Regioselective aromatic C–H silylation of five-membered heteroarenes with fluorodisilanes catalyzed by iridium(I) complexes†
Tatsuo
Ishiyama
*,
Kazuaki
Sato
,
Yukihiro
Nishio
,
Takeaki
Saiki
and
Norio
Miyaura
*
Division of Chemical Process Engineering, Graduate School of Engineering, Hokkaido University, Sapporo, 060-8628, Japan. E-mail: ishiyama@org-mc.eng.hokudai.ac.jp; Fax: +81 11 706 6562; Tel: +81 11 706 6562
First published on 21st September 2005
Abstract
The aromatic C–H silylation of five-membered heteroarenes with 1,2-di-tert-butyl-1,1,2,2-tetrafluorodisilane regioselectively proceeded at 120 °C in octane in the presence of a catalytic amount of iridium(I) complexes generated from 1/2[Ir(OMe)(COD)]2 and 2-tert-butyl-1,10-phenanthroline.
Transition metal-catalyzed aromatic C–H silylation of aromatic compounds is highly attractive as a convenient, economical, and environmentally benign process for preparing synthetically useful aromatic silicon compounds.1 Although several studies on this type of transformation with disilanes2 or hydrosilanes2a,3 have been carried out, application of the protocol has been limited to the synthesis of less-reactive aromatic triorganosilicon derivatives. Recently, we found that iridium(I) complexes comprised of an air-stable, easily available 1/2[Ir(OMe)(COD)]2 precursor and a simple, commercially available 2,2′-bipyridine (bpy) or 4,4′-di-tert-butyl-2,2′-bipyridine (dtbpy) ligand effectively catalyzed aromatic C–H silylation of neat arenes with 1,2-di-tert-butyl-1,1,2,2-tetrafluorodisilane (t-BuF2Si)2 at 120 °C to give the corresponding aryl fluorosilanes in high yields with high regioselectivities.4,5 Also, the synthetic utility of the aryl fluorosilanes was demonstrated by their palladium-catalyzed cross-coupling6 with aromatic electrophiles and rhodium-catalyzed 1,4-addition7 to enones.4 As an extension of our methodology to other aromatic substrates, we describe here the iridium(I)-catalyzed aromatic C–H silylation of five-membered heteroarenes with (t-BuF2Si)2 in octane at 120 °C (Scheme 1). Thiophene and furan derivatives were exclusively silylated at the α-position by using 1/2[Ir(OMe)(COD)]2–(2-tert-butyl-1,10-phenanthroline)8 (tbphen) catalysts. On the other hand, the reactions of 1-triisopropylsilylpyrrole and -indole catalyzed by 1/2[Ir(OMe)(COD)]2–tbphen or –dtbpy complexes selectively yielded β-silyl products.
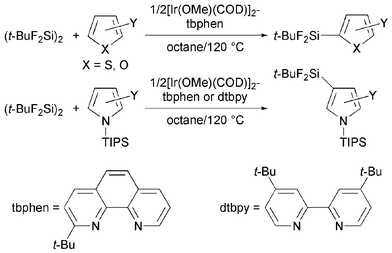 | ||
| Scheme 1 C–H silylation of five-membered heteroarenes. | ||
The silylation of 3-methylthiophene (60 mmol) with (t-BuF2Si)2 (1.0 mmol) in the presence of catalysts generated from [Ir(OMe)(COD)]2 (0.015 mmol) and dtbpy (0.03 mmol) resulted in only a 38% yield of the silylated product after 16 h at 120 °C,4 probably due to the high coordinating ability of the sulfur atom which retards the formation of a coordinatively unsaturated iridium species active for C–H activation.9 This result prompted us to use an appropriate solvent inactive toward the C–H silylation. Indeed, a silylated product was obtained in 90% yield when 5.0 mmol of the substrate was diluted in 6 mL of octane. At that time, we expected formation of regioisomerically pure 5-silyl-3-methylthiophene, because previous studies on C–H silylation of alkylated arenes demonstrated that the reactions did not occur at sterically hindered ortho positions.4 However, the present reaction gave a regioisomeric mixture of 5-, 4-, and 2-silylated products in a ratio of 69 ∶ 3 ∶ 28, indicating that the dtbpy-ligated iridium species was insufficient to recognize the steric environment of the substrate. Thus, we examined a sterically hindered 6,6′-di-Me-bpy ligand, but the reaction resulted in only an 8% yield. Presumably, twisting between two pyridyl units arising from steric hindrance around the iridium center leads to the low activity.4 Based on this hypothesis, we investigated the reaction by using structurally rigid 1,10-phenanthroline (phen) derivatives as ligands for iridium (Table 1).
| Entry | R1 | R2 | Yield (%)b | 5- ∶ 4- ∶ 2- (%)c |
|---|---|---|---|---|
|
|
||||
| a Reactions were carried out at 120 °C for 16 h by using (t-BuF2Si)2 (1.0 mmol), 3-methylthiophene (5.0 mmol), [Ir(OMe)(COD)]2 (0.015 mmol), ligand (0.03 mmol), and octane (6 mL). b GC yields based on (t-BuF2Si)2. c Isomer ratios were determined by 1H NMR. d 8 h. e 10 mmol of 3-methylthiophene, 32 h. | ||||
| 1 | H | H | 99 | 66 ∶ 4 ∶ 30 |
| 2 | Me | Me | 85d | 81 ∶ 0 ∶ 19 |
| 3 | n-Bu | n-Bu | 65 | 91 ∶ 0 ∶ 9 |
| 4 | i-Pr | i-Pr | 65 | 93 ∶ 0 ∶ 7 |
| 5 | t-Bu | t-Bu | 0 | — |
| 6 | i-Pr | H | 91 | 72 ∶ 0 ∶ 28 |
| 7 | t-Bu | H | 40 | 98 ∶ 0 ∶ 2 |
| 8 | t-Bu | H | 96e | 99 ∶ 0 ∶ 1 |
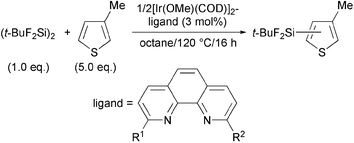
As we expected, phen ligands even having substituents at 2 and 9 positions displayed high reactivity. Although unsubstituted phen produced the desired 5-silylated products with 66% selectivity (Entry 1), the selectivity was improved to 81%, 91% and 93% when using 2,9-di-Me-phen, 2,9-di-n-Bu-phen and 2,9-di-i-Pr-phen, respectively (Entries 2–4). However, the use of sterically more hindered 2,9-di-t-Bu-phen led to no reaction (Entry 5). The result prompted us to examine monosubstituted phen derivatives. 2-i-Pr-phen resulted in a 72% selectivity (Entry 6), but 2-t-Bu-phen (tbphen) improved the selectivity to 98%, while its reactivity was low (Entry 7). Finally, we obtained the desired 5-silyl-3-methylthiophene in 96% yield with 99% regioselectivity when the reaction was carried out for 32 h by using 10 mmol of 3-methylthiophene (Entry 8).
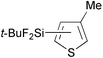
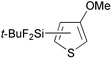
Reactions of thiophene derivatives with (t-BuF2Si)2 catalyzed by the combination of 1/2[Ir(OMe)(COD)]2 and tbphen at 120 °C in octane are summarized in Table 2. 3-Substituted (Entries 1–4), 2-substituted (Entries 5 and 6), benzo-fused (Entry 7), and unsubstituted (Entry 8) thiophenes were all viable substrates to produce the corresponding silylated products in excellent yields with high regioselectivities. The reactions were suitable for substrates possessing various functional groups, such as OMe, Cl, and CO2Me or benzylic C–H bonds10 (Entries 1–6). The regiochemistry was controlled by both electronic and steric effects. The electronegative sulfur atom caused the α-C–H bonds to be active,11 and the substituents on thiophene blocked activation of the neighboring C–H bonds.4 The low selectivity observed in the reaction of 3-methoxythiophene may be ascribed to the small steric hindrance of the MeO group (Entry 2).
| Entry | Product | Yield (%)b | Isomer ratio (%)c |
|---|---|---|---|
| a Reactions were carried out at 120 °C for 48 h by using (t-BuF2Si)2 (1.0 mmol), thiophenes (10 mmol), [Ir(OMe)(COD)]2 (0.015 mmol), tbphen (0.03 mmol), and octane (6 mL). b GC yields based on (t-BuF2Si)2. c Isomer ratios were determined by 1H NMR. d 32 h. | |||
| 1 |
|
96d | 5- ∶ 4- ∶ 2- = 99 ∶ 0 ∶ 1 |
| 2 |
|
97 | 5- ∶ 4- ∶ 2- = 87 ∶ 2 ∶ 11 |
| 3 |
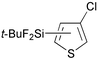
|
92 | 5- ∶ 4- ∶ 2- = 98 ∶ 0 ∶ 2 |
| 4 |
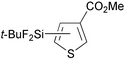
|
93 | 5- ∶ 4- ∶ 2- = 98 ∶ 0 ∶ 2 |
| 5 |
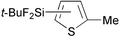
|
99d | 5- ∶ 4- ∶ 3- = 95 ∶ 5 ∶ 0 |
| 6 |
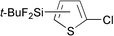
|
88 | 5- ∶ 4- ∶ 3- = 95 ∶ 5 ∶ 0 |
| 7 |
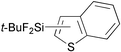
|
98 | 2- ∶ 3- = > 99 ∶ < 1 |
| 8 |
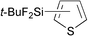
|
96d | 2- ∶ 3- = 93 ∶ 7 |
Although furan derivatives also underwent smooth C–H silylation, the regiochemistry was perplexing. Reactions of 3-methylfuran and benzofuran using dtbpy as a ligand predominantly yielded unexpected 4-silylated and 3-silylated products, respectively. In such cases, use of a tbphen ligand resulted in the expected 5-silylated and 2-silylated products with 88% and 83% selectivities (Scheme 2).
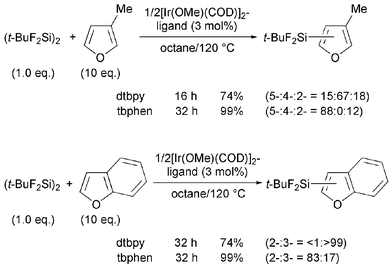 | ||
| Scheme 2 C–H silylation of furan derivatives. | ||
The C–H silylation of unsubstituted pyrrole and indole by using either dtbpy or tbphen as a ligand produced complex mixtures probably due to the presence of an acidic hydrogen in the substrates, but the reactions of 1-triisopropylsilylpyrrole and -indole successfully gave silylated products. The silylation occurred only at the β-positions when using either dtbpy or tbphen because of the large steric hindrance of the TIPS group (Scheme 3).
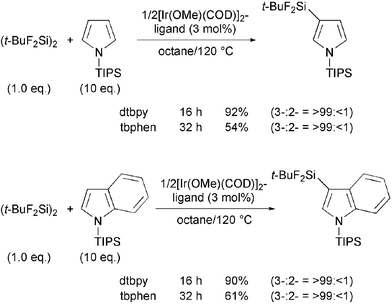 | ||
| Scheme 3 C–H silylation of pyrrole derivatives. | ||
In summary, iridium complexes comprised of 1/2[Ir(OMe)(COD)]2 and 2-tert-butyl-1,10-phenanthroline were found to be efficient catalysts for the regioselective aromatic C–H silylation of five-membered heteroarenes with 1,2-di-tert-butyl-1,1,2,2-tetrafluorodisilane. Further investigations to survey the scope and limitations of this C–H silylation, including that of other heteroarenes such as pyridines, as well as to elucidate the reaction mechanisms are in progress.
This work was partially supported by a Grant-in-Aid for Scientific Research on Priority Areas (No. 14078101, “Reaction Control of Dynamic Complexes”) from the Ministry of Education, Culture, Sports, Science and Technology, Japan. T.I. thanks The Akiyama Foundation and the Takeda Science Foundation for support of a part of his work.
Notes and references
- For a review, see: F. Kakiuchi and N. Chatani, Adv. Synth. Catal., 2003, 345, 1077 Search PubMed.
- (a) T. Sakakura, Y. Tokunaga, T. Sodeyama and M. Tanaka, Chem. Lett., 1987, 2375 CAS; (b) M. Ishikawa, S. Okazaki, A. Naka and H. Sakamoto, Organometallics, 1992, 11, 4135 CrossRef CAS; (c) M. Ishikawa, A. Naka and J. Ohshita, Organometallics, 1993, 12, 4987 CrossRef CAS; (d) N. A. Williams, Y. Uchimaru and M. Tanaka, J. Chem. Soc., Chem. Commun., 1995, 1129 RSC; (e) A. Naka, K. K. Lee, K. Yoshizawa, T. Yamabe and M. Ishikawa, Organometallics, 1999, 18, 4524 CrossRef CAS; (f) N. A. Williams, Y. Uchimaru and M. Tanaka, Dalton Trans., 2003, 236 RSC.
- (a) W. A. Gustavson, P. S. Epstein and M. D. Curtis, Organometallics, 1982, 1, 884 CrossRef CAS; (b) Y. Uchimaru, A. M. M. El Sayed and M. Tanaka, Organometallics, 1993, 12, 2065 CrossRef CAS; (c) K. Ezbiansky, P. I. Djurovich, M. LaForest, D. J. Sinning, R. Zayes and D. H. Berry, Organometallics, 1998, 17, 1455 CrossRef CAS; (d) F. Kakiuchi, K. Igi, M. Matsumoto, N. Chatani and S. Murai, Chem. Lett., 2001, 422 CrossRef CAS; (e) F. Kakiuchi, K. Igi, M. Matsumoto, T. Hayamizu, N. Chatani and S. Murai, Chem. Lett., 2002, 396 CrossRef CAS; (f) F. Kakiuchi, M. Matsumoto, K. Tsuchiya, K. Igi, T. Hayamizu, N. Chatani and S. Murai, J. Organomet. Chem., 2003, 686, 134 CrossRef CAS.
- T. Ishiyama, K. Sato, Y. Nishio and N. Miyaura, Angew. Chem., Int. Ed., 2003, 42, 5346 CrossRef CAS.
- The catalysts were also effective for aromatic C–H borylation, see: (a) T. Ishiyama, J. Takagi, K. Ishida, N. Miyaura, N. R. Anastasi and J. F. Hartwig, J. Am. Chem. Soc., 2002, 124, 390 CrossRef CAS; (b) J. Takagi, K. Sato, J. F. Hartwig, T. Ishiyama and N. Miyaura, Tetrahedron Lett., 2002, 43, 5649 CrossRef CAS; (c) T. Ishiyama, J. Takagi, J. F. Hartwig and N. Miyaura, Angew. Chem., Int. Ed., 2002, 41, 3056 CrossRef CAS; (d) T. Ishiyama, J. Takagi, Y. Yonekawa, J. F. Hartwig and N. Miyaura, Adv. Synth. Catal., 2003, 345, 1103 CrossRef; (e) T. Ishiyama, Y. Nobuta, J. F. Hartwig and N. Miyaura, Chem. Commun., 2003, 2924 RSC; (f) T. Ishiyama and N. Miyaura, J. Organomet. Chem., 2003, 680, 3 CrossRef CAS; (g) T. Ishiyama and N. Miyaura, Chem. Rec., 2004, 3, 271 Search PubMed; (h) K. Kurotobi, M. Miyauchi, K. Takakura, T. Marufuji and Y. Sugihara, Eur. J. Org. Chem., 2003, 3663 CrossRef CAS; (i) A. Datta, A. Köllhofer and H. Plenio, Chem. Commun., 2004, 1508 RSC; (j) D. N. Coventry, A. S. Batsanov, A. E. Goeta, J. A. K. Howard, T. B. Marder and R. N. Perutz, Chem. Commun., 2005, 2172 RSC; (k) G. A. Chotana, M. A. Rak and M. R. Smith, III, J. Am. Chem. Soc., 2005, 127, 10539 CrossRef CAS. For a theoretical study, see: (l) H. Tamura, H. Yamazaki, H. Sato and S. Sakaki, J. Am. Chem. Soc., 2003, 125, 16114 CrossRef CAS.
- (a) T. Hiyama and E. Shirakawa, Top. Curr. Chem., 2002, 219, 61 CAS; (b) S. E. Denmark and R. F. Sweis, Acc. Chem. Res., 2002, 35, 835 CrossRef CAS.
- (a) S. Oi, M. Moro and Y. Inoue, Organometallics, 2001, 20, 1036 CrossRef CAS; (b) T.-S. Huang and C.-J. Li, Chem. Commun., 2001, 2348 RSC; (c) K. Fagnou and M. Lautens, Chem. Rev., 2003, 103, 169 CrossRef CAS.
- M. P. T. Sjögren, S. Hansson, B. Åkermark and A. Vitagliano, Organometallics, 1994, 13, 1963 CrossRef.
- J. J. Li and G. W. Gribble, in Tetrahedron Organic Chemistry Series, ed. J. E. Baldwin and R. M. Williams, Pergamon, Amsterdam, 2000, vol. 20, pp. 233 Search PubMed.
- D. P. Curran, in Comprehensive Organic Synthesis, ed. B. M. Trost, Pergamon, Oxford, 1991, vol. 4, pp. 715 Search PubMed.
- (a) L. Dong, S. B. Duckett, K. F. Ohman and W. D. Jones, J. Am. Chem. Soc., 1992, 114, 151 CrossRef CAS; (b) T. Morikita, M. Hirano, A. Sasaki and S. Komiya, Inorg. Chim. Acta, 1999, 291, 341 CrossRef CAS; (c) S. N. Ringelberg, A. Meetsma, B. Hessen and J. H. Teuben, J. Am. Chem. Soc., 1999, 121, 6082 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: experimental procedures and spectral analyses of products. See http://dx.doi.org/10.1039/b511171d |
| This journal is © The Royal Society of Chemistry 2005 |