mL × 3). After drying over anhydrous Na2SO4, the dichloromethane extract was concentrated to dryness. The product was reprecipitated from benzene upon addition of hexane to afford a purple powder. It is noteworthy that DEPC is a superior reagent for this condensation reaction compared to other reagents, such as N,N′-dicyclohexylcarbodiimide. The product 1 was characterized by UV–VIS, NMR, IR and HR mass (FAB) spectroscopies.† The absorption spectrum of 1 in dichloromethane showed the typical shape for the dicyano compound, λmax/nm 279, 317, 371, 422, 550 and 589. Thus, further one equivalent mole of the cyanide ion from DEPC is trapped by the cobalt complex as an axial ligand.
 | ||
| Scheme 1 | ||
Interestingly, the dicyanated form of the precursor; dicyanocobyrinic acid hexamethyl ester, (CN)2Cob(III)6C1ester, does not react with 3-(trimethoxysilyl)propylamine. It is known that there are differences in various physicochemical properties between the dicyano-form and the cyanoaqua-form of the hydrophobic vitamin B12 derivative.12 The microenvironmental properties apparently play a vital role in the reactivity of the carboxylic group on the B-pyrrole ring.
Immobilization of 1 onto a platinum electrode was carried out as follows. Anodization of the bare platinum plate or mesh in 0.5 M H2SO4 at +1.9 V vs. Ag/AgCl for 5 min gives a platinum oxide layer.13 The anodized plate was immersed in a 5 mM solution of 1 in dry toluene for 30 min under nitrogen atmosphere using Schlenk apparatus. After the plate was taken from the solution, it was rinsed with toluene (2 times), dichloromethane (2 times), and ethanol (2 times) to remove any physisorbed material. The modified electrode was dried (aged) for 12 h at 80 °C.
The electrode surface was characterized by X-ray photoelectron spectroscopy (XPS) experiments. XPS peaks at 153.5, 284.8, 398.8, 531.8 and 779.8 eV were observed, which could be attributed to silicon, carbon, nitrogen, oxygen, and cobalt, respectively. The distribution of these atoms is coincident with that for the hydrophobic vitamin B12 on the electrode surface.‡ This XPS result, combined with the subsequent electrochemical evidence, indicated clearly that the hydrophobic vitamin B12 was successfully immobilized onto the platinum plate.
Before the electrochemical study, we tried to remove the axially coordinated cyanide ion from the complex in the following manner, since the dicyano form of hydrophobic vitamin B12 is poorly redox-active. The modified electrode was successively scanned in the potential range from 0 to −1.8 V vs. Ag/AgCl in DMF containing benzyl bromide at 40 °C. In this condition, the in situ formed Co(I) species reacts with benzyl bromide to form an alkylated complex since the Co(I) form of hydrophobic vitamin B12 is a supernucleophile and has high reactivity toward organic halides.14 The alkylated complex was thermally unstable and the cobalt–carbon bond cleaved at 40 °C to form the Co(II) and organic radical species. Thus, the formed Co(II) species of hydrophobic vitamin B12 is electrochemically active and exhibits a reversible Co(II)/Co(I) redox couple at −0.72 V vs. Ag/AgCl in DMF as shown in Fig. 1.§ As the corresponding redox potential for heptamethyl cobyrinate perchlorate, [Cob(II)7C1ester]ClO4, in DMF is −0.62 V vs. Ag/AgCl,15 the Co(II)/Co(I) redox potential of the modified electrode slightly shifts towards the cathodic side. The apparent coverage of the surface with the immobilized 1 was evaluated by the charge under the reductive Co(II)/Co(I) wave with a sweep rate of 100 mV s−1 in CV, 1.60 × 10−10 mol cm−2, and a surface coverage of 80% was calculated from a CPK model for full surface coverage.
 | ||
| Fig. 1 Cyclic voltammograms of 1 immobilized platinum electrode in DMF containing of 0.1 M nBu4NClO4 at different scanning rates. Scan rate (a) 400, (b) 300, (c) 200, (d) 100, (e) 50 mV s−1. | ||
As described above, the Co(I) form of hydrophobic vitamin B12 is a supernucleophile and reacts with various organic halides to form cobalt–carbon bonds with dehalogenation.14 Thus, the controlled potential electrolysis of phenethyl bromide was carried out using the B12 modified electrode at −1.4 V vs. Ag/AgCl in DMF under irradiation with visible light, and the products were analyzed by GC-MS.¶ After an electrical charge of 1 F mol−1 was passed, based on the initial concentration of the substrate, 56% of styrene was selectively formed. The turnover number based on the B12 catalyst immobilized onto the electrode was 6260 for 1 h. The reaction did not proceed when a bare platinum electrode was used under the same conditions. Upon addition of α-phenyl N-(t-butyl)nitrone (PBN) as a spin-trapping reagent, the formation of styrene was inhibited and the corresponding PBN spin adduct was observed by EPR spectroscopy; g = 2.01, AH = 2.6 G, AN = 13.9 G (104 G = 1 T). This result indicates that the reaction proceed via radical species. A proposed mechanism is shown in Fig. 2. When the controlled-potential electrolysis of phenethyl bromide was carried out in the presence of [Cob(II)7C1ester]ClO4 in DMF at −1.4 V vs. Ag/AgCl, the cobalt complex was transformed into the corresponding photo-sensitive complex with absorption maxima at 305, 369sh and 466 nm. These absorption maxima and the photochemical behaviour are characteristic of those for the complex with a cobalt–carbon bond. Therefore, it is expected that the compound with a cobalt–carbon bond is formed as a catalytic intermediate even in the modified electrode.
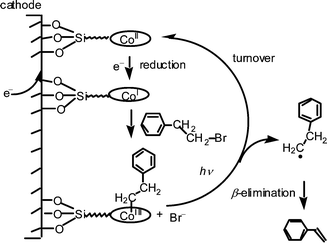 | ||
| Fig. 2 Proposed mechanism for the electrolysis of phenethyl bromide catalyzed by a vitamin B12 modified electrode. | ||
In summary, hydrophobic vitamin B12 derivative is covalently immobilized onto an oxidised platinum electrode surface efficiently, and the complex maintained the electrochemical activity. Some organic halides are known as environmental pollutants; therefore, the degradation of such compounds is important from the viewpoint of green chemistry. Further work on the reactivities of the modified electrode for dechlorination of various organic halides is in progress in our laboratory.
The present work was partly supported by a Grant-in-Aid on Scientific Research from the Ministry of Education, Science, Sports and Culture of Japan. The authors are also grateful for financial support from the Salt Science Research Foundation, the Yazaki Memorial Foundation for Science and Technology and Tokuyama Science Foundation.
Notes and references
- A. J. Bard, Integrated Chemical Systems, Wiley Interscience, New York, 1994 Search PubMed.
- F. Bedioui, J. Devynck and C. B. Charreton, Acc. Chem. Res., 1995, 28, 30 CrossRef CAS.
- H. Aga, A. Aramata and Y. Hisaeda, J. Electroanal. Chem., 1997, 437, 111 CrossRef CAS.
- K. Ariga, K. Tanaka, K. Katagiri, J. Kikuchi, H. Shimakoshi and Y. Hisaeda, Phys. Chem. Chem. Phys., 2001, 3, 3442 RSC.
- H. Shimakoshi, A. Nakazato, M. Tokunaga, K. Katagiri, K. Ariga, J. Kikuchi and Y. Hisaeda, Dalton Trans., 2003, 2308 RSC.
- A. Ruhe, L. Walder and R. Scheffold, Helv. Chim. Acta, 1985, 68, 1301 CAS.
- Y. Murakami, Y. Hisaeda, T. Ozaki and Y. Matsuda, J. Chem. Soc., Chem. Commun., 1989, 1094 RSC.
- C. J. Campbell, C. K. Njue, B. Nuthakki and J. F. Rusling, Langmuir, 2001, 17, 3447 CrossRef CAS.
- Y. Hisaeda, T. Nishioka, Y. Inoue, K. Asada and T. Hayashi, Coord. Chem. Rev., 2000, 198, 21 CrossRef CAS.
- Y. Murakami, Y. Hisaeda, T. Ohno, H. Kohno and T. Nishioka, J. Chem. Soc., Perkin Trans. 2, 1995, 1175 RSC.
- S. Yamada, Y. Kasai and T. Shioiri, Tetrahedron Lett., 1973, 14, 1595 CrossRef.
- M. J. Pfammatter, T. Darbre and R. Keese, Helv. Chim. Acta, 1998, 81, 1105 CAS.
- J. R. Lenhard and R. W. Murray, J. Electroanal. Chem., 1977, 78, 195 CrossRef CAS.
- H. Shimakoshi, A. Nakazato, T. Hayashi, Y. Tachi, Y. Naruta and Y. Hisaeda, J. Electroanal. Chem., 2001, 507, 170 CrossRef CAS.
- Y. Murakami, Y. Hisaeda, A. Kajihara and T. Ohno, Bull. Chem. Soc. Jpn., 1984, 57, 405 CAS.
- P. K. Ghosh and T. G. Spiro, J. Am. Chem. Soc., 1980, 102, 5543 CrossRef CAS.
Footnotes |
† 1: Yield 88%; selected 1H NMR (CDCl3, 500 MHz): δ
= 0.50 [m, 2H, SiCH2–], 1.49 [s, 2H, SiCH2CH2–], 2.71 [m, 1H, NHCH2], 3.35 [m, 1H, NHCH2], 3.47 [s, 9H, OCH3], 3.61 [s, 3H, CO2CH3], 3.66 [s, 3H, CO2CH3], 3.67 [s, 3H, CO2CH3], 3.68 [s, 3H, CO2CH3], 3.69 [s, 3H, CO2CH3], 3.75 [s, 3H, CO2CH3], 5.52 [s, 1H, corrin-10-H], 7.00 [s, 1H, NH]. HRMS (FAB, m/z): Calc. for C58H86N6CoO16Si: [M − CN]+, 1209.5202. Found: [M − CN]+, 1209.5175. IR, ν/cm−1: 2940 (C–H, str.), 1740 (ester C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, str.), 1660 (amide CO, str.). O, str.), 1660 (amide CO, str.). |
| ‡ Obsd.: C1s (52%), N1s (6.6%), O1s (15%), Si2s (0.98%), Co2p (1.0%), Calc.: C (56%), N (7.0%), O (16%), Si (1.0 %), Co (1.0%). |
| § The anodic and cathodic peaks are expected to be at the same voltage for a rigidly surface-immobilized species, but the peak separation was observed in the present system. The value of the voltammetric peak current depends on the square root of the scan rate. Therefore, it is conceivable that the electron transfer of the hydrophobic vitamin B12 derivative immobilized onto the electrode is a diffusion-driven process, because the cobalt complex can move by means of a flexible linker. Another possibility is the formation of a multi-layer film. It is known that trimethoxysilyl substituted compounds form multiple layers on the electrode surface (ref. 16). If a multi-layer film is produced in the present system, the sweep rate dependence is expected by slow electron transfer through the film. |
| ¶ The controlled potential electrolysis was carried out at −1.4 V vs. Ag/AgCl in a divided cell equipped with a B12-modified platinum cathode (mesh, 42.3 cm2) and a magnesium anode (rod) in DMF; [phenethyl bromide] = 5.0 mM, [nBu4NClO4] = 0.1 M under an Ar atmosphere with irradiation by a 500 W tungsten lamp. The GC-MS were obtained using a Shimadzu GC-QP5050A equipped with a J&W Scientific DB-1 column. |
| This journal is © The Royal Society of Chemistry 2004 |