Catalytic asymmetric hydroamination of non-activated olefins
Kai C.
Hultzsch
*
Institut für Organische Chemie, Friedrich-Alexander Universität Erlangen-Nürnberg, Henkestr. 42, D-91054 Erlangen, Germany. E-mail: hultzsch@chemie.uni-erlangen.de; Fax: +49-9131-8526865; Tel: +49-9131-8526866
First published on 7th April 2005
Abstract
Hydroamination is a highly atom-economical process in which an amine N–H functionality is added to an unsaturated carbon–carbon linkage. This potentially highly useful process gives access to various nitrogen-containing basic and fine chemicals as well as naturally occurring alkaloid skeletons. Asymmetric hydroamination reactions promoted by chiral catalysts are particularly attractive. This review highlights recent progress in the development of early transition metal catalysts for the asymmetric hydroamination of non-activated alkenes.
 Kai C. Hultzsch | Kai Carsten Hultzsch studied chemistry at the University of Mainz and Toronto from 1991 to 1996. He finished his PhD in 1999 under the guidance of Jun Okuda in Mainz. After two years as a Feodor Lynen postdoctoral fellow in the group of Richard R. Schrock at MIT, he began his independent research career in 2001 as a DFG Emmy Noether fellow at the University of Erlangen-Nürnberg. He is the recipient of the Wöhler young investigator award and the ADUC award for habilitands. His main areas of interest are transition metal catalysed stereoselective olefin heterofunctionalisations, as well as (co)polymerisation of non-polar and polar monomers. |
Introduction
The efficient synthesis of nitrogen-containing compounds has sparked significant research efforts, due to their high relevance in biological systems and industrially important basic and fine chemicals. Although many synthetic methods have been devised over the last century,1 one of the simplest synthetic approaches, the hydroamination, has become only the focus of attention with the advent of transition metal catalysts. The addition of amine N–H functionalities to unsaturated carbon–carbon bonds, either in an intermolecular [eqn. (1)] or intramolecular [eqn. (2)] fashion, generates amines in a waste-free, highly atom-economical manner starting from simple and inexpensive precursors.2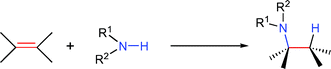 | (1) |
 | (2) |
Although the direct addition of amines to alkenes is thermodynamically feasible (ΔH![[circle, cut, short horiz bar]](https://www.rsc.org/images/entities/char_e0eb.gif) ≈
−52.7 kJ mol−1, ΔS
≈
−127.3 J K−1 mol−1 for the addition of ammonia to ethylene),2a,b the hydroamination usually requires assistance from a catalyst:
≈
−52.7 kJ mol−1, ΔS
≈
−127.3 J K−1 mol−1 for the addition of ammonia to ethylene),2a,b the hydroamination usually requires assistance from a catalyst:
• an electrostatic repulsion between the nitrogen lone pair of the approaching amine and the π-bond of the electron-rich olefin results in a high activation barrier;
• a concerted [2 + 2] addition of the N–H bond to the alkene is an orbital-symmetry forbidden process and is unfavourable due to the large difference in energy between the carbon–carbon π-bond and the N–H σ-bond;
• the equilibrium is shifted towards the starting materials at elevated temperatures due to the negative reaction entropy.
Therefore, uncatalysed additions are commonly only observed in hydroamination reactions of activated, electron-deficient alkenes.3 Increasing research effort has led to the development of many transition metal based catalyst systems (early transition metals of groups 3–5, lanthanides and actinides,2g,h and late transition metals of groups 8–10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 2f
2f![[hair space]](https://www.rsc.org/images/entities/char_200a.gif) ) over the last two decades. Despite this progress, substrate diversity for most catalyst systems has remained limited.
) over the last two decades. Despite this progress, substrate diversity for most catalyst systems has remained limited.
Because of the chirality of many hydroamination products, asymmetric hydroamination (AHA) carried out with chiral catalysts is an attractive, but highly challenging task.4 This review will focus in particular on the progress in the catalytic asymmetric hydroamination of non-activated alkenes, a domain for rare earth metal complexes and more recently also group 4 metal catalysts. Asymmetric hydroamination of activated alkenes (vinyl arenes,5a–c 1,3-dienes,5d norbornene5e) or alkynes,5f a typical domain of late transition metal catalysts, is out of the scope of this review. Furthermore, asymmetric aza-Michael reactions,6 in which the catalyst merely plays the role of a chiral Lewis acid, will not be covered.
Rare earth metal based catalysts
The synthesis and handling of organometallic compounds of the rare earth metals (including Sc, Y, La to Lu) is complicated by their high sensitivity towards air and moisture. Nevertheless, their unique chemical behaviour and high activity in a wide variety of catalytic applications7 has fuelled intensive research efforts. With improved and simplified synthetic protocols, e.g. alkane or amine elimination routes, these catalysts can often be prepared in situ using standard Schlenk line and glove box techniques. However, major drawbacks remain, such as their reduced catalytic performance in the presence of strong coordinating groups (e.g. ethers) and incompatibility with protic functional groups (e.g. alcohols, acids).In their seminal work, Marks and co-workers showed that organometallic rare earth metal complexes are particularly suitable for the hydroamination/cyclisation of terminal alkenes, 1,3-dienes, allenes and alkynes.2h Internal olefins react under more forcing elevated reaction temperatures.8 Although most intramolecular hydroamination reactions proceed readily at room temperature, intermolecular hydroamination reactions are usually more problematic. Rates for the intermolecular process are usually two to three orders of magnitude slower,9 due to entropic disadvantages and unfavourable competition between strongly binding amines and weakly coordinating olefins.
Common rare earth metal based precatalysts comprise of at least one amido or alkyl ligand which is protolytically substituted for an aminoalkene substrate during the catalyst activation step of the hydroamination/cyclisation (Scheme 1). The key step of the catalytic cycle involves rate-limiting insertion of the olefin into the rare earth metal amido bond in a chair-like transition state.10 Thus, reactions are usually zero order in substrate concentration and first order in catalyst concentration. Subsequent protonation of the resulting rare earth metal alkyl species by a second amine molecule regenerates the rare earth metal amido species and releases the heterocyclic product.
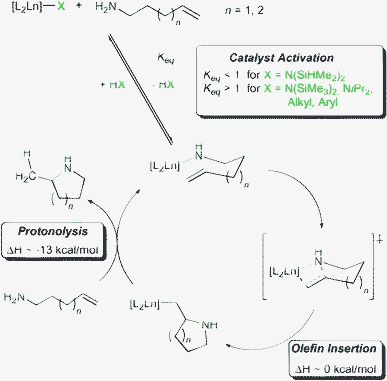 | ||
| Scheme 1 Simplified mechanism for rare earth metal catalysed hydroamination/cyclisation. | ||
Asymmetric hydroaminations were first reported in 1992 using C1-symmetric chiral ansa-lanthanocenes (Fig. 1).11 Cyclisation of aminopentene substrates generated pyrrolidine products in up to 74% ee (Table 1) at low temperatures. Enantioselectivities for piperidines were much less favourable until the ‘extended wingspan’ catalysts (S)-211c were introduced recently, which allowed the facile synthesis of enantioenriched (+)-coniine via aminodiene cyclisation (Scheme 2).11e Although most precatalysts could be prepared in diastereomeric pure form, the catalysts underwent facile epimerisation under the conditions of catalytic hydroamination via reversible protolytic cleavage of the metal cyclopentadienyl bond (Scheme 3).8d,11b–e Therefore, enantiomeric excess and sense of optical rotation of the cyclised products were independent of the diastereomeric purity of the lanthanocene precatalyst.
![Chiral lanthanocene precatalysts for asymmetric hydroamination [R* = (+)-neomenthyl (1a), (−)-menthyl (1b), (−)-phenylmenthyl (1c); Ln = La, Nd, Sm, Y, Lu; E = CH, N].](/image/article/2005/OB/b418521h/b418521h-f1.gif) | ||
| Fig. 1 Chiral lanthanocene precatalysts for asymmetric hydroamination [R* = (+)-neomenthyl (1a), (−)-menthyl (1b), (−)-phenylmenthyl (1c); Ln = La, Nd, Sm, Y, Lu; E = CH, N]. | ||
|
|
||||||
|---|---|---|---|---|---|---|
| Cat. | Substr. | [cat]/[s] (%) | T/°C | t/h; conv. (%) | TOF/h−1 | ee (%) (config.) |
| (S)-1b-Sm | 11 | 25 | 84 | 53 (S) | ||
| (S)-1b-Sm | 11 | −30 | 74 (S) | |||
| 4a | 11 | 4 | 70 | 22; 77 | 2.5 | 36 (S) |
| 4b | 11 | 2 | 25 | 1; 98 | 61 | 8 (S) |
| 5 | 11 | 4 | 60 | 7; 92 | 13.5 | 28.3 (S) |
| 6a | 11 | 4 | 22 | 3; ![[greater than or equal, slant]](https://www.rsc.org/images/entities/char_2a7e.gif) 98 98 |
8 | 43 (S) |
| 6b | 11 | 4 | 22 | 2; 98 |
14 | 53 (S) |
| 7a | 11 | 3 | 60 | 336; 100 | 50 | |
| 7b | 11 | 3 | 35 | 120; 100 | 45 | |
| 8 | 11 | 1 | 70 | 40; 100 | 61 | |
| 9 | 11 | 5 | 23 | —; >98 | 25 | 67 (R) |
| (S)-1b-Sm | 13 | 25 | 33 | 62 (S) | ||
| (S)-1b-Sm | 13 | 0 | 72 (S) | |||
| 5 | 13 | 4 | 70 | 43; 91 | 57 (S) | |
| 6a | 13 | 4 | 22 | 24; 98 |
1.2 | 69 (S) |
| 6b | 13 | 4 | 22 | 20; 98 |
2.2 | 83 (S) |
| 9 | 13 | 5 | 23 | 0.09 | 40 (R) | |
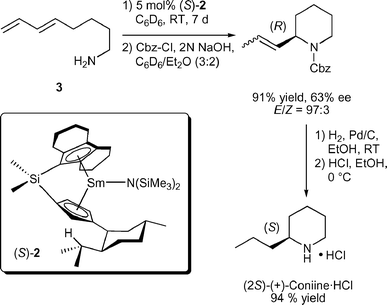 | ||
| Scheme 2 Synthesis of (+)-coniine·HCl via enantioselective aminodiene hydroamination/cyclisation (Cbz = benzyloxycarbonyl). | ||
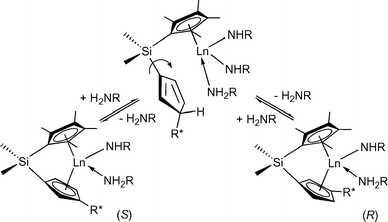 | ||
| Scheme 3 Epimerisation of chiral lanthanocene complexes during the hydroamination reaction. | ||
The limitations in enantioselectivity and configurational stability of chiral lanthanocene hydroamination catalysts made the development of catalyst systems based on non-cyclopentadienyl ligand sets imminent. The desired catalyst system should have comparable catalytic activity to the lanthanocene catalyst systems, while allowing a facile catalyst tuning through modular ligand structures in order to achieve efficient enantioselectivities. Furthermore, the catalyst should be prepared conveniently, without the need for elaborate purification techniques, and should be configurationally stable under the conditions of catalytic hydroamination.
Indeed, several new chiral rare earth metal hydroamination catalysts were introduced recently (Fig. 2, Table 1) utilising various non-metallocene ligand frameworks, such as biphenolate (4),12a,b binaphtholate (5 and 6a,b),12a,c biaryl diamido (7a,b),13a,c aminophenolate (8),13b bisoxazolinato (9)14 and diamidobinaphthyl (10)15 ligands.
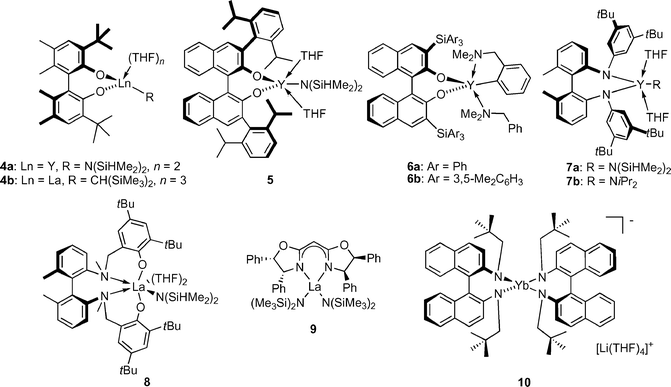 | ||
| Fig. 2 Chiral, non-metallocene, rare earth metal catalysts for asymmetric hydroamination. | ||
Catalyst systems 4a, 5, 7a and 8 displayed only low catalytic activity and required elevated reactions temperatures (60 °C). Here, the low basicity of the N(SiHMe2)2 ligand hampers catalyst activation (Scheme 1, Keq < 1) so that the majority of the precatalyst remains unreactive in the catalytic reaction. Activation of precatalysts having more basic N(SiMe3)2, NiPr2, alkyl or aryl ligands proceeds more smoothly and significantly better catalyst performance can be observed (Scheme 1, Keq > 1). Hence, complexes 4b, 6a, 6b and 9 reached appreciable turnover rates at room temperature, which are of the same order of magnitude observed for lanthanocenes.
The second, more challenging task was tuning the ligand for optimal enantioselectivities and prevention of undesired catalyst aggregation. The biphenolate catalysts 4a,b can serve as illustrative examples: the tert-butyl groups are not sterically demanding enough and shield the metal insufficiently. This results in poor enantioselectivities and ready formation of catalytically less active phenolate-bridged dimeric species in the absence of THF.12a,b Sterically more demanding 2,6-diisopropylphenyl substituents in the binaphtholate complex 5 effectively prevented these aggregations and enantioselectivities up to 57% ee were obtained. Sterically more demanding tris(aryl)silyl substituents in complexes 6a,b further improved the selectivity. The highest turnover frequencies and enantioselectivities (up to 83% ee) were achieved with the sterically most hindered catalyst 6b.12c
The biaryl diamido complexes 7a were only moderately selective.13a Increasing the denticity of the ligand by introducing ortho-anisole substituents on nitrogen did not improve enantioselectivity.13c The aminophenolate catalyst 8 was more selective (61% ee for 11), due to a better ‘reach-around’ of the chelating aminophenolate ligand.13b Shorter linkages between the phenol unit and the biaryl amine resulted in a significantly less selective stereodifferentiation.
The bisoxazolinato compexes 9 displayed ligand accelerated behaviour,14,16 with maximum rates and highest enantioselectivities observed with a 1 : 1 ligand-to-metal ratio. Variation of the bisoxazoline ligand substitution pattern revealed that an aromatic group in the 4-position and an alkyl or aryl substitution in the 5-position were highly beneficial for catalyst activity and selectivity. A model explaining the observed stereochemistry was proposed based on molecular modelling studies (Fig. 3). Most interestingly, substitution of the aryl group in the 4-position of the ligand for an alkyl substituent led to inversion of product configuration, possibly due to a change in the mode of approach of the olefin to the La–N bond.
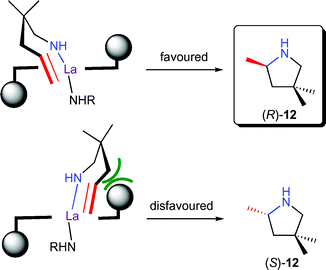 | ||
| Fig. 3 Stereomodel for enantioselective hydroamination/cyclisation of aminopentene 11 by 9 with an equatorial approach of the olefin. | ||
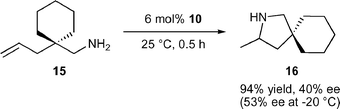
The ate-complex 1015 is an unusual hydroamination catalyst system as it is the only ate-complex reported to catalyse hydroamination and lacks a reactive amido or alkyl group. It seems feasible that at least one of the diamidobinaphthyl amido groups is protonated, which could ultimately result in loss of one diamidobinaphthyl ligand. Cyclisation of 15, which is highly activated due to the Thorpe–Ingold effect, to the spirocyclic pyrrolidine 16 proceeded in good activity but with only moderate enantioselectivity [eqn. (3)]. The role of the counter-cation with respect to activity and selectivity is currently unclear.
 | (3) |
Note that there are two opposite trends in the dependence of enantioselectivity on the ionic radius of the metal. For systems based on biphenolate (4a,b), binaphtholate (6a,b), biaryl diamido (7a), and diamidobinaphthyl (10) ligands the selectivity increased with decreasing ionic radius, whereas the opposite was true for aminophenolate (8) and bisoxazolinato (9) complexes. Chiral lanthanocene complexes (1 and 2) on the other hand were most selective with samarium as the central ion.
Kinetic resolution of chiral aminoalkenes
Kinetic resolution processes play a pivotal role in many asymmetric syntheses.17 Early attempts to perform kinetic resolution of 17a using chiral lanthanocene complexes were frustrated by low enantiomeric excess (<20% ee) at various extents of conversion.11b However, significant higher efficiencies were observed using binaphtholate complexes 6a,b (Table 2) as indicated by krel values17 as high as 16.12c The 2,5-disubstituted pyrrolidines were obtained in good to excellent trans diastereoselectivity, depending on the steric hindrance of the α-substituent. The sterically more hindered binaphtholate catalyst 6b was more effective in the kinetic resolution of the sterically less demanding 17a, whereas sterically more encumbered 17b was more efficiently resolved using the sterically less hindered 6a.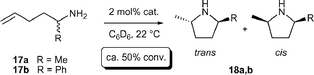
The preferred formation of (2S,5S)-17a using catalyst 6b can be explained with an impediment of the cyclisation of (R)-17a by a sterically unfavourable interaction of the substrate with a trisarylsilyl substituent in the chair-like transition state (Fig. 4).
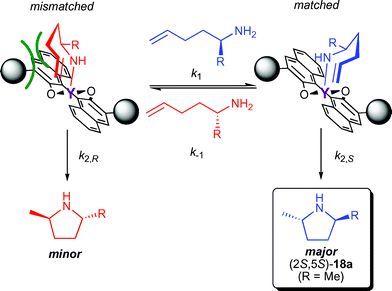 | ||
| Fig. 4 Proposed stereomodel for the kinetic resolution of chiral aminoalkenes with an equatorial approach of the alkene. | ||
Group 4 metal based catalysts
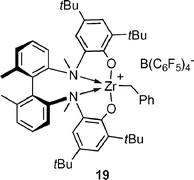
The application of group 4 metal catalysts in asymmetric hydroamination reactions would be highly desirable as they are easier to prepare and less sensitive than rare earth metal complexes. Their importance in polyolefin synthesis18 has led to the development of many chiral group 4 metal complexes, some of which are even commercially available.Unfortunately, neutral group 4 metal complexes are commonly restricted to the catalytic hydroamination of alkynes and allenes only.2g The mechanism involves a [2 + 2] cycloaddition of an alkyne or allene to the catalytically active metal imido species, whereas simple alkenes do not react. Although asymmetric cyclisation of aminoallenes to vinylpyrrolidines is possible,19 the decisive breakthrough was reported by Scott and co-workers using the cationic aminophenolate complex 19,
|
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Substr. | [cat]/[s] (%) | T/°C |
t/ha |
ee (%) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| a Time to 100% conversion of substrate. b 70% conversion to hydroamination product and 30% double bond isomerised aminoalkene. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 20a | 10 | 100 | 4 | 64 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 20b | 5 | 70 | 48 | 14b | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 20c | 10 | 100 | 3 | 82 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
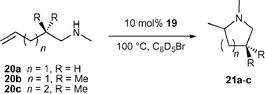
However, cyclisation of 20b at 70 °C was significantly less enantioselective and partial double bond isomerisation of the starting material was observed.
The mechanism is believed to be similar to that proposed for rare earth metal catalysts, with a catalytically active cationic zirconium amido species. No reaction was observed with primary aminoalkenes, because cationic zirconium amido species are readily deprotonated to yield catalytically inactive zirconium imido species (Scheme 4).
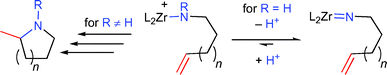 | ||
| Scheme 4 Reaction pathways for primary and secondary aminoalkenes with cationic zirconium catalysts (n = 1, 2). | ||
Future perspectives
Despite significant recent progress many challenges still lie ahead. So far, only relative simple substrates leading to pyrrolidines and piperidines have been investigated with chiral catalysts, but larger rings and more complex (e.g. bicyclic) skeletons are in principle accessible too.8a,b,9b,21 Another test case will be the application of these systems in intermolecular hydroamination reactions, which might require further catalyst tuning. In spite of the low functional group tolerance and higher air- and moisture-sensitivity of early transition metal catalyst systems, chiral catalyst systems based on late transition metals may be highly desirable, but so far only activated substrates do react.5,6 The ultimate and final goal, from an atom-economical and cost-efficiency point of view, remains the direct asymmetric hydroamination of non-activated alkenes using ammonia as sole nitrogen source.Research groups around the globe have accepted the challenge to develop the asymmetric hydroamination into a highly useful catalytic technique in the toolbox for synthetic organic chemists. The payoff for these endeavours will be a facile access to highly valued target compounds starting from simple feed-stocks.
Acknowledgements
Generous financial support by the Deutsche Forschungsgemeinschaft (DFG) and the Fonds der Chemischen Industrie is gratefully acknowledged. K. C. H. is a DFG Emmy Noether fellow (2001–2005) and thanks Professor John A. Gladysz for his generous support.References
- J. R. Malpass, Comprehensive Organic Chemistry, D. Barton and W. D. Ollis, eds., Pergamon Press: Oxford, 1979, vol. 2, p. 1 Search PubMed.
- (a) D. Steinborn and R. Taube, Z. Chem., 1986, 26, 349 CAS; (b) R. Taube in Applied Homogeneous Catalysis, B. Cornils and W. A. Herrmann, eds., Wiley-VCH, Weinheim, 1996, vol. 1, p. 507 Search PubMed; (c) T. E. Müller and M. Beller, Chem. Rev., 1998, 98, 675 CrossRef; (d) J. J. Brunet and D. Neibecker in Catalytic Heterofunctionalization from Hydroamination to Hydrozirconation, A. Togni and H. Grützmacher, eds., Wiley-VCH, Weinheim, 2001, p. 91 Search PubMed; (e) J. Seayad, A. Tillack, C. G. Hartung and M. Beller, Adv. Synth. Catal., 2002, 344, 795 CrossRef CAS; (f) M. Beller, C. Breindl, M. Eichberger, C. G. Hartung, J. Seayad, O. R. Thiel, A. Tillack and H. Trauthwein, Synlett, 2002, 1579 CrossRef CAS; (g) S. Doye, Synlett, 2004, 1653 CrossRef CAS; (h) S. Hong and T. J. Marks, Acc. Chem. Res., 2004, 37, 673 CrossRef CAS.
- M. E. Jung in Comprehensive Organic Synthesis, B. M. Trost and I. Fleming, eds., Pergamon Press, Oxford, 1991, vol. 4, p. 1 Search PubMed.
- P. W. Roesky and T. E. Müller, Angew. Chem., Int. Ed., 2003, 42, 2708 CrossRef CAS.
-
(a) M. Kawatsura and J. F. Hartwig, J. Am. Chem. Soc., 2000, 122, 9546 CrossRef CAS;
(b) K. Li, P. N. Horton, M. B. Hursthouse and K. K. Hii, J. Organomet. Chem., 2003, 665, 250 CrossRef CAS;
(c) M. Utsunomiya and J. F. Hartwig, J. Am. Chem. Soc., 2003, 125, 14286 CrossRef CAS;
(d) O. Löber, M. Kawatsura and J. F. Hartwig, J. Am. Chem. Soc., 2001, 123, 4366 CrossRef CAS;
(e) R. Dorta, P. Egli, F. Zürcher and A. Togni, J. Am. Chem. Soc., 1997, 119, 10857 CrossRef CAS;
(f) L. M. Lutete, I. Kadota and Y. Yamamoto, J. Am. Chem. Soc., 2004, 126, 1622 CrossRef CAS.
- W. Zhuang, R. G. Hazell and K. A. Jørgensen, Chem. Commun., 2001, 1240 RSC; L. Fadini and A. Togni, Chem. Commun., 2003, 30 RSC; K. Li and K. K. Hii, Chem. Commun., 2003, 1132 RSC; K. Li, X. Cheng and K. K. Hii, Eur. J. Org. Chem., 2004, 959 CrossRef CAS.
- G. A. Molander and E. D. Dowdy, Topics Organomet. Chem., 1999, 2, 119 Search PubMed; G. A. Molander and J. A. C. Romero, Chem. Rev., 2002, 102, 2161 CrossRef CAS.
- (a) G. A. Molander and E. D. Dowdy, J. Org. Chem., 1998, 63, 8983 CrossRef CAS; (b) G. A. Molander and E. D. Dowdy, J. Org. Chem., 1999, 64, 6515 CrossRef CAS; (c) J.-S. Ryu, T. J. Marks and F. E. McDonald, Org. Lett., 2001, 3, 3091 CrossRef CAS; (d) J.-S. Ryu, T. J. Marks and F. E. McDonald, J. Org. Chem., 2004, 69, 1038 CrossRef CAS.
-
(a) Y. Li and T. J. Marks, Organometallics, 1996, 15, 3770 CrossRef CAS;
(b) J.-S. Ryu, G. Y. Li and T. J. Marks, J. Am. Chem. Soc., 2003, 125, 12584 CrossRef CAS.
- M. R. Gagné, C. L. Stern and T. J. Marks, J. Am. Chem. Soc., 1992, 114, 275 CrossRef CAS; A. Motta, G. Lanza, I. L. Fragalà and T. J. Marks, Organometallics, 2004, 23, 4097 CrossRef CAS.
-
(a) M. R. Gagné, L. Brard, V. P. Conticello, M. A. Giardello, T. J. Marks and C. L. Stern, Organometallics, 1992, 11, 2003 CrossRef CAS;
(b) M. A. Giardello, V. P. Conticello, L. Brard, M. R. Gagné and T. J. Marks, J. Am. Chem. Soc., 1994, 116, 10241 CrossRef CAS;
(c) M. R. Douglass, M. Ogasawara, S. Hong, M. V. Metz and T. J. Marks, Organometallics, 2002, 21, 283 CrossRef CAS;
(d) S. Hong and T. J. Marks, J. Am. Chem. Soc., 2002, 124, 7886 CrossRef CAS;
(e) S. Hong, A. M. Kawaoka and T. J. Marks, J. Am. Chem. Soc., 2003, 125, 15878 CrossRef CAS.
- (a) D. V. Gribkov, K. C. Hultzsch and F. Hampel, Chem. Eur. J., 2003, 9, 4796 CrossRef CAS; (b) D. V. Gribkov, F. Hampel and K. C. Hultzsch, Eur. J. Inorg. Chem., 2004, 4091 CrossRef CAS; (c) D. V. Gribkov and K. C. Hultzsch, Chem. Commun., 2004, 730 RSC.
- (a) P. N. O'Shaughnessy and P. Scott, Tetrahedron: Asymmetry, 2003, 14, 1979 CrossRef CAS; (b) P. N. O'Shaughnessy, P. D. Knight, C. Morton, K. M. Gillespie and P. Scott, Chem. Commun., 2003, 1770 RSC; (c) P. N. O'Shaughnessy, K. M. Gillespie, P. D. Knight, I. Munslow and P. Scott, Dalton Trans., 2004, 2251 RSC.
- S. Hong, S. Tian, M. V. Metz and T. J. Marks, J. Am. Chem. Soc., 2003, 125, 14768 CrossRef CAS.
- J. Collin, J.-D. Daran, E. Schulz and A. Trifonov, Chem. Commun., 2003, 3048 RSC.
- D. J. Berrisford, C. Bolm and K. B. Sharpless, Angew. Chem., Int. Ed. Engl., 1995, 34, 1059 CrossRef CAS.
- H. B. Kagan and J. C. Fiaud, Topics Stereochem., 1988, 18, 249 Search PubMed.
- H. H. Brintzinger, D. Fischer, R. Mühlhaupt, B. Rieger and R. M. Waymouth, Angew. Chem., Int. Ed. Engl., 1995, 34, 1143 CrossRef CAS.
- However, so far only 16% ee has been achieved, see: J. M. Hoover, J. R. Petersen, J. H. Pikul and A. R. Johnson, Organometallics, 2004, 23, 4614 Search PubMed.
- P. D. Knight, I. Munslow, P. N. O'Shaughnessy and P. Scott, Chem. Commun., 2004, 894 RSC ; see also: D. V. Gribkov and K. C. Hultzsch, Angew. Chem., Int. Ed., 2004, 43, 5542 Search PubMed.
- Y. Li and T. J. Marks, J. Am. Chem. Soc., 1998, 120, 1757 CrossRef CAS; V. M. Arredondo, S. Tian, F. E. McDonald and T. J. Marks, J. Am. Chem. Soc., 1999, 121, 3633 CrossRef CAS; G. A. Molander and S. K. Pack, Tetrahedron, 2003, 59, 10581 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2005 |