The design and synthesis of inhibitors of pantothenate synthetase†
Kellie L. Tucka, S. Adrian Saldanhaa, Louise M. Bircha, Alison G. Smithb and Chris Abella
aUniversity Chemical Laboratory, Lensfield Road, Cambridge, UK CB2 1EW. E-mail: ca26@cam.ac.uk; Fax: +44 1223 3336362; Tel: +44 1223 333405
bDepartment of Plant Sciences, Downing Street, Cambridge, UK CB2 3EA. E-mail: as25@plant.sciences.cam.ac.uk; Fax: +44 1223 333953; Tel: +44 1223 333900
First published on 30th August 2006
Abstract
Pantothenate synthetase catalyses the ATP-dependent condensation of D-pantoate and β-alanine to form pantothenate. Ten analogues of the reaction intermediate pantoyl adenylate, in which the phosphodiester is replaced by either an ester or sulfamoyl group, were designed as potential inhibitors of the enzyme. The esters were all modest competitive inhibitors, the sulfamoyls were more potent, consistent with their closer structural similarity to the pantoyl adenylate intermediate.
Introduction
Pantothenate 1 (vitamin B5) is a key precursor of the 4′-phosphopantetheine moiety of coenzyme A (CoA) and the acyl carrier protein (ACP). Both CoA and ACP are necessary cofactors for cell growth and are involved in essential biosynthetic pathways. Pantothenate 1 (Scheme 1) is biosynthesised in micro-organisms, plants, and fungi, but not in animals,1 and the enzymes of the pantothenate pathway are considered to be potential herbicide and antimicrobial targets.The pathway to pantothenate 1 is best understood in Escherichia coli, where it comprises four enzymatic reactions.2,3 The final transformation, to produce pantothenate 1, is catalysed by pantothenate synthetase (EC 6.3.2.1, Scheme 1), encoded by the panC gene. Pantothenate is biosynthesised by the condensation of D-pantoate and β-alanine. One equivalent of ATP is used, resulting in the formation of AMP and pyrophosphate (PPi). The Mg2+-dependent reaction consists of two sequential steps, initial pantoyl adenylate 2 formation, followed by subsequent nucleophilic attack on the activated carbonyl by β-alanine (Scheme 1).4 The kinetic mechanism of Mycobacterium tuberculosis pantothenate synthetase has been shown to be a BiUniUniBiPingPong mechanism.4,5 Pantothenate synthetase is a member of the aminoacyl-tRNA synthetase superfamily and the mechanism for formation of the pantoyl adenylate 2 is similar to that for formation of the acyl adenylate intermediate.6 Several inhibitors of aminoacyl-tRNA synthetases are known which mimic the aminoacyl adenylate intermediate.7–9
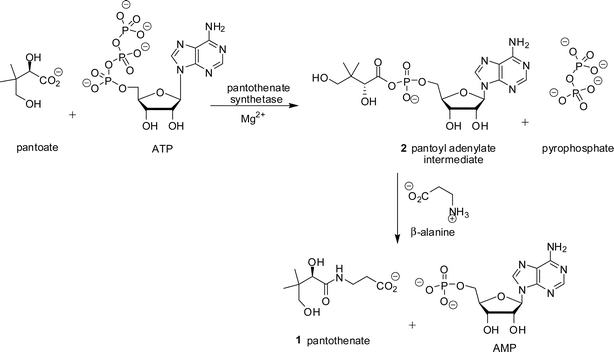 | ||
| Scheme 1 Reaction catalysed by pantothenate synthetase. | ||
No potent inhibitors of pantothenate synthetase have been reported. Several potential inhibitors, pantoate analogues or phosphonate analogues of pantoate/pantothenate (Fig. 1) have been synthesised, however none showed significant inhibition of pantothenate synthetase under a variety of assay conditions.10,11 A non-hydrolysable analogue of ATP, AMPCPP, has been shown to inhibit M. tuberculosis pantothenate synthetase, with a Ki of 290 µM.4 No mimics of the pantoyl adenylate 2 have been described.
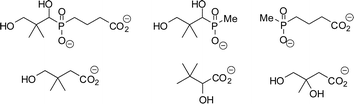 | ||
| Fig. 1 Compounds reported as potential inhibitors of pantothenate synthetase.10,11 | ||
The crystal structures of the apoenzyme forms of E. coli pantothenate synthetase (PDB code: 1iho)6 and M. tuberculosis pantothenate synthetase have been published, as well as several very informative complexes of the M. tuberculosis enzyme including the pantoyl adenylate 2 bound at the active site (PDB code: 1n2h).12–14 Pantothenate synthetase exists as a homodimer. Each subunit has two well-defined domains, an N-terminal Rossmann fold which contains the active site cavity and a C-terminal domain which acts as a hinged lid for this cavity. ATP binds into the N-terminal domain,6,13 and pantoate binds into a pocket on the other domain. For E. coli pantothenate synthetase it has been proposed that the binding of ATP is accompanied by a hinge bending action that moves the two domains of the enzyme closer together from an open apo-structure to a closed structure.6 The ternary M. tuberculosis pantothenate synthetase structure adopts a closed conformation, believed to be catalytically relevant.
In this paper we describe the preparation of a closed model of E. coli pantothenate synthetase, the synthesis of a number of pantoyl adenylate analogues (compounds 3–12, Fig. 2), molecular docking studies of these analogues into the active site of E. coli pantothenate synthetase, and their inhibition of E. coli pantothenate synthetase.
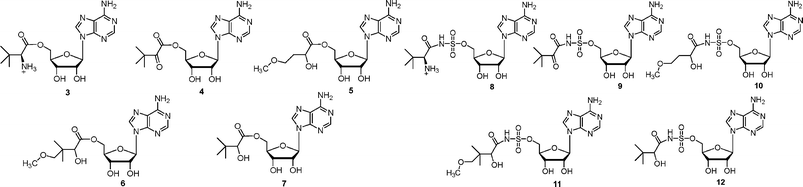 | ||
| Fig. 2 Analogues of pantoyl adenylate 2 described in this paper. | ||
Results
Preparation of the closed model of E. coli pantothenate synthetase
A sequence alignment of E. coli and M. tuberculosis pantothenate synthetase was performed with the CLUSTALW 1.82 software program (http://www2.ebi.ac.uk/clustalw).15 This revealed that the active sites of the two enzymes possess a high degree of sequence similarity. The residues M30, H37, N58, Q61, G149, D152 and Q155 in E. coli are conserved as M40, H47, N69, Q72, G158, D161 and Q164 in M. tuberculosis.The high degree of similarity in the active sites of E. coli and M. tuberculosis pantothenate synthetase, the number of conserved residues and the fact that the M. tuberculosis crystal structures were in the required conformation to be catalytic, led to the construction of a model for the closed structure of E. coli pantothenate synthetase using the M. tuberculosis pantothenate synthetase (PDB code: 1n2h) structure. The position of pantoyl adenylate 2 was modelled into the E. coli active site. An overlay of a subunit of the two structures is shown in Fig. 3A (the protein-ligand interactions for M. tuberculosis pantothenate synthetase (PDB code: 1n2h) and the modelled closed form of E. coli pantothenate synthetase are shown as LIGPLOT diagrams16 in the Supplementary Information (ESI)†). Of note is a molecule of water bound 3.3 Å away from the C2′ ribose hydroxyl of the pantoyl adenylate 2 which mediates hydrogen bonds from the ribose hydroxyl(s) to the protein backbone. This key structural water molecule is present in all pantothenate synthetase crystal structures that include ribose-containing ligands. The LIGPLOT diagrams show the importance of the hydrogen bonds between the water molecule and the hydroxyl group at the C2′, the aspartate residue and the glycine residue.
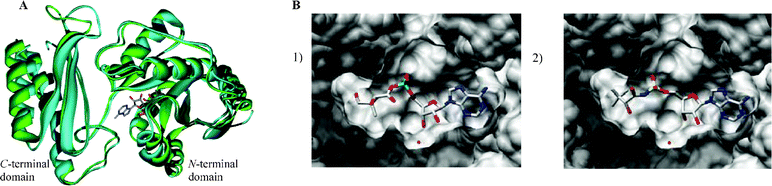 | ||
| Fig. 3 A) Overlay of M. tuberculosis pantothenate synthetase from the crystal structure (2.0 Å, PDB code: 1n2h, shown in blue) with the modelled closed conformation of E. coli pantothenate synthetase (shown in green). The bound pantoyl adenylate 2 is also shown. The diagram was generated using Weblab Viewer Pro (Molecular Simulations Inc) and Povray (http://www.povray.org/). B) GOLD docking results of the ligands in the active site of the closed model of E. coli pantothenate synthetase (1) pantoyl adenylate 2, (2) inhibitor (2R)-12. These representations were produced using the UCSF Chimera package from the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco (supported by NIH P41 RR-01081).28 | ||
Docking studies and design of inhibitors
The structure of M. tuberculosis pantothenate synthetase (PDB code: 1n2h), was prepared using SYBYL 6.5.17 The pantoyl adenylate 2, the glycerol molecule, the ethanol molecules and all the water molecules (except the key structural water, see above) were removed from the crystal structure. Hydrogens were added to the amino acid residues and the structure was minimised using a Tripos force field.17 The closed model structure of E. coli pantothenate synthetase was prepared as described in the Experimental section. GOLD (version 2.1.1)18 was used to dock small molecules into the protein binding site. The structures of the ligands were prepared using the program SYBYL 6.517 and energy-minimised using the Tripos force field. A maximum of 25 independent GOLD runs were executed for each ligand.18 The dockings were allowed to terminate early if the root-mean-squared deviation (RMSD) between the heavy atoms of the top five ranked conformations was less than 1.5 Å. The GoldScore scoring function was used in all cases.Pantoyl adenylate 2 was initially docked as a control into the active sites of E. coli and M. tuberculosis pantothenate synthetase, and then compared to either the pantoyl adenylate–enzyme complex (M. tuberculosis) or the pantoyl adenylate–enzyme model (E. coli). The docked conformation corresponded to that of the crystallographically observed pantoyl adenylate 2 to within 0.6 Å RMSD.
The inhibitors were designed to be analogues of the pantoyl adenylate 2. Ten compounds were chosen (3–12), where the labile phosphodiester is replaced with either an ester or sulfamoyl group and the pantoyl functionality is modified. The (2S)-diastereomer of ligands 3 and 8 and each stereoisomer of the ligands 4–7 and 10–12 were docked into the active site of M. tuberculosis pantothenate synthetase and the active site of the closed model of E. coli pantothenate synthetase. The docking results suggested that the ligands would bind in the same orientation as the pantoyl adenylate 2 in the active site. The top docking results of the pantoyl adenylate 2 and the ligand (2S)-12 in the active site of E. coli pantothenate synthetase are shown in Fig. 3B.
Synthesis of ester analogues 3–7
The amino ester analogue 3 was prepared from commercially available N-Boc-L-tert-leucine. The N-Boc protected amino acid was coupled with 2′,3′-isopropylideneadenosine, under Mitsunobu conditions (Scheme 2).19 Deprotection of both the acetonide and N-Boc protecting groups was accomplished under acidic conditions to give the ester 3.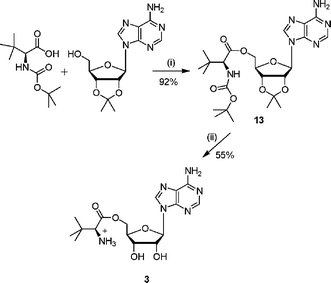 | ||
| Scheme 2 Reagents and conditions: (i) DEAD, PPh3, THF, 16 h; (ii) TFA–H2O, 3 h. | ||
Ester analogue 4 was synthesised in three steps from pinacolone (Scheme 3). The required ketoacid 14 was synthesised by permanganate oxidation of the methyl group.20 Coupling of the ketoacid 14 with 2′,3′-isopropylideneadenosine gave the protected ester 15, which was deprotected under acidic conditions to give 4.
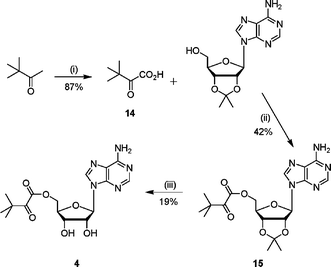 | ||
| Scheme 3 Reagents and conditions: (i) KMnO4, NaOH(aq), 0 °C, 1 h, 22 °C, 2 h; (ii) DCC, CH2Cl2, 16 h; (iii) TFA–H2O, 3 h. | ||
The methoxy ether ester analogues 5 and 6 were synthesised in five steps from α-hydroxy-χ-butyrolactone and (±)-pantolactone respectively (Scheme 4). In both cases the secondary alcohol of the lactone was protected as a 2-methoxyethoxymethyl (MEM) ether to give compounds 16 and 17, and 15-crown-5 was added as a co-catalyst to improve the solubility of NaH.21 The required acids, 18 and 19, were obtained after ring opening of the protected lactone and derivatisation to the corresponding methoxy ether. Coupling of the acids 18 and 19 with 2′,3′-isopropylideneadenosine, under Mitsunobu conditions, gave the protected esters (20 or 21 respectively). The final compounds, analogues 5 and 6, were obtained after removal of the acetal protecting group.
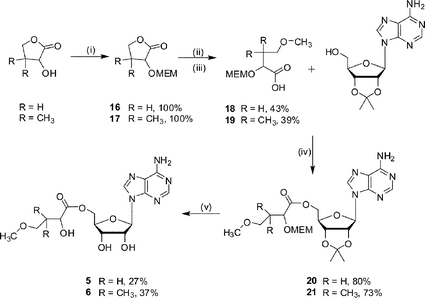 | ||
| Scheme 4 Reagents and conditions: (i) NaH, 15-crown-5, MEMCl, 0 °C, warm to 22 °C, 15 h (ii) H2O–MeOH, KOH, 80 °C, 4 h; (iii) NaH, 15-crown-5, MeI, 0 °C, warm to 22 °C, 15 h; (iv) DEAD, PPh3, THF, 16 h; (v) TFA–H2O, 3 h. | ||
Ester analogue 7 was prepared from 3,3-dimethyl-1,2-butanediol in four steps (Scheme 5). The required acid 22 was obtained after oxidation of 3,3-dimethyl-1,2-butanediol with Pt–C in the presence of oxygen. Protection of the secondary alcohol with a dichloroacetate group gave the dichloroacetate ester 23 in good yield. This protected 3-dimethyl-2-hydroxybutanoic acid 23 was coupled with 2′,3′-isopropylideneadenosine under Mitsunobu conditions to give the protected ester 24. Deprotection under acidic conditions gave the corresponding ester 7 in good yield.
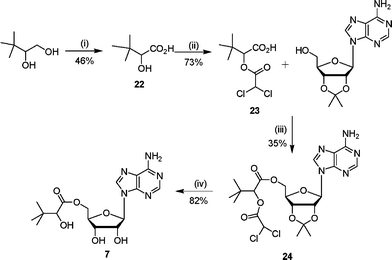 | ||
| Scheme 5 Reagents and conditions: (i) Pt–C, NaOH(aq), CH2CH2, O2(g), 70 °C, 5 h; (ii) ClCOCHCl2, 80 °C, 4 h; (iii) DEAD, PPh3, THF, 16 h; (iv) TFA–H2O, 3 h. | ||
Synthesis of sulfamoyl analogues 8–12
The synthesis of N-acylsulfamoyls 8–12 was based on literature procedures for the preparation of isoleucyl- and tyrosyl-tRNA synthetase inhibitors.7,9,22 The required 2′,3′-protected adenosinesulfamate 25 was prepared from 2′,3′-isopropylideneadenosine and sulfamyl chloride using a modification of literature conditions.7,22,23 Previous work had shown that the 5′-O–SO2NH2 group was more reactive than the 6-NH2 amino group on the adenine ring.24 Hence the adenosinesulfamate derivative 25 was directly acylated with appropriately activated carboxylic acid derivatives to give the N-acylsulfamates.Sulfonamide analogues 8 and 9 were prepared by coupling the adenosinesulfamate 25 with the hydroxysuccinimide ester of N-Boc-L-tert-leucine and the diketone 14 respectively (Schemes 6 and 7). The hydroxysuccinimide ester was formed by reacting N-Boc-L-tert-leucine or the diketone 14 with N-hydroxysuccinimide to give the activated acid (26 or 28).25 The hydroxysuccinimide esters were unstable to purification by chromatography. Deprotection of the N-acylsulfamates (27 or 29) under acidic conditions gave the required sulfamoyls.
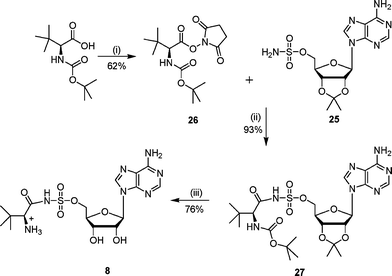 | ||
| Scheme 6 Reagents and conditions: (i) hydroxysuccinimide, DME, DCC, 0 °C, 15 h; (ii) DBU, DCM, 16 h; (iii) TFA–H2O, 16 h. | ||
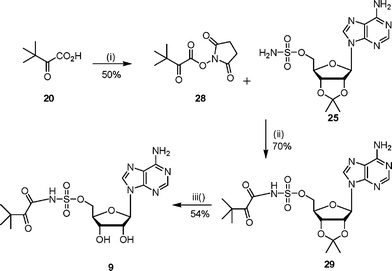 | ||
| Scheme 7 Reagents and conditions: (i) hydroxysuccinimide, DME, DCC, 0 °C, 15 h; (ii) DBU, DCM, 16 h; (iii) TFA–H2O, 3 h. | ||
Due to the instability of the hydroxysuccinimide esters a different activating group was tried. Consequently, the sulfamoyl analogues 10 and 11 were prepared by coupling 2′,3′-protected adenosinesulfamate 25 with the pentafluorophenol esters of the hydroxy acids 18 or 19 (Scheme 8). Purification by chromatography resulted in an oil which was a mixture in which the desired product (32 or 33) was identified by NMR spectroscopy and LCMS analysis. Removal of both the acetonide and MEM protecting groups was accomplished under acidic conditions. The vigorous conditions required to remove the MEM protecting group led to a mixture of products from which the sulfamoyls (10 or 11) were purified.
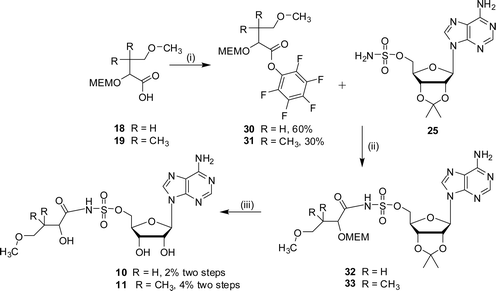 | ||
| Scheme 8 Reagents and conditions: (i) EDC, DMAP, CH2Cl2, HOC6F5, 15 h; (ii) DBU, DMF, 16 h; (iii) TFA–H2O, 3 h. | ||
Sulfonamide analogue 12 was also prepared by coupling 2′,3′-protected adenosinesulfamate 25 with the pentafluorophenol ester of the protected acid 23 (Scheme 9). Purification by chromatography led to a mixture of products including some loss of the diacetyl protecting group. Deprotection of the acetonide protecting group was accomplished under acidic conditions which resulted in several products, from which 12 was purified.
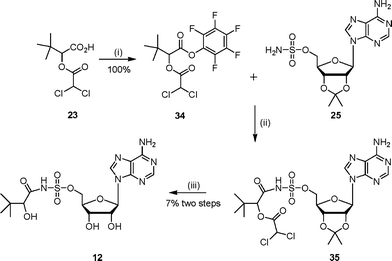 | ||
| Scheme 9 Reagents and conditions: (i) EDC, DMAP, CH2Cl2, HOC6F5, 15 h; (ii) DBU, DMF, 16 h (iii) TFA–H2O, 16 h. | ||
Assay results
E. coli pantothenate synthetase was assayed using a coupled assay that measures the formation of AMP spectrophotometrically.26 The kinetic parameters were obtained by plotting the rate as a function of concentration of varied substrate and curve fitting to the Michaelis–Menten equation. Control experiments were carried out to show that the compounds did not inhibit the coupling enzymes.The ester analogues, (2S)-3 and the racemic compounds 4–7, were tested as inhibitors against E. coli pantothenate synthetase. In all cases the inhibition studies were carried out using a constant concentration of pantoate and β-alanine and varying concentrations of ATP. The Ki values were obtained by plotting the rate as a function of the concentration of ATP at different inhibitor concentrations and curve fitting to the equation for competitive inhibition (Table 1).27 All the compounds were found to be modest reversible inhibitors of E. coli pantothenate synthetase, and were competitive with ATP. The degree of inhibition is relatively insensitive to the structure of the acyl group, with the Kis ranging from 1.9 mM (for 4) to 18.2 mM (for 6). This suggests that most of the binding is coming from the adenine and ribose groups in the ATP pocket, and that the varied acyl group cannot reach far enough into the pantoate binding pocket to pick up the specific binding interactions associated with recognition of the pantoyl group in the reaction intermediate 2.
| Compound | Ki/mM | Inhibition |
|---|---|---|
| a [E. coli pantothenate synthetase] 50 nM. Km ATP 1.75 mM, (±)-pantoate 1.45 mM, β-alanine 0.31 mM, kcat = 1.4 s−1 at 25 °C. | ||
| 3 | 9.5 ± 1.2 | Competitive |
| 4 | 1.9 ± 0.22 | Competitive |
| 5 | 3.5 ± 0.4 | Competitive |
| 6 | 18.2 ± 4.2 | Competitive |
| 7 | 2.5 ± 0.2 | Competitive |
Inhibition studies with the sulfamoyl analogues, (2S)-8 and the racemic compounds 10–12, were carried out against E. coli pantothenate synthetase. The inhibition by these compounds was measured against varying concentrations of ATP, pantoate and β-alanine. In all assays the concentration of two of the substrates were kept constant and that of the third substrate was varied. The Ki values were obtained by plotting the rate as a function of the concentration of the varied substrate at different inhibitor concentrations, and curve fitting to equations for competitive, non-competitive or un-competitive inhibition (Table 2).27 The inhibition data for compound 12versus ATP, pantoate and β-alanine are shown in Fig. 4.
| Compound | Varying substrate | Ki/µM | Inhibition |
|---|---|---|---|
| a [E. coli pantothenate synthetase] 50 nM, ND—not determined, insufficient compound, Km (ATP) 1.75 mM, (±)-pantoate 1.45 mM, β-alanine 0.31 mM, kcat = 1.4 s−1 at 25 °C. | |||
| 8 | ATP | 65 ± 7 | competitive |
| pantoate | 160 ± 13 | competitive | |
| β-alanine | 250 ± 23 | uncompetitive | |
| 9 | ATP | 14 ± 2 | competitive |
| pantoate | 15 ± 2 | competitive | |
| β-alanine | 120 ± 9 | uncompetitive | |
| 10 | ATP | 0.8 ± 0.1 | competitive |
| pantoate | ND | competitive | |
| β-alanine | ND | uncompetitive | |
| 11 | ATP | 30 ± 4 | competitive |
| pantoate | 54 ± 5 | competitive | |
| β-alanine | 144 ± 17 | uncompetitive | |
| 12 | ATP | 0.3 ± 0.05 | competitive |
| pantoate | 0.8 ± 0.05 | competitive | |
| β-alanine | 1.1 ± 0.08 | uncompetitive | |
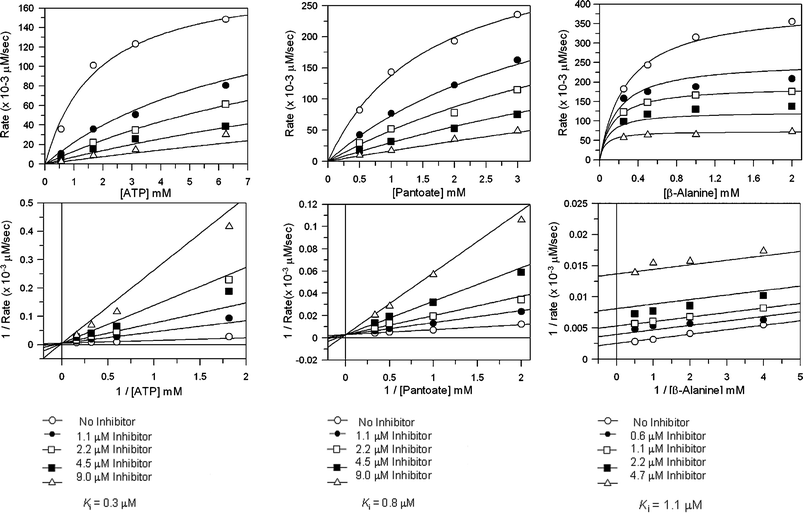 | ||
| Fig. 4 Steady state inhibition studies of E. coli pantothenate synthetase with inhibitor (2SR)-12, A) versus ATP, data were fitted to the equation for competitive inhibition, B) versus pantoate, data were fitted to the equation for competitive inhibition, C) versus β-alanine, data were fitted to the equation for uncompetitive inhibition. GraFit software (version 5.0.6, Erithacus Software Limited, http://www.erithacus.com/grafit/) was used to construct Michaelis–Menten plots of the kinetic data and to calculate Ki values for competitive inhibition. | ||
Conclusions
The esters 3–7 and sulfamoyl compounds 8–12 were designed as analogues of the intermediate in the pantothenate synthetase reaction, pantoyl adenylate 2. The compounds were synthesised and assayed against E. coli pantothenate synthetase. The sulfamoyls 8–12 were approximately 100-fold more potent inhibitors than their corresponding ester analogues, reflecting their closer structural similarity with pantoyl adenylate, and suggesting that the interactions that the phosphate group makes with the enzyme contribute significantly to binding. The inhibition values for these compounds ranged from 300 nM (compound 12, versus ATP) to 250 µM (compound 8, versus β-alanine).The sulfamoyl groups 8–12 were competitive inhibitors against ATP and pantoate and uncompetitive inhibitors against β-alanine. This was somewhat unexpected as it has previously been reported that the kinetic catalytic mechanism of M. tuberculosis pantothenate synthetase involves ordered binding of ATP followed by pantoate.4,5 It is interesting however that pantoate can be soaked into a crystal of apoenzyme, suggesting that binding of ATP is not absolutely required for pantoate binding.13 Sulfamoyl analogue 12, the most potent inhibitor, is structurally most similar to the pantoyl adenylate 2. The pantoate fragment differs only by replacement of the primary hydroxyl group with a proton. The inclusion of a primary methyoxy ether in 11 results in a hundred-fold decrease in potency. This result was consistent with docking studies which revealed that 11 was too large to bind in the same orientation as the pantoyl adenylate 2, due to the combined presence of the methoxy ether and the gem-dimethyl groups. Docking studies of compound 10, which had the gem-dimethyl groups removed, showed that this compound could bind in the same orientation as the pantoyl adenylate 2 and this was mirrored by an increase in potency. Replacement of the secondary hydroxyl in compound 12 with a ketone (compound 9) or amino group (compound 8) resulted in less potent molecule, indicating that the secondary hydroxyl group is required for activity. All compounds were found to be poor inhibitors of pantothenate kinase and ketopantoate reductase (data not showm).
This paper reports the first synthesis of a submicromolar inhibitor of pantothenate synthetase. The structure of the inhibitor is consistent with the mechanism of the enzyme proceeding through a pantoyl adenylate intermediate. The kinetics of the inhibition suggest that a random order binding of the two substrates, ATP and pantoate, is possible for the E. coli enzyme.
Experimental
Design of the closed model of E. coli pantothenate synthetase
The N-terminal domains of E. coli pantothenate synthetase (PDB code: 1iho) and M. tuberculosis pantothenate synthetase complexed with the pantoyl adenylate 2 (PDB code: 1n2h) were superimposed using Swiss-PDBViewer. The backbone RMSD was found to be 1.0 Å for this region. This domain corresponds to the Rossmann fold and is assumed to stay rigid. Particular attention was then paid to the hinge region between the domains.The program SYBYL was used to modify the torsion angles along the backbone around the central proline (P176 in E. coli and P180 in M.tuberculosis) so that the C-terminal domains overlapped. The proline ring conformation was changed in order to accommodate the backbone torsion angle changes. The backbone torsion angles were modified near residues 175–177, with no amide torsion (ω) being moved by more than 4 degrees. The region between residues 174–180 was then minimised in SYBYL to allow for reorientation of the sidechains. The active site of the protein was then modelled by extracting the position observed in the (superimposed) M. tuberculosis pantothenate synthetase structure, merging the pantoyl adenylate into the new active site and then conducting a minimisation of the intermediate and the surrounding sidechains using a substructure minimisation in SYBYL (hydrogens were added to both the protein and the intermediate). The key water molecule from the M. tuberculosis pantothenate synthetase structure was retained in the active site.
GOLD docking
All ligands and the receptor were prepared using SYBYL 6.5 and used as MOL2 files. The structures of the ligands were energy-minimised using the Tripos force-field prior to docking. Each ligand was docked into the active site of pantothenate synthetase (M. tuberculosis. and E. coli) using GOLD 2.1.1 in 25 independent genetic algorithm (GA) runs, and for each of these a maximum of 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 GA operations were performed on a single population of 50 individuals. Operator weights for crossover, mutation and migration in the entry box were used as default parameters (95, 95 and 10, respectively), as well as the initial hydrogen bonding (4.0 Å) and external van der Waals (2.5 Å) parameters. The position of the active site was introduced and the radius was set to 10 Å, with the automatic active site detection on. The “flip ring corners”, “flip amide bonds” and “flip planer N” flags were switched on, while “internal H-bonds” was switched off.
000 GA operations were performed on a single population of 50 individuals. Operator weights for crossover, mutation and migration in the entry box were used as default parameters (95, 95 and 10, respectively), as well as the initial hydrogen bonding (4.0 Å) and external van der Waals (2.5 Å) parameters. The position of the active site was introduced and the radius was set to 10 Å, with the automatic active site detection on. The “flip ring corners”, “flip amide bonds” and “flip planer N” flags were switched on, while “internal H-bonds” was switched off.Protein expression
The panC gene was subcloned into SmaI–EcoRI-digested pUC19, such that the panC gene was under the control of the lacZ promoter. For overexpression of pantothenate synthetase, E. coli cells were grown overnight at 37 °C on LB medium containing ampicillin (100 mg ml−1). The lacZ gene was induced by the addition of IPTG (70 mg ml−1) at the start of the culture. Pantothenate synthetase was purified as described previously,6 and stored in aliquots as concentrated solutions in glycerol–Tris buffer at −20 °C.Pantothenate synthetase assay
Pantothenate synthetase activity was assayed by coupling the formation of AMP to the reactions of myokinase, pyruvate kinase, and lactate dehydrogenase as described previously26 except that Tris buffer was used. The decrease in absorbance of NADH at 340 nm (ε340 = 6220 M−1 cm−1) was measured in a 96-well plate using a Biotek Powerwave XS plate reader running KC4 v3.2 Biotek instrument software. Conversion of (±)-pantolactone to (±)-sodium pantoate was achieved by treatment of (±)-pantolactone with 1 molar equivalent of sodium hydroxide.The standard assay contained 100 mM Tris, pH 7.5, 10 mM MgCl2, 5 mM ATP, 3 mM β-alanine, 1.5 mM potassium phosphoenolpyruvate, 150 µM NADH, 5 units of myokinase, 10 units of pyruvate kinase, and 10 units of lactate dehydrogenase and E. coli pantothenate synthetase (50 nM) in a total volume of 160 µL at 25 °C. After incubation for 5 min at 25 °C, reactions were initiated by the addition of sodium pantoate (4 mM, 40 µL, preincubated to 25 °C). The rate of pantothenate formation is proportional to the rate of oxidation of NADH, two molecules of NADH are oxidized for each molecule of pantothenate formed. The apparent KM values for (±)-pantoate, ATP and β-alanine were 1.45 ± 0.38 mM, 1.75 ± 0.05 mM and 0.31 ± 0.04 mM, respectively, and kcat was 1.4 s−1. GraFit software (version 5.0.6, Erithacus Software Limited, http://www.erithacus.com/grafit/) was used to construct Michaelis–Menten plots of the kinetic data. All experiments were performed in duplicate and the mean values are reported.
Inhibition studies
The pantothenate synthetase assay was repeated in the presence of inhibitor. When ATP was the varied substrate the enzymatic reaction was initiated by the addition of sodium pantoate (4 mM, 40 µL, preincubated to 25 °C). When sodium pantoate was the substrate varied the enzymatic reaction was initiated by the addition of ATP (5 mM, 40 µL, preincubated to 25 °C). When β-alanine was the substrate varied the enzymatic reaction was initiated by the addition of (±)-pantoate (3 mM, 40 µL, preincubated to 25 °C).GraFit software (version 5.0.6, Erithacus Software Limited, http://www.erithacus.com/grafit/) was used to generate Michaelis–Menten plots of the kinetic data and calculate Ki values for competitive inhibition, non-competitive inhibition, uncompetitive inhibition and mixed inhibition.27 The type of inhibition was determined by statistical analysis (F test) of the Ki. The results are shown in Tables 1 and 2. All experiments were performed in duplicate, unless otherwise stated, and the mean values are reported.
Chemistry—General Methods
1H NMR spectra were recorded on either a Bruker Avance DPX-250 MHz spectrometer, a Bruker DPX-400 MHz spectrometer, or a Bruker DPX-400 MHz spectrometer in deuterated solvents, as specified. Splitting patterns are abbreviated as follows: s, singlet, d, doublet, t, triplet, q, quartet and m, multiplet. 13C NMR spectra were recorded on a Bruker DPX-400 MHz spectrometer operating at 100 MHz or Bruker DPX-500 spectrometer operating at 125 MHz. Samples were referenced with respect to the residual solvent signal.Infrared spectra were recorded on an ATI Mattson Genesis FTIR infrared spectrometer using NaCl plates or on a Perkin Elmer Spectrum One FTIR spectrometer using attenuated transmittance reflectance (ATR).
Mass spectrometry was carried out using a Micromass Quadrapole-time of flight (Q-Tof) spectrometer. Liquid chromatography mass spectrometry (LCMS) was carried out using an Alliance HT Waters 2795 Separations Module coupled to a Waters Micromass ZQ Quadrapole Mass Analyzer. Samples were detected using a photomultiplier detection system. Samples were run on a gradient from 10 mM ammonium acetate containing 0.1% formic acid to 95% acetonitrile over a period of 8 minutes.
Column chromatography was performed with Kieselgel 60 (230–400 mesh). Where indicated, compounds were purified with diaion HP-20SS resin (Supelco). TLC was performed using commercially prepared silica gel (Kieselgel 60 0.25 mm) plates which were visualized either with UV light (254 nm) or by immersion in acidic ammonium molybdate solution.
HPLC purification was carried out on a Gilson HPLC system fitted with a Gilson 118 UV–vis detector, with detection at 254 nm. Where noted, compounds were purified using a Hichrom KR-100 C18 5 µm 250 × 4.6 mm column. Columns were eluted with a gradient of methanol–water.
Organic solutions were dried over Na2SO4 or MgSO4, as indicated. All organic solvents were freshly distilled prior to use. Dichloromethane (DCM), ethyl acetate (EtOAc), methanol, hexane and acetonitrile were distilled over calcium hydride. Tetrahydrofuran (THF) and diethyl ether (Et2O) were distilled over a mixture of lithium aluminium hydride and calcium hydride. All other reagents were purchased from Aldrich, Acros, or Fisher Scientific, and were used without further purification. Moisture sensitive reactions were carried out under a nitrogen atmosphere in pre-dried glassware.
The protected ester 13 (600 mg, 1.15 mmol) was dissolved in water–TFA (2 : 4 v/v) and stirred at 22 °C for 3 h. The solvent was removed in vacuo by azeotroping with ethanol and toluene. Purification by column chromatography (methanol–DCM gradient 2–15% methanol) gave the ester as a white solid (244 mg, 55%). δH (250 MHz, DMSO): 0.90 (s, 9H, CH3), 4.15 (m, 1H, H3′), 4.24 (m, 1H, H4′), 4.38 (m, 2H, H5′), 4.78 (m, 1H, H2′), 5.52 (br s, 2H, NH2), 5.90 (d, 1H, J = 5.5 Hz, H1′), 7.25 (br s, 2H, NH2), 8.12 (s, 1H, H2″), 8.31 (s, 1H, H8″). δC (62.5 MHz, DMSO): 26.2, 33.3, 61.0, 65.7, 70.6, 72.7, 81.5, 87.8, 119.4, 140.2, 149.5, 152.7, 156.2, 169.5. HRMS (ES) for C16H24N6O5: (MH+) calcd: 381.1808, found: 381.1805.
The protected ester 15 (184 mg, 0.43 mmol) was dissolved in water (0.4 mL) and cooled to 0 °C. TFA (1 mL) was added dropwise and the reaction mixture was stirred and allowed to 22 °C over 3 h. The solvent was removed in vacuo by azeotroping with ethanol and toluene. Purification by column chromatography (methanol–EtOAc gradient 1 : 20 v/v) gave the ester as a white solid (30 mg, 19%).δH (400 MHz, MeOD): 1.20 (s, 9H, t-Bu), 4.34 (m, 2H, H5′), 4.41 (t, 1H, J = 5.0 Hz, H4′), 4.57 (m, 1H, H3′), 4.73 (t, 1H, J = 4.9 Hz, H2′), 6.07 (d, 1H, J = 4.7 Hz, H1), 8.23 (s, 1H, H2″), 8.33 (s, 1H, H8″). δC (100 MHz, MeOD): 26.0, 43.6, 65.6, 71.9, 75.4, 83.3, 90.2, 120.5, 141.6, 150.6, 152.6, 156.5, 164.3. νmax (ATR): 3449 (m), 1677 (s), 1615 (s), 1454 (m), 1203 (s), 1088 (s) cm−1. HRMS (ES) calcd for C16H21N5O6: (MNa+) calcd: 380.1570, found: 380.1583.
The protected ester 20 (600 mg, 1.2 mmol) was stirred for 3 h at 22 °C in water–TFA (2 : 4 v/v). The solvent was removed in vacuo by azeotroping with ethanol and toluene. Purification by column chromatography (methanol–DCM gradient 2–30% methanol) gave the ester as a white solid (125 mg, 27%). δH (250 MHz, CDCl3): 1.74 (m, 2H, H3), 3.15 (s, 2H, H5), 3.25 (m, 2H, H4), 4.25 (m, 3H, H2, H4′and H5′), 4.60 (m, 1H, H3′), 5.51 (m, 2H, OH, H2′), 5.90 (d, 1H, J = 5.5 Hz, H1′), 7.00 (m, 2H, NH2), 8.13 (s, 1H, H2″), 8.32 (s, 1H, H8″). δC (125 MHz, DMSO): 34.0, 58.6, 61.8, 64.3, 67.7, 70.5, 73.5, 81.7, 88.0, 119.4, 140.0, 149.5, 152.7, 156.2, 174.0. νmax (ATR): 3321 (w), 1643 (s), 1603 (s), 1478 (m), 1203 (m), 1075 (s) cm−1. HRMS (ES) for C15H19N5O7: (MH+) calcd: 384.1519, found: 384.1508.
The protected ester 21 (385 mg, 0.73 mmol) was stirred for 3 h at 22 °C in water–TFA (2 : 4 v/v). The solvent was removed in vacuo by azeotroping with ethanol and toluene. Purification by column chromatography (methanol–DCM gradient 2–30% methanol) gave the ester as a white solid (110 mg, 37%). δH (250 MHz, DMSO): 0.83 (s, 6H, H4), 3.16 (s, 2H, H5), 3.17 (s, 3H, H6), 3.90 (s, 1H, H2), 4.19 (m, 4H, H3′, H4′ and H5′), 4.67 (m, 1H, H2′), 5.93 (d, 1H, J = 5.2 Hz, H1′), 7.42 (br s, 2H, NH2), 8.17 (s, 1H, H2″), 8.39 (s, 1H, H8″). δC (62.5 MHz, DMSO): 20.5, 21.2, 55.0, 58.6, 64.1, 70.5, 73.1, 74.2, 78.0, 81.8, 87.5, 119.1, 139.9, 149.4, 152.2, 155.7, 172.8. νmax (ATR): 3434 (m), 1732 (m), 1644 (s), 1601 (m), 1477 (w), 1150 (s) cm−1. HRMS (ES) for C17H25N5O7: (MH+) calcd: 412.1824, found: 412.1828.
The protected ester 24 (150 mg, 0.28 mmol) was dissolved in pyridine–water (1 : 1 v/v, 2 mL, pH 7.5) and stirred for 16 h at 22 °C. The solvent was removed in vacuo and the residue was dissolved in water–TFA (2 : 4 v/v) and stirred at 22 °C for 3 h. The solvent was removed in vacuo by azeotroping with ethanol and toluene. Purification by column chromatography (methanol–DCM gradient 2–20% methanol) gave the ester 7 as a white solid (90 mg, 82%). δH (250 MHz, MeOD): 0.91 (s, 9H, CH3), 3.82 (s, 1H, H2), 4.39 (m, 4H, H3′, H4′ and H5′), 4.74 (m, 1H, H2′), 6.03 (d, 1H, J = 5.7 Hz, H1′), 8.21 (s, 1H, H2″), 8.34 (s, 1H, H8″). δC (62.5 MHz, DMSO): 25.0, 34.5, 63.6, 70.6, 73.9, 78.6, 82.2, 88.9, 122.2, 140.7, 150.6, 154.7, 157.1, 173.1. HRMS (ES) for C16H24N6O5: (MH+) calcd: 382.1726, found: 382.1737.
To a solution of 2′,3′-isopropylideneadenosine (1.0 g, 3.3 mmol), DCM (10 ml) and DBU (1 mL) was added a solution of chlorosulfonylamine (1.4 g) in DCM (5 mL) over 5 min. The solution was stirred for 15 h, and then the solvent was removed in vacuo. Column chromatography (methanol–ethyl acetate 10 : 90 v/v) gave the sulfamoyl as a white solid (751 mg, 58%). δH (400 MHz, MeOD): 1.32 (s, 3H, CH3, H6), 1.53 (s, 3H, CH3, H7), 4.26–4.39 (AB of ABX, 2H, H5), 4.54 (X of ABX, 1H, H4), 5.12 (dd, J = 2.8 and 6.0 Hz, 1H, H3), 5.41 (dd, 1H, J = 2.4 and 6.0 Hz, H2), 6.24 (d, 1H, J = 2.4 Hz, H1), 8.21 (s, 1H, H2′), 8.27 (s, 1H, H8′). δC (100 MHz, MeOD): 26.0, 28.0, 70.5, 83.5, 86.0, 86.3, 92.3, 116.2, 121.0, 142.0, 150.8, 154.6, 157.9. νmax (ATR): 3122 (m), 2100 (w), 1686 (s), 1650 (s), 1374 (s), 1105 (s) cm−1. HRMS (ES) for C13H18N6O6SNa: (MNa+): calcd 409.0906, found 409.0927.
To a solution of isopropylidene sulfamoyl adenoside 25 (350 mg, 0.90 mmol) and DBU (500 µL) in DCM (5 mL) was added 2-tert-butoxycarbonylamino-3,3-dimethylbutyric acid 2,5-dioxopyrrolidin-1-yl ester 26 (330 mg, 1.00 mmol) portionwise. The reaction was stirred at 22 °C for 16 h, then the solvent was removed in vacuo and the residue was purified by column chromatography (EtOAc). Compound 27 was obtained as a white solid (263 mg) which included DBU impurities (5% by 1H NMR spectroscopy). δH (400 MHz, MeOH): 1.28 (s, 9H, t-Bu), 1.37 (s, 3H, CH3), 1.51 (s, 9H, t-Bu), 1.59 (s, 3H, CH3), 4.21 (br s, 1H, CH), 4.53 (br s, 2H, H5′), 4.59 (m, 1H, H4′), 5.16 (m, 1H, H3′), 5.33 (dd, 1H, J = 3.2, 5.8 Hz, H2′), 6.21 (d, 1H, J = 3.1 Hz, H1′), 8.36 (s, 1H, H2″), 8.55 (s, 1H, H8″). νmax (ATR): 3118 (m), 2101 (m), 1686 (s), 1647 (s), 1604 (s), 1374 (s), 1178 (s), 1079 (s) cm−1. LCMS: Rt 4.0, (MH+) 600.
The protected compound 27 (82 mg, 0.14 mmol) was stirred for 16 h at 22 °C in water–TFA (2 : 4 v/v). The solvent was removed in vacuo by azeotroping with ethanol and toluene. Purification by diaion HP-20SS chromatography (THF–water gradient 0 to 5%) gave the sulfamoyl as a white solid (48 mg, 76% yield). δH (250 MHz, D2O): 1.07 (s, 9H, H4), 4.38 (m, 4H, H2, H4′ and H5′), 4.63 (m, 1H, H3′), 4.79 (m, 1H, H2′), 6.09 (m, 1H, H1′), 8.18 (s, 1H, H2″), 8.51 (s, 1H, H8″). δC (125 MHz, D2O): 25.7, 32.8, 64.4, 67.5, 70.6, 74.6, 82.8, 88.1, 118.7, 139.8, 149.3, 152.5, 155.8, 174.1. νmax (ATR): 2990 (m), 1634 (s), 1600 (s), 1335 (m), 1150 (s), 1044 (s) cm−1. HRMS (ES) for C16H25N7O7SNa: (MNa+) calcd: 482.1434, found: 482.1428.
To a solution of 2′,3′-isopropylidene-5′-O-sulfamoyladenosine 25 (598 mg, 1.5 mmol) and DBU (500 µL) in DCM (10 mL) was added 3,3-dimethyl-2-oxobutyric acid 2,5-dioxopyrrolidin-1-yl ester 28 (820 mg, 3.6 mmol) portionwise. After the reaction was stirred at 22 °C for 16 h, the solvent was removed in vacuo. The residue was purified by column chromatography (EtOAc–MeOH 90 : 10 v/v) and 29 was obtained as a white solid (524 mg, 70%). δH (400 MHz, MeOH): 1.22 (s, 9H, t-Bu), 1.39 (s, 3H, CH3), 1.62 (s, 3H, CH3), 4.31 (t, 2H, J = 4.3 Hz, H5′), 4.56 (m, 1H, H4′), 5.16 (dd, 1H, J = 2.2 and 6.1 Hz, H3′), 5.39 (dd, 1H, J = 3.0 and 6.1 Hz, H2′), 6.25 (d, 1H, J = 2.9 Hz, H1′), 8.23 (s, 1H, H2″), 8.42 (s, 1H, H8″). δC (125 MHz, MeOD): 25.6, 27.1, 27.5, 30.0, 33.8, 50.0, 70.2, 83.3, 85.7, 115.4, 120.2, 141.4, 150.5, 154.1, 157.4, 167.5, 174.6. LCMS: Rt 4.1, (MH+) 499.
The protected compound 29 (50.2 mg, 0.10 mmol) was stirred for 3 h at 22 °C in water–TFA (2 : 4 v/v). The solvent was removed in vacuo by azeotroping with ethanol and toluene to give a white solid (40 mg, 87%). Purification by diaion HP-20SS chromatography (THF–water gradient 0 to 5%) gave the sulfamoyl as a white solid (25 mg, 54%). δH (400 MHz, D2O): 1.10 (s, 9H, t-Bu), 4.36 (m, 2H, H5′), 4.77 (m, 3H, H4′, H3′, H2′), 4.98 (m, 1H, H2′), 6.10 (d, 1H, J = 5.1 Hz, H1′), 8.35 (s, 1H, H2″), 8.50 (s, 1H, H8″). δC (125 MHz, D2O): 24.6, 30.6, 57.9, 69.6, 74.7, 82.3, 92.6, 130.6, 139.4, 144.1, 148.8, 152.6, 175.9, 198.8. νmax (ATR): 3324 (w), 2928 (m), 1684 (s), 1436 (m), 1134 (s) cm−1. HRMS (ES) for C16H23N6O8S: (MH+) calcd: 459.1298, found: 459.1284.
2′,3′-Isopropylidene-5′-O-sulfamoyladenosine 25 (386 mg, 1 mmol), DMF (10 mL), and DBU (230 µL, 1.5 mmol) were combined and stirred at 22 °C for 10 min. 4-Methoxy-2-(2-methoxyethoxymethoxy)-butyric acid pentafluorophenol ester 30 (529 mg, 1.36 mmol) was added and the reaction was stirred at 30 °C for 16 h. The residue was diluted with ethyl acetate (30 mL), washed with water (20 mL), dried (Na2SO4) and concentrated in vacuo. Purification by column chromatography (methanol–DCM 2–30% methanol) gave an oil (100 mg), which was a mixture of products including the desired product 32 by LCMS.
The protected compound 32 (50 mg by LCMS analysis, max. 0.08 mmol) was stirred for 40 h at 22 °C in water–TFA (2 : 4 v/v). The solvent was removed in vacuo by azeotroping with ethanol and toluene. Purification by HPLC gave the sulfamoyl 10 as a white solid (10 mg, 2% yield from 2′,3′-isopropylidene-5′-O-sulfamoyladenosine 25). δH (400 MHz, MeOD): 2.01 (m, 2H, H3), 3.22 (t, 2H, J = 6.9 Hz, H4), 3.30 (s, 3H, H5), 4.35 (m, 1H, H2), 4.43 (m, 4H, H3′, H4′ and H5′), 4.65 (m, 1H, H2′), 6.14 (d, 1H, J = 4.8 Hz, H1′), 8.40 (s, 1H, H2″), 8.55 (s, 1H, H8″). δC (125 MHz, D2O): 35.8, 37.5, 52.6, 58.9, 69.9, 71.9, 76.2, 84.3, 90.4, 120.4, 143.6, 145.7, 150.1, 152.0, 174.4. HRMS (ES) for C15H23N6O9S: (MH+) calcd: 463.1247, found: 463.1263.
2′,3′-Isopropylidene-5′-O-sulfamoyladenosine 25 (232 mg, 0.6 mmol), DMF (10 mL), and DBU (179 µL, 1.2 mmol) were combined and stirred at 22 °C for 10 min. 4-Methoxy-2-(2-methoxyethoxymethoxy)-3,3-dimethylbutyric acid pentafluorophenol ester 31 (180 mg, 0.45 mmol) was added and the reaction mixture was stirred at 30 °C for 16 h. The solvent was removed in vacuo and the residue was dissolved with ethyl acetate (30 mL), washed with water (20 mL), dried (Na2SO4) and concentrated in vacuo. Purification by column chromatography (methanol–DCM 2–30% methanol) gave an oily residue (77 mg). This was a mixture in which the desired product 33 was detected by LCMS.
The protected compound 33 (max. 77 mg, 0.12 mmol) was stirred for 16 h at 22 °C in water–TFA (2 : 4 v/v). The solvent was removed in vacuo by azeotroping with ethanol and toluene. Purification by HPLC gave the sulfamoyl 11 as a white solid (8 mg, 4% yield from 2′,3′-isopropylidene-5′-O-sulfamoyladenosine 25). δH (250 MHz, MeOD): 0.94 (m, 3H, H4), 0.97 (m, 3H, H5), 3.15 (m, 2H, H6), 3.29 (s, 3H, H7), 3.89 (s, 1H, H2), 4.38 (m, 4H, H3′, H4′ and H5′), 4.63 (m, 1H, H2′), 6.05 (m, 1H, H1′), 8.25 (s, 1H, H2″), 8.29 (s, 1H, H8″). δC (125 MHz, D2O): 20.8, 21.0, 38.2, 58.6, 68.1, 70.2, 73.9, 75.6, 77.4, 82.4, 87.1, 118.7, 139.7, 149.1, 152.8, 155.7, 180.3. νmax (ATR): 3257 (w), 1672 (s), 1648 (s), 1439 (w), 1325 (w), 1180 (s) cm−1. HRMS (ES) for C17H27N6O9S: (MH+) calcd: 491.1156, found: 491.1577
The protected compound 35 (386 mg, max. 0.77 mmol) was stirred for 16 h at 22 °C in water–TFA (2 : 4 v/v). The solvent was removed in vacuo by azeotroping with ethanol and toluene. Purification by diaion HP-20SS chromatography (THF–water gradient 0 to 5%) gave the sulfamoyl 12 as a white solid (33 mg, 7% yield from isopropylidene sulfamoyl adenoside 25). δH (250 MHz, D2O): 0.79 (s, 9H, H4), 3.58 (m, 1H, H2), 4.30 (m, 4H, H3′, H4′ and H5′), 4.65 (m, 1H, H2′), 6.05 (m, 1H, H1′), 8.25 (s, 1H, H2″), 8.29 (s, 1H, H8″). δC (125 MHz, D2O): 25.8, 36.8, 70.7, 72.8, 76.5, 83.5, 85.1, 89.7, 121.2, 142.3, 151.6, 155.4, 158.1, 183.3. νmax (ATR): 3110 (w), 1633 (s), 1599 (s), 1376 (m), 1150 (s), 1040 (s) cm−1. HRMS (ES) for C16H25N6O8S: (MH+) calcd: 461.1454, found: 461.1460.
Acknowledgements
We gratefully acknowledge financial support from the BBSRC and Christ's College, Cambridge.References
- F. Neidhardt, Escherichia coli and Salmonella typhimurium: cellular and molecular biology, American Society for Microbiology, Washington, DC, 2nd edn, 1996, vol. 1, p. 687 Search PubMed.
- J. E. Cronan, Jr., K. J. Little and S. Jackowski, J. Bacteriol., 1982, 149, 916 CAS.
- M. E. Webb, A. G. Smithh and C. Abell, Nat. Prod. Rep., 2004, 21, 695 RSC.
- R. Zheng and S. Blanchard, Biochemistry, 2001, 40, 12904 CrossRef CAS.
- K. Miyatake, Y. Nakano and S. Kitaoka, J. Nutr. Sci. Vitaminol., 1978, 24, 243 Search PubMed.
- F. von Delft, A. Lewendon, V. Dhanaraj, T. L. Blundell, C. Abell and A. G. Smith, Structure, 2001, 9, 439 CrossRef CAS.
- (a) J. Lee, S. U. Kang, S. Y. Kim, S. E. Kim, M. K. Kang, Y. J. Jo and S. Kim, Bioorg. Med. Chem. Lett., 2001, 11, 961 CrossRef; (b) J. Lee, S. U. Kang, S. Y. Kim, S. E. Kim, Y. J. Jo and S. Kim, Bioorg. Med. Chem. Lett., 2001, 11, 965 CrossRef CAS; (c) L. Koroniak, M. Ciustea, J. A. Gutierrez and N. G. J. Richards, Org. Lett., 2003, 5, 2033 CrossRef CAS.
- D. Heacock, C. J. Forsyth, S. Kiyotaka and K. Musier-Forsyth, Bioorg. Chem., 1996, 24, 273 CrossRef CAS.
- J. Lee, S. E. Kim, J. Y. Lee, S. Y. Kim, S. U. Kang, S. H. Seo, M. W. Chun, T. Kang, S. Y. Choi and H. O. Kim, Bioorg. Med. Chem. Lett., 2003, 13, 1087 CrossRef CAS.
- A. C. Baillie, C. L. Cornell, B. J. Wright and K. Wright, Tetrahedron Lett., 1992, 33, 5133 CrossRef CAS.
- V. H. Cheldelin and C. A. Schink, J. Am. Chem. Soc., 1947, 69, 2625 CrossRef CAS.
- Pdb database http://www.rcsb.org crystal structure 1n2h.
- S. S. Wang and D. Eisenberg, Protein Sci., 2003, 12, 1097 CrossRef CAS.
- S. Wang and D. Eisenberg, Biochemistry, 2006, 45, 1554 CrossRef CAS.
- J. D. Thompson, D. G. Higgins and T. J. Gibson, Nucleic Acids Res., 1994, 22, 4673 CrossRef CAS.
- A. C. Wallace, R. A. Laskowski and J. M. Thornton, Protein Eng., 1995, 8, 127 CrossRef CAS.
- Tripos SYBYL v6.5, Tripos, Inc, South Hanley Road, St Louis, Missouri, 63144, USA, 1699 Search PubMed.
- (a) G. Jones, P. Willet and R. C. Glen, J. Mol. Biol., 1995, 245, 43 CrossRef CAS; (b) G. Jones, P. Willet, R. C. Glen, A. R. Leach and R. Taylor, J. Mol. Biol., 1997, 267, 727 CrossRef CAS.
- For a general review see: D. L. Hughes, The Mitsunobu reaction, Org. React., 1992, 42, 337 Search PubMed.
- C. J. Koch, S. Simonyiová, J. Pabel, A. Kärtner, K. Polborn and K. T. Wanner, Eur. J. Org. Chem., 2003, 7, 1244 CrossRef.
- H. C. Aspinall, N. Greeves, W. Lee, E. G. McIver and P. M. Smith, Tetrahedron Lett., 1997, 38, 4679 CrossRef CAS.
- See: A. K. Forrest, R. L. Jarvest, L. M. Mensah, P. J. O'Hanlon, A. J. Pope and R. J. Sheppard, Bioorg. Med. Chem. Lett., 2000, 10, 1871 Search PubMed , and references therein.
- I. D. Jenkins, P. H. Verheyden and J. G. Moffatt, J. Am. Chem. Soc., 1976, 98, 3346 CrossRef CAS.
- J. Castro-Pichel, M. T. Garcia-López and F. G. De las Heras, Tetrahedron, 1987, 43, 383 CrossRef CAS.
- G. W. Anderson, J. E. Zimmerman and F. M. Callahan, J. Am. Chem. Soc., 1964, 86, 183.
- K. Miyatake, Y. Nakano and S. Kitaoka, Agric. Biol. Chem., 1973, 37, 1205 CAS.
- A. Cornish-Bowden, Fundamentals of Enzyme Kinetics, Portland Press, London, 2004, pp. 113–140 Search PubMed.
- E. F. Pettersen, T. D. Goddard, C. C. Huang, G. S. Couch, D. M. Greenblatt, E. C. Meng and T. E. Ferrin, J. Comput. Chem., 2004, 25, 1605 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Diagram of binding interactions made by pantoyl adenylate in the active site of M. tuberculosis pantothenate synthetase based on the crystal structure of pantothenate synthetase in complex with pantoyl adenylate 2. Figure generated using LIGPLOT15 (from PDB coordinates 1n2h). Diagram of binding interactions made by pantoyl adenylate 2 in the model of the close structure of the active site of E. coli pantothenate synthetase. Figure generated using LIGPLOT.15. See DOI: 10.1039/b609482a |
| This journal is © The Royal Society of Chemistry 2006 |