Perfusion and chemical monitoring of living cells on a microfluidic chip
Jonathan G.
Shackman
a,
Gabriella M.
Dahlgren
a,
Jennifer L.
Peters
a and
Robert T.
Kennedy
*ab
aDepartment of Chemistry, University of Michigan, Ann Arbor, MI, USA
bDepartment of Pharmacology, University of Michigan, Ann Arbor, MI, USA
First published on 22nd July 2004
Abstract
A microfluidic device that incorporates continuous perfusion and an on-line electrophoresis immunoassay was developed, characterized, and applied to monitoring insulin secretion from single islets of Langerhans. In the device, a cell chamber was perfused with cell culture media or a balanced salt solution at 0.6 to 1.5 µL min−1. The flow was driven by gas pressure applied off-chip. Perfusate was continuously sampled at 2 nL min−1 by electroosmosis through a separate channel on the chip. The perfusate was mixed on-line with fluorescein isothiocyanate-labeled insulin (FITC-insulin) and monoclonal anti-insulin antibody and allowed to react for 60 s as the mixture traveled down a 4 cm long reaction channel. The cell chamber and reaction channel were maintained at 37 °C. The reaction mixture was injected onto a 1.5 cm separation channel as rapidly as every 6 s, and the free FITC-insulin and the FITC-insulin-antibody complex were separated under an electric field of 500 to 600 V cm−1. The immunoassay had a detection limit of 0.8 nM and a relative standard deviation of 6% during 2 h of continuous operation with standard solutions. Individual islets were monitored for up to 1 h while perfusing with different concentrations of glucose. The immunoassay allowed quantitative monitoring of classical biphasic and oscillatory insulin secretion with 6 s sampling frequency following step changes in glucose from 3 to 11 mM. The 2.5 cm × 7.6 cm microfluidic system allowed for monitoring islets in a highly automated fashion. The technique should be amenable to studies involving other tissues or cells that release chemicals.
Introduction
Studies of cellular physiology require maintaining cells in a life-supporting environment while electrical, chemical, optical, or mechanical measurements are made on the cells. Such studies are typically performed with cells bathed in a physiological medium while adhered to culture plates or immobilized in a macroscale perfusion system.1–5 The advent of microfabricated fluidic systems offers the potential to develop sophisticated and automated cellular physiology experimental stations that incorporate multi-parameter, highly parallel, or complex measurements on cells. Initial cell studies utilizing microfluidic systems focused on cytometry,6–8 sorting,9–11 and cell lysis followed by extraction and analysis of intracellular contents12–14 using microfluidic devices. More recent microfluidic work has demonstrated culturing of cells on chips15 and in vitro cellular measurements of Ca2+ flux16 and oxygen consumption.17Another important physiological function is secretion of chemical products from cells. A variety of chemicals are released from cells, including signaling molecules such as hormones or neurotransmitters, trophic factors, and metabolic products. Temporally resolved measurements of cell releasates are important in studying the regulation of the secretory process as well as technological pursuits such as drug development. Such measurements are typically performed by perfusing cells, collecting fractions, and then performing off-line analysis by immunoassays or other appropriate methods. The development of a microfabricated device that could miniaturize and automate such measurements would be a valuable tool in physiological studies. Recently, initial efforts toward such systems have been reported including a system to measure dopamine release that used microfluidics to trap a cell while an external microelectrode was manually positioned over the cell for measurement.18
In this work we report on a microfluidic device that perfuses a cluster of cells and monitors secretion at 6–10 s intervals using a rapid electrophoresis-based immunoassay. The system is applied to monitoring insulin secretion from single islets of Langerhans. Islets are 75–200 µm diameter spheroid microorgans located in the pancreas that contain 2000–4000 endocrine cells each. 70–80% of islet cells are β-cells that secrete insulin as part of the glucose homeostasis mechanism (for a review see ref. 19). Insulin secretion, stimulated primarily by glucose, has complex dynamics upon exposure to step increases in glucose concentration that include an initial burst of insulin release (first phase) followed by a lower rate of release (second phase) that is frequently oscillatory (for reviews see ref. 20 and 21). Insulin secretion regulation is of interest because impaired secretion is associated with type 2 diabetes, and drugs that treat diabetes target this process.
In previous work we demonstrated a microfabricated device that could detect insulin secreted from single islets using an electrophoresis-based immunoassay.22 The islet was housed in a small chamber on the chip that was continuously sampled by electroosmotic flow into a narrow channel. The resulting sample stream was mixed online with fluorescein isothiocyanate-labeled insulin (FITC-insulin) and anti-insulin antibody (Ab). The mixture was allowed to react as it was electroosmotically pumped along a heated channel prior to injection onto an electrophoresis channel where the bound (B) and free (F) FITC-insulin were separated. The ratio of the bound to free FITC-insulin (B/F) was used to quantify insulin. This device allowed electropherograms to be acquired at 15 s intervals and had a detection limit of 3 nM. Although this system demonstrated the feasibility of using microfabricated devices for chemical monitoring of live cells, it was limited in that cells were maintained in a quiescent solution and not perfused. Lack of perfusion resulted in serious limitations including inability to: (1) obtain truly dynamic measurements of secretion because insulin secreted from the islet continually increased in concentration within the chamber, (2) rapidly raise and lower concentration of drugs or secretagogues on the cell as required for many experiments, and (3) continually provide fresh nutrients to the cells, which limited the time that cells could be viable on the chip. In this work we describe modification of the chip that enables perfusion of cells while maintaining sampling. It is demonstrated that this modification enables vastly improved temporal resolution for monitoring, facile control of the cellular environment, and compatibility with longer-term measurements. The system is also demonstrated to be compatible with different types of physiological media.
Experimental
Chemicals and reagents
Cell culture reagents were from Invitrogen (Carlsbad, CA). FITC-insulin was purchased from Molecular Probes (Eugene, OR). Monoclonal Ab to human insulin (Ka = 109 M−1) was from Biodesign International (Saco, ME). All solutions were made from Milli-Q (Millipore, Bedford, MA) ≥ 18 MΩ cm−1 deionized water. Tween-20, insulin, collagenase type XI, 4-(2-hydroxyethyl)piperazine-1-ethanesulfonic acid (HEPES), Ficoll, ethylenediaminetetraacetic acid (EDTA), and bovine serum albumin (BSA) were from Sigma (St. Louis, MO). All other chemicals were obtained from Fisher (Pittsburgh, PA) and were of the highest purity available. Insulin standard solutions were prepared daily. Stock Ab was maintained at 4 °C in the manufacturer provided phosphate buffer solution; stock FITC-insulin was diluted to 166 µM with the working immunoassay reagent buffer and stored at −32 °C. Solutions were filtered using 0.2 µm nylon syringe filters (Fisher).Buffer and physiological solutions
Several buffer and salt solutions were used as physiological saline or reagent solvents. The buffer composition and abbreviations used throughout text are: HEAT40 consisting of 40 mM NaCl, 20 mM HEPES, 1 mM EDTA, pH 7.4, supplemented with 1 mg mL−1 BSA and 0.1% (w/v) Tween 20; PETA consisting of 20 mM NaH2PO4, 1 mM EDTA, pH 7.4, supplemented with 0.1% (w/v) Tween 20 and 0.7 mg mL−1 BSA; RPMI 1640, a standard cell culture medium containing L-glutamine (Invitrogen), supplemented with 25 mM HEPES and either 3 or 11 mM glucose, adjusted to pH 7.4; and BSS (balanced salt solution) consisting of 125 mM NaCl, 5.9 mM KCl, 1.2 mM MgSO4, 2.4 mM CaCl2, 25 mM HEPES, pH 7.4, and 1 mg mL−1 BSA.Instrumentation
A Zeiss Axiovert 100 inverted microscope with epi-fluorescence optics was used for imaging the islet and for electrophoresis detection. The 488 nm line of a 20 mW Ar+ laser (Melles Griot, Carlsbad, CA) was directed onto a 500 nm longpass dichroic mirror and through a 40×, 0.6 numerical aperture, long working distance objective (Carl Zeiss, Inc., Thornwood, NY). After passing through the dichroic mirror, the emission light was further filtered by a 520 ± 10 nm bandpass and 1 mm pinhole spatial filter. The emission was detected by a photon counting detector (Thermo Oriel, Stratford, CT) at 100 Hz. Instrument control and data collection were performed using LabVIEW software written in-house (National Instruments, Austin, TX). Electropherograms were analyzed using Cutter software.23 Visual images were collected using a PDC2300Z digital camera (Polaroid Corp., Waltham, MA) through a microscope eyepiece using a 10× objective and were processed using in-house written LabVIEW software.Fabrication of microfluidic chips
Fig. 1a and b illustrate the design of the device used in all experiments. Microfluidic chips were fabricated using a modified method described previously.22 Briefly, 1 mm thick Borofloat photomask blanks (2.5 cm × 7.6 cm) with a 530 nm layer of AZ1518 positive photoresist on a 120 nm chrome layer were purchased from Telic Co. (Santa Monica, CA). A collimated UV light source (Optical Associates, Inc., Milpitas, CA) was used to expose the blank through a patterned photomask (Digidat, Pasadena, CA) for 5 s at 26 mW cm−2. The exposed photomask was developed in AZ915 MIF developer (Clariant Corp., Summerville, NJ) for 15 s, followed by 45 s developing in CEP-200 chrome etchant (Microchrome Technologies, Inc., San Jose, CA). The exposed glass was etched for 20 min in 14:20:66 (v/v/v) HNO3:HF:H2O resulting in 6 µm deep channels that were 32 µm wide at the top. 300 µm diameter access holes were formed using diamond-tipped drill bits (Tartan Tool Co., Troy, MI). The etched chip and a blank coverplate were cleaned in piranha solution (3:1, v/v, H2SO4:H2O2) for 20 min followed by RCA solution (5:1:1, v/v/v, H2O:NH4:H2O2) at 60 °C for 20 min. The two plates were bonded at 640 °C for 8 h under vacuum in a Neytech Centurian Qex furnace (Pacific Combustion, Los Angeles, CA), during which time 400 g of stainless steel were applied.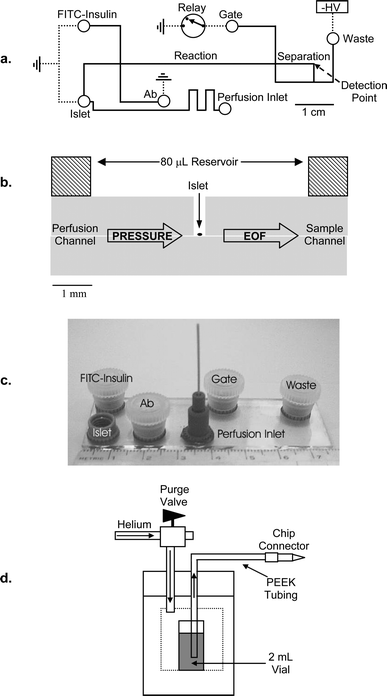 | ||
| Fig. 1 Microfluidic device to continuously monitor insulin release from islets of Langerhans with perfusion. (a) Layout of the device. Channels (lines) and access holes (circles) are drawn to scale. All channels were 6 µm deep. Electrical connections are shown as dashed lines. (b) Side-on, cutaway view of chip at the islet chamber. A single islet was contained in a 300 µm diameter chamber with an 80 µL fluid reservoir above. Pressure-driven fluid bathed the islet via the perfusion channel and flowed into the reservoir above. Solution around the islet was sampled by EOF through the sample channel. (c) Photograph of a completed 2.5 cm × 7.5 cm device. The larger ports contained 80 µL of solution and were capped during experimentation. The high pressure perfusion inlet (lower center port) was connected via PEEK™ tubing to a stainless steel pressure bomb containing a 2 mL vial of the perfusion solution (d). | ||
Microfluidic reservoirs applied after bonding were purchased from Upchurch Scientific (Oak Harbor, WA). 80 µL reservoirs (Upchurch N-131) were used for all ports except the high-pressure perfusion inlet, which used a 1/32″ tubing interconnect port (Upchurch N-124H). A photograph of the completed device is shown in Fig. 1c. A thin-film resistive heater (Minco Products, Inc., Fridley, MN) was taped to the chip underside to maintain the islet, immunoassay reagents, and the reaction channel at 37 °C, as monitored by digital thermometer (Fisher). During calibration and islet monitoring, 80 µL of the appropriate solution were placed in all reservoirs (excluding the perfusion inlet) and covered with plastic caps drilled with Pt electrode access holes. The cell chamber was perfused using a gas-pressure system off-chip illustrated in Fig. 1d. The perfusion inlet was connected to the high-pressure bomb via 80 cm PEEK™ tubing (0.20″ id, 1/32″ od, Upchurch); the PEEK™ tubing was flushed with water followed by the perfusion solution prior to chip connection. To change the perfusion media, the pressure was released from the pressure bomb via the purge valve, the fluid changed, and the PEEK™ tubing flushed with perfusion solution prior to reapplying pressure. Any air within the perfusion lines would cause the islets to be blown off the glass and disperse; hence, utmost care was taken when making the perfusion connections to maintain the outlet lower than the pressure bomb to sustain siphon flow.
Microfluidic chip operation
The device utilized a previously described flow gate method of injecting sample onto the separation channel.22,24 During the experiment, the immunoassay reagent reservoirs and islet were held at ground. Voltage was applied to the waste reservoir via a high voltage power supply (Spellman High Voltage Electronics, Hauppauge, NY). The gate reservoir was connected to ground via a high voltage relay (Kilovac, Santa Barbara, CA). When the relay was opened, sample was allowed to load onto the separation channel. The gate was then returned to ground and separation was performed, after which time another sample was loaded for separation. Detection occurred 1 cm from the injection point.Chips were conditioned daily by electroosmotically flowing 1 M NaOH through all channels, followed by 150 mM HEPES, pH 7.4, followed by the experimental solutions. After calibration or islet experiments, the chip was conditioned with 150 mM HEPES, pH 7.4, followed by the experimental solutions. This procedure allowed the same chip to be repeatedly used on successive days. Experiments on islets were performed using two different perfusion media. One medium was RPMI 1640 (a cell culture media) supplemented with 25 mM HEPES and either 3 or 11 mM glucose, adjusted to pH 7.4. When this media was used, 150 nM FITC-insulin was placed in the FITC-insulin reservoir and 75 nM antibody placed in the Ab reservoir (Fig. 1a). Both of these reagents were dissolved in PETA buffer (see composition above). Electrophoresis buffer in the gate and waste reservoirs consisted of 150 mM HEPES adjusted to pH 7.4 with NaOH. The applied voltage was 4 kV, with an electric field of 500 V cm−1 in the separation channel and an immunoassay reaction time of 60 s. Sample injection time was 1 s applied at 9 s intervals. Cells were perfused at 1.5 µL min−1 by applying 100 psi (He) to the pressure bomb.
Experiments were also performed with cells perfused with BSS (see composition above). For these experiments the immunoassay reagent concentrations were 100 nM for FITC-insulin and 50 nM for antibody. These reagents were dissolved in HEAT40 buffer (see composition above). Electrophoresis buffer was 150 mM HEPES adjusted to pH 7.4 with NaOH. The applied voltage was 5 kV, with an electric field of 600 V cm−1 in the separation channel and an immunoassay reaction time of 60 s. Sample injection time was 0.5 s applied at 5.5 s intervals. Cells were perfused at 0.6 µL min−1 by applying 50 psi (He) to the pressure bomb.
Concentrations of insulin were determined from each electropherogram by comparing B/F peak height ratios to a calibration curve. The calibration curve was obtained by pumping different concentrations of standard insulin in the perfusion media via the high-pressure bomb to the islet reservoir. Perfusion media flow rates were determined by weighing the chip (n = 3) before and after 30 min of perfusion and converting to volume using the density of the perfusion buffer.
Isolation and protocol of islets measurements
Islets of Langerhans were obtained by a previously described method.25 Briefly, 20–30 g male CD-1 mice were sacrificed by cervical dislocation followed by ductal injection of collagenase type XI. The pancreas was dissected and incubated in 5 mL of collagenase solution at 37 °C for 10 min. Endocrine tissue was separated from exocrine tissue using a Ficoll gradient, and islets were picked by hand under a stereomicroscope. Islets were selected that had an oblong to spherical shape, a smooth surface (indicative of an intact islet membrane), and a diameter of 100–200 µm. The islets were placed in RPMI 1640 cell culture media supplemented with 10% fetal bovine serum, 100 units mL−1 penicillin, and 100 µg mL−1 streptomycin and incubated at 37 °C, 5% CO2. Islets were used 1–6 days following isolation. To monitor secretion, isolated islets were transferred from culture medium in 4 µL aliquots and washed in 3 mL of the perfusion buffer containing 3 mM glucose. The islet was then placed in the cell chamber using a pipette under a stereomicroscope, where it quickly settled to the glass surface and adhered. The device was then transferred to the inverted microscope workstation for monitoring the electrophoresis immunoassay, where the islet was perfused with 3 mM glucose for at least 20 min, during which time it was monitored to establish basal levels of secretion. Different concentrations of glucose were then applied using the gas-pressure system as described above. All values for secretion are reported as the average ± standard error of the mean.Results and discussion
Construction of perfusion system
In designing the perfusion system it was desirable to keep the cell chamber open to the atmosphere in order to minimize the difficulty of loading cells into the chip, maintain access for sensors or electrophysiological probes, and minimize pressure drops across the cells that might damage the cells. In addition, high temporal resolution monitoring of the cells required that the perfusion system could rapidly replace solution in the cell chamber (approximately 70 nL volume) even though the sampling rate by electroosmotic flow (EOF) was only 2 nL min−1. The design shown in Fig. 1 allowed fluid to be pumped into the islet chamber at microliter per minute flow rates, sufficient to wash-out the cell chamber in a few seconds even as a small fraction of the perfusate is sampled by EOF. The majority is pumped out the low flow-resistance opening above the cell chamber.Several methods were considered for pumping media into the chip. EOF was inadequate to produce the necessary flow rates, especially when using high ionic strength islet media, and would place a damaging electric field across the islet. A second method was use of a mechanical syringe pump (Model 55–3206, Harvard Apparatus, Inc., Holliston, MA) to drive the perfusion buffer. This technique failed due to high irreproducibility of flow. We then investigated the use of gas-pressure driven flow. Helium was used to minimize degassing and avoid oxidation of buffer constituents or insulin. As some physiological buffers, including the RPMI 1640 cell culture media, are based on bicarbonate and intended to be used in a 5% CO2 atmosphere, it was necessary to add a different buffering agent to these solutions (25 mM HEPES). The use of gas pressure proved to be simple and reliable route to pumping into the chip.
The perfusion inlet to the 300 µm diameter cell chamber was a comparatively small 6 µm deep channel, leading to the possibility of heterogeneous distribution of molecules that flow into the chamber. This situation was expected to be exacerbated by the presence of a 100–200 µm diameter islet within the cell chamber. To test this possibility, the cell chamber was imaged with and without an islet as 100 nM fluorescein was pumped into it from the perfusion channel at 1.5 µL min−1 (Fig. 2). Fig. 2a and b illustrates that the fluorescence intensity was homogenous across the cell chamber within the limits of the measurement technique. (Imaging was performed at 10× magnification resulting in a roughly 250 µm laser spot illuminating the 300 µm cell chamber. The Gaussian laser beam profile results in a peaked fluorescent intensity in the center of the chamber.) With an islet present, the fluorescence distribution was still relatively even, although it was apparent that the side of the islet opposite the flow input received a slightly lower concentration of fluorescein than the inlet side (Fig. 2d and e).
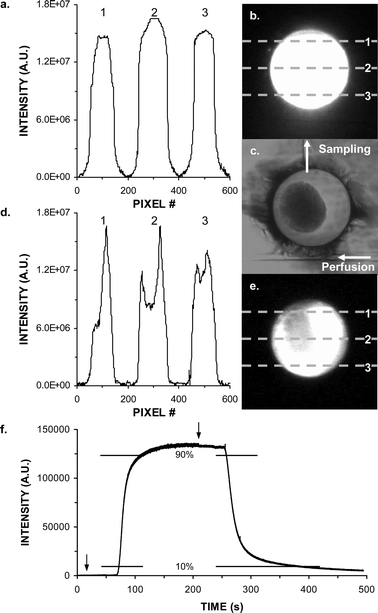 | ||
| Fig. 2 Images and analysis of solution replacement within the islet chamber. (a) Fluorescence intensity across a cell chamber perfused with 100 nM fluorescein at 1.5 µL min−1. Numbers correspond to the pixel row scanned. Lines correspond to different locations in chamber as depicted in the fluorescent image in (b). (c) Brightfield image of chamber containing an islet. Perfusion inlet channel, sample outlet channel, and fluid flow directions are marked. (d) Same as (a) except with an islet present in the chamber. Scan line positions are shown in fluorescent image in (e). (f) Fluorescence intensity detected in reaction channel as perfusion media switched from 0 nM to 250 nM to 0 nM fluorescein in BSS. Solution changes are marked with arrows. 10% and 90% of maximal intensity are marked with vertical bars. | ||
In order to determine the efficiency of sampling a rapid change in the cell chamber, a 250 nM fluorescein solution in BSS was pumped into the chamber followed by a 0 nM fluorescein solution at 0.6 µL min−1 (Fig. 2f) while monitoring fluorescence in the reaction channel adjacent to the intersection of the immunoassay reagents and islet sampling channels. A delay time of 80 s was observed from commencing flow to detecting a fluorescence signal that was 10% of the maximum intensity. The rise time of the fluorescence increase from 10% to 90% maximum intensity was 30 s. Upon switching to blank solution the 80 s delay was again observed and the fluorescence dropped from 90% to 10% intensity over 70 s. These times represent the upper limit of temporal resolution because at least part of the spread of signal was due to the time required to rinse out the perfusion channel and cell chamber. As illustrated with actual islet measurements (see below), faster responses can be measured when the analyte molecule is produced directly within the chamber. These results also confirm that the perfusion system was adequate for rapidly changing concentration of drugs or nutrients (e.g. glucose) that were applied to the islet chamber.
On-line immunoassay with perfusion
Calibration of the chip was performed by perfusing different concentrations of insulin into the sample chamber while continuously sampling from the cell chamber. A typical calibration curve from 5 nM to 1 µM with insulin dissolved in RPMI 1640 is shown in Fig. 3a. The limit of detection (LOD), calculated as the concentration required to obtain a B/F change 3 times the standard deviation of the B/F ratio at 0 nM insulin, was 0.8 nM. This LOD was an improvement of about 4-fold over our previous system.22 The improved LOD is attributed to several small refinements in the system including use of different buffers and fresh reagents. Calibrations were reproducible on consecutive days as long as the same reagent stocks were used; however, over a two-week period the response drifted enough to require re-calibration (see Fig. 3b). For all islet experiments calibrations were performed daily.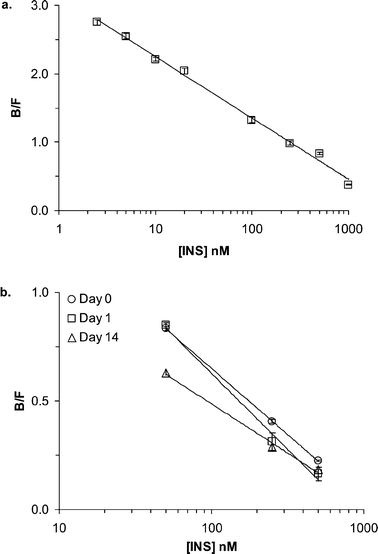 | ||
| Fig. 3 Calibration curves plotting B/F peak height ratios versus insulin concentrations obtained on the device while perfusing RPMI 1640 at 1.5 µL min−1. (a) Calibration of 5 nM to 1 µM insulin standards. Points were fitted by a simple logarithmic function with R2 = 0.995. (b) Calibrations obtained on consecutive days (Day 0 and Day 1) and two weeks later (Day 14) on the same device. | ||
We also determined the stability of immunoassay during continuous operation in a single day as this will limit the time that a single islet can be monitored. Fig. 4a illustrates the B/F calculated from 720 electropherograms collected at 10 s intervals for 2 h while perfusing the chip with 50 nM insulin. After an initial increase in B/F associated with the device reaching a stable temperature, the relative standard deviation (RSD) of B/F was 6%. Example electropherograms acquired at 20 min and 2 h are compared in Fig. 4b. At the end of 2 h the baseline in the electropherograms had begun to rise, apparently due to degradation of HEPES in the electrophoresis buffer; the electropherograms in Fig. 4b and c have been normalized to zero, resulting in the apparent lower absolute intensities in Fig. 4c relative to 4b due to the elevated baseline after 2 h. In addition, evaporation of all solutions except the perfusion media became a factor after this length of time. (The reservoirs storing the solutions were covered; however, they were only loosely capped to prevent negative pressure build-up in the reservoir as reagents were consumed.) This performance was a significant improvement over the non-perfusion system which could only be maintained for 30 min without intervention.22 The non-perfusion device may have suffered from degradation of insulin standards over time at atmospheric conditions, whereas the new device constantly replenished the standards that were maintained in a pressurized helium environment. Continual or periodic replenishment of the immunoassay reagent and electrophoresis solutions would likely allow operation of the perfusion chip even longer. Such replenishment could be achieved automatically by adding perfusion lines to the appropriate reservoirs or manually by pipetting fresh solutions into the chip.
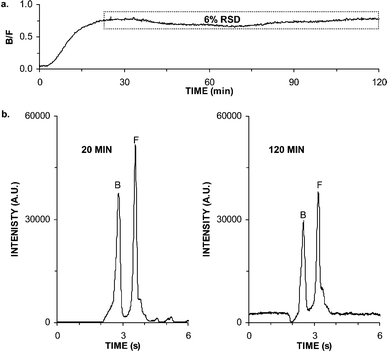 | ||
| Fig. 4 Stability for continuous operation. (a) Bound to free ratios (B/F) during a continuous 2 h perfusion of 50 nM insulin. Dashed box shows region used to calculate relative standard deviation (RSD). (b) Electropherograms collected during the experiments depicted in (a) at 20 min and 2 h. Peaks corresponding to free FITC-insulin (F) and bound to antibody (B) are marked. | ||
In addition to long term operation over a single day, we found that it was possible to re-use individual chips for multiple experiments. A single chip was utilized for over 6 months and several islets (discussed below) as part of this study. Such long-term operation was attributed to the use of the cleaning procedures outlined in the experimental section and using purified solutions within the chip.
Monitoring insulin secretion from single islets in RPMI 1640
The chip was used to monitor insulin secretion induced by step changes from 3 to 11 to 3 mM glucose at single islets perfused with RPMI 1640. The addition of an islet to the cell chamber did not appear to affect the separation of the immunoassay reagents (Fig. 5a). All islets tested (n = 3) exhibited a biphasic response with an initial high burst of secretion followed by a lower second phase period (see example in Fig. 5b), which has been previously demonstrated with isolated mouse islets using traditional methods.21 Average basal levels prior to increasing the glucose concentration were 82 ± 29 pg min−1. The average maximal response of the first phase (a rise in insulin release of >10% from basal after stimulation) was 400 ± 81 pg min−1, which is comparable to previous reports. For example, the maximal secretion response of single islets from ob/ob mice was ca. 600 pg min−1 as monitored by ELISA.26 Two of the islets exhibited regular oscillatory patterns during the second phase with a period of 2.7 ± 0.1 min, similar to previously reported values of 2–3 min cycles of secretion.27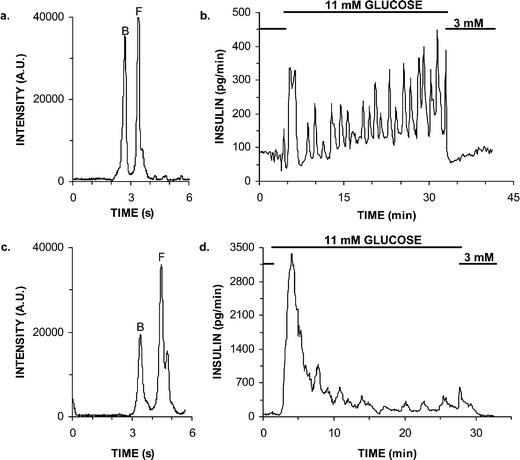 | ||
| Fig. 5 Monitoring insulin release during glucose step changes during perfusion of various buffers. (a) Electropherogram collected during on-line monitoring of a single islet during RPMI 1640 perfusion. (b) Insulin release of an islet as glucose was raised from 3 to 11 mM, followed by a return to 3 mM. Islet was perfused RPMI 1640 cell culture media at 1.5 µL min−1. (c) Electropherogram collected during on-line monitoring of a single islet during balanced salt solution (BSS) perfusion. (d) Insulin release of an islet as glucose was raised from 3 to 11 mM, followed by a return to 3 mM. Islet was perfused at BSS at 0.6 µL min−1. | ||
The results of monitoring single islets in RPMI 1640 demonstrate that the system has both the sensitivity and temporal resolution to detect oscillations in insulin secretion. After first phase and during oscillations, insulin levels returned to nearly basal levels in as little as 30 s, demonstrating that secreted insulin was effectively removed by the perfusion. Additionally, when islets were returned to 3 mM glucose from 11 mM, the levels returned to basal amounts within 60 s without any further pulses of secretion demonstrating that the glucose was rapidly washed out of the cell chamber as expected from the results in Fig. 2. Detection of such rapid changes in secretion also illustrate that sampling occurs with high temporal resolution.
Islets were monitored for up to an hour without any morphological signs of damage, such as cell lysis or dispersion of the islet membrane. Additionally, unregulated insulin secretion was not observed; islets maintained either regular oscillatory or stable, raised second phase secretion upon glucose stimulation, and steady basal levels of insulin release following glucose stimulation were observed, providing further evidence of the lack of cellular damage during the course of the experiments. The stability of islets indicates that the constant flow of buffer and electrical effects within the cell chamber are not detrimental to the cells over the time scale of the measurements used here. The possibility of electrical effects within the islet chamber arises from the use of electroosmotic flow, achieved by grounding the islet chamber and applying voltage at a downstream reservoir, for sampling. We estimate that the voltage dropped within the islet chamber is inconsequential (3 × 10−18 V) and the current density is just 38 µA mm−2. Given these low values, it is not surprising that the islets retained structural integrity and normal physiological responses within the chamber.
It is reasonable to expect that islet monitoring would be limited only by the reagent stability (2 h as mentioned above), because the islets are maintained on the device in conditions similar to those in an incubator, i.e. at 37 °C with a constant supply of fresh cell culture media. Longer-term measurements may be possible with chips that incorporate continual immunoassay reagent and separation solution replacement.
Monitoring insulin secretion from single islets in BSS
Although islets are commonly cultured in RPMI 1640, it is also important to be able to study islets in minimal, well-defined buffered saline solutions. An example of such a solution used for mammalian studies is modified Krebs Ringer buffer (KRB)28 containing (in mM): 118 NaCl, 5.4 KCl, 2.4 CaCl2, 1.2 MgSO4, 1.2 KH2PO4, 3 glucose, and 20 HEPES. Experiments with KRB flowing through the cell chamber and immunoassay reagents dissolved in PETA buffer failed due to CaHPO4 precipitating within the channel leading from the islet to the reaction channel. Precipitation of this salt is not surprising given that the saturation concentration of HPO42− within KRB containing 2.4 mM Ca2+ is estimated to be 1 mM according to:29| Ksp = [Ca2+]γCa2+[HPO42−]γHPO42− = 2.6 × 10−7 | (1) |
We then explored phosphate-free balanced salt solution (BSS), which is also commonly used as a defined media for physiological experiments.26,27 Utilization of BSS in combination with HEAT40 as the reagent solvent eliminated precipitation in the channels even during extended operation. Under these conditions it was necessary to thoroughly rinse out NaOH, used to clean the chips, during conditioning to prevent Ca(OH)2 from forming when BSS was introduced to the chip.
Upon switching to the BSS solution for perfusion and HEAT40 as reagent solvent, several smaller changes in the operation of the device were made including: lowering the perfusion flow rate from 1.5 µL min−1 to 0.6 µL min−1, reducing the immunoassay reagent concentrations from 150 nM to 100 nM FITC-insulin and 75 nM to 50 nM Ab, and increasing the separation field strength from 500 V cm−1 to 600 V cm−1. With these modifications, resolution of B and F was still adequate (see Fig. 5c); however, electropherograms could be stably acquired at 6 s intervals instead of 10 s.
Using the new operation parameters, the chip was used to monitor insulin secretion from individual islets perfused with BSS while the glucose concentration was stepped from 3 to 11 to 3 mM. Similar to RPMI 1640, biphasic responses were observed in all islets studied. Average basal levels prior to increasing the glucose concentration were 47 ± 34 pg min−1. The maximal response during first phase was 2360 ± 400 pg min−1, which was much higher than the first phase response observed with RPMI 1640. This greater initial burst of insulin release was likely due to the higher concentration of Ca2+ in BSS (2.4 mM) relative to RPMI 1640 ([Ca2+] = 0.424 mM),30 because entry of extracellular Ca2+ is a primary trigger of insulin secretion. The second phase of secretion was similar in the two buffers, both in terms of absolute level and oscillations. With BSS, 2 of 3 islets displayed oscillations with a period of 2.3 ± 0.2 min. (It is not uncommon for only a fraction of islets to display oscillations.)
These results demonstrate that the microfluidic device can perfuse and sample from both complex cell culture media and a minimal salt solution, while still performing an on-line immunoassay. The primary problem in changing buffers was clogging of microfluidic channels associated with precipitation of phosphate salts. These problems can be circumvented with appropriate tailoring of solutions.
Conclusions
We have demonstrated a novel system for high-resolution monitoring of insulin secretion from single islets of Langerhans. The results, in terms of temporal resolution, were comparable to those of the best previous off-line methods utilizing ELISA, but were obtained in a highly automated, on-line device with real-time read-out. The method was an advancement over a previously reported microfluidic chip for islet monitoring in that it allowed the cells to be continually perfused thus allowing temporally resolved measurements, longer-term cell survival in the chip, and rapid changing of solutions contacting the cells. Furthermore, minor modifications in chip operation improved detection limits 4-fold to 0.8 nM, sampling frequency 3-fold to 6 s, and long term stability 4-fold to 2 h compared to the previously reported chip. Maintenance-free operation time could likely be extended by automated replenishment of buffer and reagent reservoirs. The device was shown to be compatible with both simple salt media and complex cell culture media. A limitation of the use of microchannels is that precipitation of certain salts, typically not problematic in macroscale devices, must be avoided. Although microfluidic chips are hailed for their disposability, the device could be reused with appropriate care and cleaning. While the system was demonstrated for routine monitoring of insulin secretion, it seems feasible to extend it to other cell types and hormones with minor modification.Acknowledgements
This work was supported by National Institutes of Health grant DK46960. J.G.S. received partial support from the Eastman Chemical Company through an analytical focus fellowship.References
- M. Valdeolmillos, R. M. Santos, D. Contreras, B. Soria and L. M. Rosario, FEBS Lett., 1989, 259, 19–23 CrossRef CAS.
- P. Bergsten, E. Grapengiesser, E. Gylfe, A. Tengholm and B. Hellman, J. Biol. Chem., 1994, 269, 8749–8753 CAS.
- Y. J. Liu, A. Tengholm, E. Grapengiessar, B. Hellman and E. Gylfe, J. Physiol., 1998, 508.2, 471–481 CAS.
- M. A. Ravier and J. C. Henquin, FEBS Lett., 2002, 530, 215–219 CrossRef CAS.
- S. Zraika, M. Dunlop, J. Proietto and S. Andrikopoulos, Arch. Biochem. Biophys., 2002, 405, 275–279 CrossRef CAS.
- D. P. Schrum, C. T. Culbertson, S. C. Jacobson and J. M. Ramsey, Anal. Chem., 1999, 71, 4173–4177 CrossRef CAS.
- S. Gawald, L. Schild and Ph. Renaud, Lab Chip, 2001, 1, 76–82 RSC.
- M. A. McClain, C. T. Culbertson, S. C. Jacobson and J. M. Ramsey, Anal. Chem., 2001, 73, 5334–5338 CrossRef CAS.
- S. Fiedler, S. G. Shirley, T. Schnelle and G. Fuhr, Anal. Chem., 1998, 70, 1909–1915 CrossRef CAS.
- A. Y. Fu, C. Spence, A. Scherer, F. H. Arnold and S. R. Quake, Nat. Biotechnol., 1999, 17, 1109–1111 CrossRef CAS.
- A. Wolff, I. R. Perch-Nielsen, U. D. Larsen, P. Friis, G. Goranovic, C. R. Poulsen, J. P. Kutter and P. Telleman, Lab Chip, 2003, 3, 22–27 RSC.
- P. C. H. Li and D. J. Harrison, Anal. Chem., 1997, 69, 1564–1568 CrossRef CAS.
- P. Wilding, L. J. Kricka, J. Cheng, G. Hvichia, M. A. Shoffner and P. Fortina, Anal. Biochem., 1998, 257, 95–100 CrossRef CAS.
- J. Gao, X. F. Yin and Z. L. Fang, Lab Chip, 2004, 4, 47–52 RSC.
- I. Inoue, Y. Wakamoto, H. Moriguchi, K. Okano and K. Yasuda, Lab Chip, 2001, 1, 50–55 RSC.
- A. R. Wheeler, W. R. Throndset, R. J. Wheelan, A. M. Leach, R. N. Zare, Y. H. Liao, K. Farrell, I. D. Manger and A. Daridon, Anal. Chem., 2003, 75, 3581–3586 CrossRef CAS.
- M. Brischwein, E. R. Motrescu, E. Cabala, A. M. Otto, H. Grothe and B. Wolf, Lab Chip, 2003, 3, 234–240 RSC.
- W. H. Huang, W. Cheng, Z. Zhang, D. W. Pang, Z. L. Wang, J. K. Cheng and D. F. Cui, Anal. Chem., 2004, 76, 483–488 CrossRef CAS.
- R. N. Kulkarni, Int. J. Biochem. Cell Biol., 2004, 36, 365–371 Search PubMed.
- R. T. Kennedy, L. M. Kauri, G. M. Dahlgren and S. K. Jung, Diabetes, 2002, 51, S152–S161 Search PubMed.
- J. C. Henquin, N. Ishiyama, M. Nenquin, M. A. Ravier and J. C. Jonas, Diabetes, 2002, 51, S60–S67 Search PubMed.
- M. G. Roper, J. G. Shackman, G. M. Dahlgren and R. T. Kennedy, Anal. Chem., 2003, 75, 4711–4717 CrossRef CAS.
- J. G. Shackman, C. J. Watson and R. T. Kennedy, J. Chromatogr., A, 2004, 1040, 273–282 CAS.
- S. C. Jacobson, S. V. Ermakov and J. M. Ramsey, Anal. Chem., 1999, 71, 3273–3276 CrossRef CAS.
- W. F. Pralong, C. Bartley and C. B. Wollheim, EMBO, 1990, 9, 53–60 Search PubMed.
- P. Bergsten and B. Hellman, Diabetes, 1993, 42, 670–674 Search PubMed.
- P. Bergsten and B. Hellman, Biochem. Biophys. Res. Commun., 1993, 192, 1182–1188 CrossRef CAS.
- R. M. C. Dawson, D. C. Elliott, W. H. Elliott, and K. M. Jones, Data for Biochemical Research, Oxford University Press, New York, 3rd edn., 1986 Search PubMed.
- D. C. Harris, Quantitative Chemical Analysis, W. H. Freeman and Co., New York, 4th edn., 1995 Search PubMed.
- G. E. Moore, R. E. Gerner and H. A. Franklin, JAMA, 1967, 199, 519–524 Search PubMed.
| This journal is © The Royal Society of Chemistry 2005 |