Selective polymorph transformation via solvent-drop grinding†
Andrew V.
Trask
a,
Ning
Shan
*b,
W. D. Samuel
Motherwell
c,
William
Jones
*a,
Shaohua
Feng
b,
Reginald B. H.
Tan
b and
Keith J.
Carpenter
b
aPfizer Institute for Pharmaceutical Materials Science, Department of Chemistry, University of Cambridge, Lensfield Road, Cambridge, UK CB2 1EW. E-mail: wj10@cam.ac.uk; Fax: +44 1223 336362; Tel: +44 1223 336468
bInstitute of Chemical and Engineering Sciences, 1 Pesek Road, Jurong Island, Singapore 627833. E-mail: shan_ning@ices.a-star.edu.sg; Fax: +65 6873 4805; Tel: +65 6796 3865
cCambridge Crystallographic Data Centre, 12 Union Road, Cambridge, UK CB2 1EZ
First published on 19th January 2005
Abstract
A method of inducing specific polymorph transformations is exemplified with two single-component systems, whereby a given crystal form undergoes conversion when subjected to solid state grinding in the presence of a minor quantity of a certain solvent.
Each crystal packing arrangement of a particular chemical compound will possess a unique set of physicochemical properties such as solubility, melting point and hygroscopicity. The discovery, selection and preparation of such polymorphs is important to a variety of disciplines including pharmaceuticals, dyes and pigments. Because polymorph selection enables physical property optimization, novel methods of generating different crystal forms garner interest from both a scientific and an intellectual property perspective.1,2 We report herein a novel method of achieving specific polymorphic transformations, which is of interest both from the viewpoint of high-throughput crystal form screening and from the perspective of green chemistry methodology.
By employing a variety of crystallization approaches and conditions, one may maximize the likelihood of discovering novel polymorphs.3 Polymorphs are often obtained by evaporating or cooling a saturated solution of the target compound. Other crystallization approaches include sublimation or crystallization from the melt. Researchers will often vary parameters such as temperature, pressure, solvent and crystallization rate in order to effect crystallization of a particular polymorph. Recently, the pore size in which crystal nucleation occurred was varied in order to provide polymorph selectivity.4
Solid state grinding is generally used as a particle size reduction technique, and may be carried out manually with a mortar and pestle or mechanically in a mill. The energetic input of solid state grinding is known to induce a variety of solid state transformations, including crystalline to amorphous,5 amorphous to crystalline,6 as well as polymorph conversion.7 One study demonstrated that 11 out of 32 polymorphic drugs exhibited some polymorph conversion upon solid state grinding or compression, and that in general, the number of different polymorphs in a polymorphic mixture of a given substance decreased upon grinding.8 In the report, the three drugs that were studied in detail all converted from a low melting metastable polymorph to a higher melting stable form.
Solid state grinding has also been employed in the formation of crystalline molecular complexes, or cocrystals.9–11 Recently it was shown that solid state grinding in the presence of several drops from a pipette of a particular solvent increased the kinetics of cocrystallization12 and directed a solid state cocrystallization product to a specific polymorph.13 Given these results, we chose to determine whether this ‘solvent-drop grinding’ method may offer improvement or selectivity in the crystallization of polymorphs of single-component systems, thereby offering much wider applicability. We report here results with anthranilic acid (AA) and succinic acid (SA). These two systems are of interest in that, inter alia, they each have a polymorphic form which has been reported to be reproducibly obtained only from high temperature processes.
AA is a well studied polymorphic system. Form I exists as two independent molecules in the asymmetric unit: one is unionized, the other is zwitterionic. AA forms II and III each consist of only unionized molecules.14 AA I is available commercially, while II may be obtained by crystallization from a variety of solvents.15 AA III is commonly prepared by the high temperature processes of sublimation and crystallization from the melt, although it was reported that III grew ‘occasionally’ from aqueous solution.16 Selective growth of AA polymorphs as thin films on certain molecular substrates has also been demonstrated.17 It is not clear from the literature which is the thermodynamically stable polymorph in this system.
The transformations of AA polymorphs upon neat grinding are not clearly established. Ojala and Etter described the results of neat grinding with a mortar and pestle, but mention that their findings contradict those of previous researchers, specifically with regard to the stability of Form I to grinding.15 Our experiments of neat (solvent-free) grinding revealed that AA II and III were stable with respect to manual as well as mechanical grinding. Interestingly, the stability of AA I was found to be grinding rate dependent: it was stable in an experiment at the low grinding rate of 15 Hz in the mechanical mill, but it was observed to convert partly to III at 30 Hz. Powder X-ray diffraction (PXRD) was used to distinguish among the AA polymorphs.18,19
Implementing the technique of solvent-drop grinding for 60 min with a mechanical mill20 allowed for several AA polymorph interconversions that could not be achieved by neat grinding (Fig. 1). Form I (commercially obtained), which converted partly to III when ground without solvent, was found to be stable with respect to grinding when ground with several drops of acetonitrile. Further, I converted to II when ground in the presence of heptane. Form II (obtained by acetonitrile recrystallization), which was stable under neat grinding, could be converted to III by grinding with chloroform. Interestingly, no solvent was found which converted II into I by grinding, despite the fact that some samples of II converted to I on standing for several days under ambient conditions. Form III (obtained from the melt), which was stable with respect to neat grinding, could be converted to I by grinding with water and to II by grinding with heptane (Fig. 2). It is noted that not all polymorph conversions proceeded to completion after one hour of grinding.
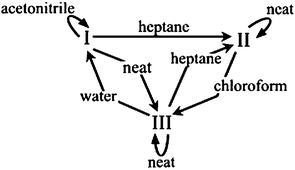 | ||
| Fig. 1 Summary of observed polymorph transformations among three AA crystal forms via solvent-drop grinding. | ||
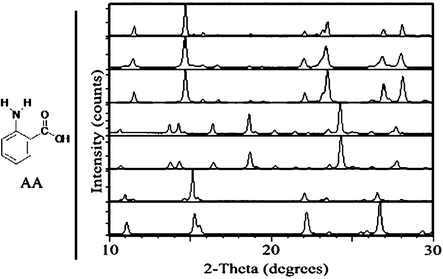 | ||
| Fig. 2 Left: chemical structure of AA. Right: PXRD comparison of AA samples, from top to bottom: III starting material for grinding experiments; after neat grinding of III; simulated III; after solvent-drop grinding of III with water; simulated I; after solvent-drop grinding of III with heptane; simulated II. | ||
SA is known to be dimorphic. The stable and commercially available form is termed the β polymorph,21 which is reported to crystallize from solutions of water and isopropanol (IPA).22 The crystal structure of a metastable polymorph, known as α, has also been determined.23 β-SA converts to α-SA above a phase equilibrium temperature of ca. 117 °C.24 The two forms are readily distinguishable by their PXRD patterns.
β-SA, which was prepared by recrystallization from isopropanol,25 was ground in a mechanical mill and results were monitored by PXRD. No polymorph transformation was observed upon neat grinding (Fig. 3). Similarly, when the experiment was repeated with the introduction of several drops of a polar solvent, including water, acetonitrile and methanol, no conversion to α-SA occurred. In contrast, grinding with several drops of a less polar solvent, including toluene, n-hexane and heptane, induced the formation of metastable α-SA, as observed by PXRD.
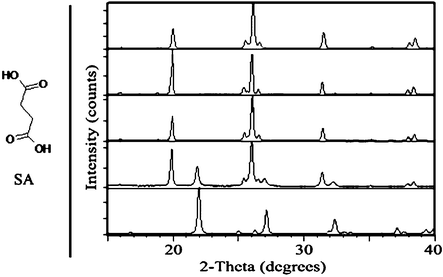 | ||
| Fig. 3 Left: chemical structure of SA. Right: PXRD comparison of SA samples, from top to bottom: simulated β-SA; β-SA starting material for grinding experiments; after 30 min neat grinding of β-SA; after 30 min solvent-drop grinding of β-SA with heptane; simulated α-SA. | ||
While the grinding process does generate heat, the temperature of the grinding jar at the conclusion of the experiments did not exceed 30 °C, and thus remained significantly below the reported transition temperature. Ambient temperature β-SA slurries in toluene, n-hexane or heptane did not exhibit detectable polymorph transformation after 1 week.
In the cases of both SA and AA, the increase in selectivity brought about by the introduction of minor amounts of solvent to the grinding process is not fully understood. The selective adsorption of particular solvents onto certain crystal faces, as described in the cocrystal case,13 is one possible rationale. In a previous study of SA transformation, the propagation of the metastable form was observed to proceed from defects in crystals of the stable form at high temperature.24 It is possible that certain solvents may promote defect production and propagation during grinding.26
Solid state grinding together with a small quantity of a certain solvent has been demonstrated to allow for selective conversion toward particular polymorphs of SA and AA. In the case of SA, a metastable polymorph that hitherto had only been obtained in significant quantities at high temperature has been prepared here by grinding near ambient temperature. With AA, it has been shown that previously unknown solid state transformations between several polymorphs are possible via solvent-drop grinding. Given the current interest in polymorphic systems, these findings suggest important industrial implications. That the transformations occur with minimal solvent addition also indicates a green chemistry application. Further, this approach may represent a novel method of revealing undiscovered metastable crystalline modifications in other systems.
We are currently extending this study to other known polymorphic systems to determine the extent to which this phenomenon is generally applicable. Further work will also be required to establish the precise mechanism by which these selective transformations occur.
We appreciate the early SA grinding experiments performed by Sharon J. Mitchell. AVT, WDSM and WJ thank the Pfizer Institute for funding.
Notes and references
- J. Bernstein, Polymorphism in Molecular Crystals, Oxford University Press, Oxford, 2002, p. 410 Search PubMed.
- S. Datta and D. J. W. Grant, Nat. Rev. Drug Discovery, 2004, 3, 42–57 CrossRef.
- S. L. Morissette, O. Almarsson, M. L. Peterson, J. F. Remenar, M. J. Read, A. V. Lemmo, S. Ellis, M. J. Cima and C. R. Gardner, Adv. Drug Delivery Rev., 2004, 56, 275–300 CrossRef.
- J. M. Ha, J. H. Wolf, M. A. Hillmyer and M. D. Ward, J. Am. Chem. Soc., 2004, 126, 3382–3383 CrossRef CAS.
- K. J. Crowley and G. Zografi, J. Pharm. Sci., 2002, 91, 492–507 CrossRef CAS.
- M. R. Caira, Y. Robbertse, J. J. Bergh, M. N. Song and M. M. De Villiers, J. Pharm. Sci., 2003, 92, 2519–2533 CrossRef CAS.
- S. R. Byrn, R. R. Pfeiffer and J. G. Stowell, Solid-State Chemistry of Drugs, 2nd edn., SSCI, Inc., West Lafayette, IN, 1999, pp. 261–278 Search PubMed.
- H. K. Chan and E. Doelker, Drug Dev. Ind. Pharm., 1985, 11, 315–332 CrossRef CAS.
- F. Toda, K. Tanaka and A. Sekikawa, J. Chem. Soc., Chem. Commun., 1987, 279–280 RSC.
- M. C. Etter and S. M. Reutzel, J. Am. Chem. Soc., 1991, 113, 2586–2598 CrossRef CAS.
- A. V. Trask and W. Jones, in Organic Solid State Reactions, Topics in Current Chemistry, ed. F. Toda, Springer, Berlin, 2005, vol. 254 Search PubMed.
- N. Shan, F. Toda and W. Jones, Chem. Commun., 2002, 2372–2373 RSC.
- A. V. Trask, W. D. S. Motherwell and W. Jones, Chem. Commun., 2004, 890–891 RSC.
- In a strict sense, form I is a mixed crystal of neutral and zwitterionic AA molecules, and a polymorphic relationship to forms II and III could be debated. It will be termed a polymorph here, according to the convention of literature regarding AA crystal forms.
- W. H. Ojala and M. C. Etter, J. Am. Chem. Soc., 1992, 114, 10288–10293 CrossRef CAS.
- H. Takazawa, S. Ohba and Y. Saito, Acta. Crystallogr., Sect. C, 1986, C42, 1880–1881 CrossRef.
- P. W. Carter and M. D. Ward, J. Am. Chem. Soc., 1994, 116, 769–770 CrossRef CAS.
- All PXRD described herein were collected at ambient temperature with a Philips X'Pert diffractometer using Cu Kα radiation and an X'Celerator detector.
- Reference PXRD patterns were simulated from the previously reported crystal structures with the software package X'Pert Plus, Philips Analytical B.V., Version 1.0, using the following structures in the Cambridge Structural Database (CSD): α-SA: SUCACB07; β-SA: SUCACB06; AA-I: AMBACO07; AA-II: AMBACO08; AA-III: AMBACO03.
- All grinding experiments described herein were performed with a Retsch MM200 Mixer Mill with 10 ml stainless steel grinding jars and one 7 mm stainless steel grinding ball per jar. 100 mg crystalline powder was ground at a rate of 30 Hz. Solvent additions were in the quantity of four drops from a pipette (ca. 0.1 ml).
- J.-L. Leviel and G. Auvert, Acta. Cryst., 1981, B37, 2185–2189 CAS.
- R. J. Davey, J. W. Mullin and M. J. L. Whiting, J. Cryst. Growth, 1982, 58, 304–312 CrossRef CAS.
- Private communication to CSD; Refcode SUCACB07, see www.ccdc.cam.ac.uk.
- N. N. Petropavlov and S. B. Yarantsev, Kristallografiya, 1983, 28, 1132–1139 CAS.
- Careful examination of PXRD patterns in conjunction with differential scanning calorimetry measurements revealed that commercially available SA contained trace amounts of the unstable α-SA polymorph. Recrystallized samples contained a smaller trace amount of α-SA, the quantity of which is too minor to be observed in Fig. 3. The limit of detection of PXRD is typically considered to be on the order of 5%.
- In addition to solvent-induced defect formation, the presence of small amounts of crystalline or amorphous impurities (below the detection limit of PXRD) may also be important.
Footnote |
| † Electronic Supplementary Information (ESI) available: PXRD patterns for the polymorph transformations of AA I and II. See http://www.rsc.org/suppdata/cc/b4/b416980h/ |
| This journal is © The Royal Society of Chemistry 2005 |