Direct conversion of methane on Mo/ZSM-5 catalysts to produce benzene and hydrogen: achievements and perspectives
Zinfer R. Ismagilov*, Ekaterina V. Matus and Lidia T. Tsikoza
Boreskov Institute of catalysis SB RAS, pr. Akademika Lavrentieva, 5, Novosibirsk, 630090, Russia. E-mail: zri@catalysis.ru; Fax: +7 383 3306219; Tel: +7 383 3306219
First published on 28th August 2008
Abstract
Development of highly effective catalysts for one-stage conversion of light hydrocarbons with high selectivity to valuable products will solve such problems as efficient utilization of natural and oil-associated gases, and environmental protection. Methane dehydroaromatization (DHA) over Mo/ZSM-5 catalysts is a promising process for direct production of valuable aromatic compounds and hydrogen from methane. This review focuses on the range of issues dealing with the effect of catalyst composition, preparation, pretreatment and operation conditions on the physicochemical properties and activities of Mo/ZSM-5 catalysts in DHA reaction. The concepts of the reaction mechanism and the nature of the active molybdenum forms are reviewed. Various aspects of the Mo/ZSM-5 deactivation under reaction conditions and methods of their regeneration are discussed. Some approaches for improvement of the Mo/ZSM-5 performance in DHA reaction are addressed in the review in detail. The perspectives of the methane dehydroaromatization process are also presented.
 Zinfer R. Ismagilov Zinfer R. Ismagilov | Zinfer R. Ismagilov is the Professor, Doctor of Chemical Sciences, Head of the Laboratory of Environmental Catalysis of the Boreskov Institute of Catalysis. His scientific activity includes the catalytic processes of hydrocarbon conversion, catalysis for environmental protection, microstructured reactors and carbon nanofibers. He is the author of over 500 scientific publications and 110 Russian patents. |
Ekaterina V. Matus is the PhD of Chemical Sciences, Researcher of the Laboratory of Environmental Catalysis. She received her PhD in 2007 and her work was devoted to the one-stage methane conversion to benzene and hydrogen. Dr Matus also has a strong interest in the elaboration of catalytic coatings for microstructured reactors. |
Lidia T. Tsikoza is the PhD of Chemical Sciences, Researcher of the Laboratory of Environmental Catalysis. She is a specialist in catalyst preparation techniques. Dr Tsikoza has been involved in the design of metal supported oxide and zeolite catalysts for different applications, catalytic coatings for microstructured reactors and honeycomb supports. |
Introduction
Methane, which is the main component of natural and oil-associated gases today, is considered to be an alternative source for synthesis of valuable products currently obtained by crude oil processing and organic synthesis.1–3 The forecasts predicting an increase of the role of gas in chemical industry are based on faster increase of the crude oil prices compared to natural gas prices.4–6Methane is mostly used as a fuel. The chemical industry consumes only 2.5–5% of natural gas.4,7,8 Wider application of methane is impeded by its high chemical and thermal stability.
The problem of processing natural gas to organic substances is usually solved by methane conversion to synthesis gas (mixture of CO and H2) in reactions with steam,9,10 carbon dioxide9,10 or oxygen11 followed by synthesis of a mixture of paraffins, olefins and alcohols by the Fischer–Tropsch method,1,12 synthesis of methanol or dimethyl ether.1,13
Some examples of one-stage methane conversion are conversion with C3–C4 hydrocarbons to form mono- and polyaromatic hydrocarbons,14 oxidative dimerization to ethylene or ethane,15–18 selective oxidation to methanol.19–23 In the presence of oxygen the formed hydrocarbons are easily oxidized to carbon dioxide and water. This side reaction decreases the selectivity at high methane conversions. This makes hydrocarbons synthesized from methane more expensive than the analogs obtained from crude oil. Overall, the processes of one-stage methane conversion are still at the laboratory stage of development. So, the search of new pathways for efficient utilization of natural gas is a very important problem.
One of such new pathways is methane dehydroaromatization (CH4 DHA). This is a method for selective conversion of methane directly to benzene and hydrogen without participation of oxygen:
| 6CH4↔ C6H6 + 9H2 | (1) |
The most effective catalyst for this process is Mo/ZSM-5.24,25 Oxygen-free conditions used for this reaction result in high benzene selectivity (up to 80%).3,24 Today several groups of researchers, mostly from China, USA, Hungary, Japan and Russia, study this process. Since the pioneering work of Wang et al.24 many research reports have been published and substantial progress in understanding the methane dehydroaromatization over Mo/ZSM-5 catalysts has been achieved.
This paper gives a review of the ongoing research concerning non-oxidative methane dehydroaromatization over Mo/ZSM-5 catalysts. The issues discussed in the review concern the dependence of the physicochemical properties and activities of Mo/ZSM-5 on the catalyst composition, preparation, pretreatment and operation conditions; Mo active forms and the DHA reaction mechanism; the nature of carbonaceous deposits, catalyst deactivation and regeneration; effects of promoters and approaches to catalyst improvement. The advancements and perspectives of DHA reaction are also presented.
1. Pathways for catalytic conversion of methane
Methane can be the starting material for synthesis of most compounds commonly produced by crude oil processing.5,15 There are two major methods for methane processing: (1) direct conversion to products; and (2) indirect conversion, most frequently, via synthesis gas.1,15 In this paragraph we will briefly overview the main methods used for catalytic transformations of methane with aim for further identification place and the role of methane dehydroaromatization (Scheme 1). Here DME is dimethyl ether, C2+ hydrocarbons are hydrocarbons containing two or more carbon atoms: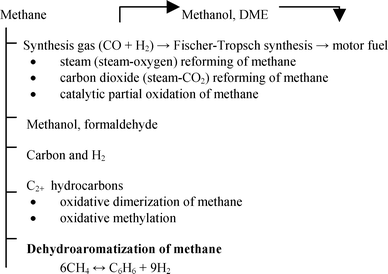 | ||
| Scheme 1 Main methods used for methane processing. | ||
Today, synthesis gas is the main primary product of methane processing. It is used for catalytic synthesis of methanol, synthetic gasoline, diesel fuel, DME.1,5,15 For example, industrial synthesis of methanol is carried out from synthesis gas with composition CO : H2 = 1 : 2 at 20 MPa pressure and T = 200–300 °C over Cu–Zn–Al oxide catalysts.5 DME production from synthesis gas is considered to be a promising process because its main characteristics (productivity, conversion of synthesis gas in one pass) are superior to those observed during methanol synthesis.5 A substantial amount of synthesis gas is also used for the synthesis of ammonia, which is a feedstock for production of such large-scale products as nitric acid and chemical fertilizers.
Oxidative conversion of methane to synthesis gas can be carried out by three methods:9–11,15
—Steam reforming
| CH4 + H2O ↔ CO + 3H2 | (2) |
| (ΔrH° = + 226 kJ mol−1, ΔrG° = −71 kJ mol−1) |
—CO2 reforming
| CH4 + CO2↔ 2CO + 2H2 | (3) |
| (ΔrH° = + 261 kJ mol−1, ΔrG° = −73 kJ mol−1) |
—Catalytic partial oxidation by oxygen
| CH4 + 1/2O2↔ CO + 2H2 | (4) |
| (ΔrH° = −44 kJ mol−1, ΔrG° = −254 kJ mol−1) |
The carbon dioxide reforming of methane (3) makes it possible to obtain synthesis gas with composition H2 : CO = 1 : 1, which is required for synthesis of formaldehyde and polycarbonates, and hydroformylation.15 This process is carried out at temperatures above 700 °C. From the thermodynamic point of view, reaction (3) is more endothermic than the steam reforming reaction (2). The ΔrG° = 0 condition is met at 680 °C. The reverse process takes place below this temperature. The conversion and selectivity values close 100% are observed at T = 1000–1100 °C. Platinum group metals29–31 and iron group metals32–34 supported on various supports show the highest activity in this reaction. The main obstacle preventing wide industrial application of this process is the catalyst carbonization due to reaction (5):5
| CH4 + 2CO2↔ C + 2CO + 2H2O | (5) |
| (ΔrH° = + 156 kJ mol−1, ΔrG° = −43 kJ mol−1) |
Catalytic partial oxidation of methane (4) is thermodynamically favorable in the whole temperature range.35 However, usually 100% conversion of methane is not achieved due to a number of side reactions:
| CH4 + 2O2↔ CO2 + 2H2O | (6) |
| (ΔrH° = −802 kJ mol−1, ΔrG° = −800 kJ mol−1) |
| CH4 + O2↔ CO2 + 2H2 | (7) |
| (ΔrH° = −305 kJ mol−1, ΔrG° = −434 kJ mol−1) |
| CH4 + 1.5O2↔ CO + 2H2O | (8) |
| (ΔrH° = −520 kJ mol−1, ΔrG° = −620 kJ mol−1) |
The presented data indicate than the syngas production is a very energy-consuming process. To a significant extent, this fact diminishes the competitiveness of producing chemical products from methane compared to their production by crude oil processing. Although the Fischer–Tropsch synthesis is the most direct method for syngas conversion to hydrocarbons, the cost of obtained synthetic fuel is still higher than of the fuel made from crude oil.5 This is caused both by the production cost of synthesis gas and by the low productivity of Fe and Co catalysts used for this process as well as the wide molecular mass distribution of the synthesis products, which requires their further processing to obtain the gasoline and diesel fractions.38
Among direct methods for methane conversion, oxidation to methanol21,39–41 and formaldehyde,21,39,42–44 and oxidative dimerization (condensation)11,45–49 are studied most intensively. However, until now no economic advantages of either of these methods have been demonstrated.
The highest methanol yields in direct methane oxidation were observed for Fe oxide catalysts under conditions close to those of the corresponding homogeneous process (T = 350–550 °C, P = 3–10 MPa).15,21 Today the CH4 conversion in one pass does not exceed 5%, whereas the selectivity can reach 70%.39–41
Methane oxidation to formaldehyde21,39,42–44 is usually carried out at higher temperatures than the methanol synthesis (550–650 °C) and atmospheric pressure. Methane conversion in this process is also not high (3–4%) with selectivity ∼80%. Two main groups of catalysts for this process can be distinguished: Mo and V oxide catalysts42,43,50 and Fe oxide catalysts.39,44 The work on this process is still at the laboratory scale, and in industry formaldehyde is produced from methanol.15
The oxidative condensation of methane results in coupling or two methane molecules in the presence of oxygen or another oxidizing agent to form C2+ hydrocarbons:
| 2CH4 + 1/2O2 = C2H6 + H2O | (9) |
| (ΔrH° = −350 kJ mol−1, ΔrG° = −223 kJ mol−1) |
| C2H6 + 1/2O2 = C2H4 + H2O | (10) |
| (ΔrH° = −105 kJ mol−1, ΔrG° = −198 kJ mol−1) |
Oxidative methylation of various organic substances is a thermodynamically feasible reaction:
RCH3 + CH4 + 1/2O2→ RCH2CH3 + H2O +1/2O2→ RCHCH2 + H2O, where R = C6H5, CH2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH, CH2C(CH3) CH, CH2C(CH3) | (11) |
Methane dehydroaromatization to benzene takes place both under oxidative53 and non-oxidative conditions.7,24,25,54–58 In the presence of oxygen the benzene selectivity over zeolite catalysts is only 3% at 600 °C.53 Low benzene selectivity is the result of formation of high quantities of CO and CO2. The possibility of methane DHA under non-oxidative conditions was first shown by Bragin et al.7,59 The process was carried out in a pulse mode at 740 °C using Cr-, Ga-, Zn- or Pt-Cr/ZSM-5 catalysts. Thermodynamically, non-oxidative dehydroaromatization of methane to benzene is not a very favorable reaction. However, despite the fact that for this reaction ΔrG° = 0 at 1075 °C,60 considerable amounts of benzene can be formed already at 700–900 °C.7 The thermodynamic equilibrium in systems 6CH4↔ C6H6 + 9H2 (ΔrH° = + 596 kJ mol−1, ΔrG° = + 146 kJ mol−1), 2CH4↔ C2H6 + H2 (ΔrH° = +73 kJ mol−1, ΔrG° = + 72 kJ mol−1), 2CH4↔ C2H4 + 2H2 (ΔrH° = +216 kJ mol−1, ΔrG° = + 81 kJ mol−1) and equilibrium concentrations of the corresponding products were calculated in the temperature range of 527–927 °C (Table 1).7 In the studied temperature range the methane DHA reaction is thermodynamically more favorable than its conversion to ethane or ethylene. For instance, at 727 °C the equilibrium conversion of methane to benzene is 13.8%.
| T/°C | Conversion CH4 (%) | Equilibrium composition (wt.%) | ||||
|---|---|---|---|---|---|---|
| C6H6 | C2H6 | C2H4 | H2 | CH4 | ||
| 6CH4↔ C6H6 + 9H2 | ||||||
| 527 | 2.5 | 2.0 | 0.5 | 97.5 | ||
| 727 | 13.8 | 11.2 | 2.6 | 86.2 | ||
| 927 | 40 | 32.6 | 7.4 | 60.0 | ||
| 2CH4↔ C2H6 + H2 | ||||||
| 527 | 1.0 | 0.9 | 0.1 | 99.0 | ||
| 727 | 3.0 | 2.8 | 0.2 | 97.0 | ||
| 927 | 5.0 | 4.7 | 0.3 | 95.0 | ||
| 2CH4↔ C2H4 + 2H2 | ||||||
| 527 | 1.0 | 0.9 | 0.1 | 99.0 | ||
| 727 | 5.0 | 4.3 | 0.6 | 95.0 | ||
| 927 | 20.0 | 17.5 | 2.5 | 80.0 | ||
As noted above, Mo/ZSM-5 is the best catalyst for CH4 DHA.55,57 According to the stoichiometry of reaction (1), this process results in the formation of hydrogen in large quantities: 9 mol H2/1 mol C6H6.61,62
Another example of a one-stage catalytic conversion of methane is its decomposition under oxygen-free conditions to filamentous carbon and hydrogen that does not contain CO and CO2 impurities.63–65 Carbon obtained by this method can be used as the catalyst support,66 whereas hydrogen can be applied in the low-temperature fuel cells.63 The process is carried out in the temperature range 600–1000 °C in the presence of catalysts containing iron group metals.64–66
2. Methods used for synthesis of Mo/ZSM-5 catalysts and their activity in CH4 DHA
2.1 Synthesis of Mo/ZSM-5 catalysts
The most widely used method for synthesis of Mo-zeolite catalysts for methane dehydroaromatization is impregnation of various zeolites, such as ZSM-5,25,24,54–58,67–72 ZSM-8,73 ZSM-11,73 MCM-41,73 FSM-16,67etc., with ammonium heptamolybdate (NH4)6Mo7O24·4H2O solution. Different initial forms of the zeolites are used: H-form,54–57,71 Na-form57,58 or NH4-form.,58,67,69,70 Typically, either incipient wetness impregnation57,58,67–70 or impregnation with excess solution25,67 are used. The concentrations of the ammonium heptamolybdate solutions used for impregnation are varied in wide range to obtain molybdenum concentrations in calcined samples varying from 1 to 20%.54,56,58,70The zeolite impregnation is carried out either at room temperature54–58,67,69,72 or at higher temperatures, e.g. 85 °C.25 The impregnation time is varied from 30 min54 to 24 h.69 Subsequent treatment of the catalysts also can be different. Usually the zeolite impregnated with the molybdenum solution is dried at 90–120 °C for 4 h,25,70 6 h56,67 or 10 h.54,58 Then, the samples are calcined in the presence of air at temperatures varied from 50025,57,69 to 720 °C.54 The calcination time can be 4 h,70 5 h,25,55,57,71 6 h58,69 or 30 h.74
Although impregnation is used most often for synthesis of Mo-zeolite catalysts, solid-phase synthesis was also used by some researchers.57,75–77 In this case a molybdenum salt (MoCl3) and a zeolite were mixed in a mortar, dried at 90 °C and calcined at 500 °C for 5 h.57 In another reported procedure a mixture of molybdenum oxide (MoO3) and a zeolite was ground in an agate mortar for 30 min. Then it was heated in a 20% O2/He flow to 700 °C with 10 °C min−1 heating rate and kept at this temperature for 30 min.76
Another approach used for synthesis of Mo-zeolite catalyst is deposition of molybdenum carbide Mo2C on H-ZSM-5 zeolite. In this case molybdenum carbide is mixed with the zeolite suspended in water. Then, water is evaporated under intensive mixing.55
The synthesis of Mo-zeolite catalysts with molybdenum in zero oxidation state has also been attempted. Molybdenum carbonyl (Mo(CO)6) was sublimed onto dehydroxylated ZSM-5 zeolite followed by treatment in hydrogen.55
An original sonochemical method has been also applied for synthesis of a Mo/ZSM-5 catalyst.78
2.2. Activity of Mo/ZSM-5 catalysts in CH4 DHA
Considering all available variety of conditions used for synthesis of Mo/ZSM-5 catalysts, it is possible to distinguish some characteristic features of their catalytic activity in CH4 DHA reaction. For instance, it was shown that H2O, CO, CO2 and H2 were formed during the first 35–45 min of CH4 reaction with a Mo/ZSM-5 catalyst.25,55,57,69,72 This time was called the induction period when the catalyst active sites are generated. C2 hydrocarbons (ethane, ethylene) and aromatic compounds (benzene, toluene) start appearing only after the induction period. Total methane conversion at 700 °C during the initial reaction period is 6–15%.25,54,55,61 Then, it decreases. Methane conversion to benzene grows during the first 60–120 min on stream and decreases after that. The selectivity to benzene initially grows. Then, it levels out at 60–85%. The selectivity to C2 hydrocarbons and their concentration increase during reaction. The naphthalene selectivity reaches a maximum at ∼120 min on stream and decreases thereafter.25,54,55,61 Hydrogen is one of the main reaction products (9–18 mol H2/1 mol C6H661,62). The dependence of the hydrogen yield on reaction time is similar to that observed for benzene.It should be noted that in some papers the selectivity to various products was calculated taking into account only the composition of the gas phase.54,55,79 Such an approach overestimates the selectivity to valuable products because carbonaceous deposits are formed during CH4 DHA, with 30–40% selectivity in addition to gaseous products.61,80
3. Physicochemical properties of Mo/ZSM-5 catalysts
The conditions used for synthesis of Mo/ZSM-5 catalysts, their pretreatment and CH4 DHA reaction are substantially different. So, it is natural to expect that the states of molybdenum and the zeolite matrix can be different as well. Therefore, below we shall discuss the physicochemical properties of Mo/ZSM-5 catalysts prepared by impregnation and solid-phase synthesis at each preparation and pretreatment stage and under reaction conditions.3.1. State of molybdenum in Mo/ZSM-5 catalysts
Many papers are devoted to investigation of the state of molybdenum in the zeolite matrix. For instance,70 it was shown by FTIR spectroscopy that after the zeolite impregnation with ammonium paramolybdate solution and drying in air at ∼100 °C, molybdenum remained in the form of ammonium paramolybdate crystallites. Calcination at 400–500 °C led to the formation of molybdenum oxides in Mo/ZSM-5 catalysts.70 At higher calcination temperature (700 °C) aluminium molybdate Al2(MoO4)356,70 was formed in addition to MoO3.58 These changes in the phase composition take place only in the catalysts with relatively high molybdenum concentrations: 10–15%. According to the EXAFS data,61 in impregnated Mo/ZSM-5 with 2–6% Mo, molybdenum exists in the form of finely dispersed oxide structures. Meanwhile, according to the results obtained by EPR81 and Raman spectroscopy,69 molybdenum may be either in the octahedral69,81 or square pyramidal oxygen environment.81There is no common opinion on molybdenum location in Mo/ZSM-5 catalysts. Apparently, this is related to the fact that molybdenum location depends on many factors: molybdenum concentration,54,70 temperature,25,70 calcination time and gas phase composition during calcination,25,74etc. It was found by NH3 TPD,811H MAS NMR74,81 and EPR81 that molybdenum migrated into the zeolite channels during calcination in air at 500 °C. It was reported25 that diffusion of some molybdenum into the zeolite channels was observed during calcination at ∼700 °C and intensified in moist air.
It was shown by FTIR spectroscopy54 that molybdenum was present in a finely dispersed state in the zeolite channels in the catalyst with Mo concentration less than 5%. At higher concentrations molybdenum oxides appeared on the external surface of the zeolite. However, in another study it was claimed57 that most molybdenum remained in the form of oxide on the external zeolite surface, even in samples with low Mo concentrations (2–4%).
It was suggested to distinguish two types of molybdenum in the zeolite—molybdenum associated with Brønsted acid sites (BAS) and the one not related to them.74 According to 1H MAS NMR data, the amount of molybdenum associated with BAS grew from 80 to 100% when the calcination was increased from 3 to 18 h. The authors assumed that one molybdenum atom interacted with one BAS.
Ultrahigh field NMR spectrometer was used to study the local structure of molybdenum species in Mo-zeolite catalysts. It was shown that 78%, 58%, and 33% Mo in 4% Mo/HZSM-5, 6% Mo/HZSM-5, and 10% Mo/HZSM-5 catalyst, respectively, were in the forms of exchanged molybdenum species, while the rest were present as molybdenum oxide crystallites.82
Adsorption of MoO3 and MoO2(OH)2 molecules on H-ZSM-5 zeolite was simulated using Monte Carlo method.83 The calculations showed that the maximum number of MoO3 molecules per zeolite cell was 15, whereas the number of MoO2(OH)2 molecules that can fit one elementary cell was only 5. MoO3 molecules fill the whole volume of the zeolite channels. Meanwhile, MoO2(OH)2 molecules were predominantly located at the intersections of the zeolite channels. It was shown that MoO2(OH)2 could be formed by decomposition of ammonium paramolybdate in air. Its vapor pressure is 4.9 Pa at 700 °C. Meanwhile, MoO3 is known to sublime at temperatures above 600 °C, and its vapor pressure reaches 56 Pa. However, mostly oligomeric molybdenum oxide molecules (MoO3)n, n = 2–5 are observed in the gas phase during sublimation. Their size substantially exceeds the diameter of the zeolite channels. The diameter of a MoO2(OH)2 molecule is about 5 Å.83 Taking into account the 1H MAS NMR data on the concentration of Brønsted acid sites before and after molybdenum introduction,71 the authors concluded that two MoO2(OH)2 molecules per zeolite cell were stabilized in 6% Mo/ZSM-5 catalyst.
According to the HRTEM data,72 molybdenum oxide was located on the external surface of the impregnated 2% Mo/ZSM-5 catalyst after pretreatment in argon at 720 °C, in the form of clusters 1–5 nm in size. In addition, better dispersed molybdenum oxide clusters with particle sizes smaller that 1 nm were present. They were not observed in the HRTEM images but detected using EDX analysis. When molybdenum concentration was increased to 10%, molybdenum oxide particles with dimensions as high as 100 nm appeared on the external zeolite surface and were observed by HRTEM.
When Mo/ZSM-5 catalyst prepared by impregnation was reduced with methane or hydrogen–methane mixture at 25–710 °C, MoO3 was first reduced to MoO2, followed by the formation of a stable molybdenum carbide phase, hcp β-Mo2C, supported on the zeolite.84,85 However, the treatment with a hydrogen-butane mixture resulted in the MoO3 reduction to a metastable molybdenum carbide phase fcc α-MoC1-x/ZSM-5 with preferential formation of molybdenum oxycarbide (MoOxHyCz) at the intermediate stages.
The investigation of the induction period of CH4 DHA reaction by TPSR86 showed that molybdenum oxide was reduced in two stages:
| 4MoO3 + CH4→ 4MoO2 + CO2 + 2H2O | (12) |
| 4MoO2 + 4CH4→ 2Mo2C + CO2 + 5H2O + CO + 3H2 | (13) |
A molybdenum concentration increase resulted in the growth of the molybdenum oxide reduction temperature, apparently, due to the presence of larger Mo oxide species requiring more rigid conditions for reduction.
It was shown by XPS that Mo2C and MoO3 coexisted on the surface of a working catalyst.87
Two types of molybdenum atoms located on the external zeolite surface and inside its channels were observed after reaction in Mo/ZSM-5 catalysts prepared by impregnation.72,81,88,89 The hyperfine splitting observed by EPR proves that molybdenum strongly interacts with the lattice aluminium.81,88,90 Molybdenum carbides were observed in the catalysts after reaction by XPS,25,55,79 EXAFS combined with CH4 TPR (temperature-programmed reaction),61,67 XRD84 and HRTEM.72,89 The formation of molybdenum carbide is an endothermic process (TDTA = 680 °C) accompanied by weight loss.61
Using HRTEM, it was shown that molybdenum carbide (α-Mo2C) was formed during the reaction on the ZSM-5 surface, and was characterized by the lattice parameters of d002 = 0.235 nm, d400 = 0.26 nm and particle size of 2–15 nm.72 According to the measured particle size distribution and the energy-dispersive X-ray (EDX) spectroscopic analysis, some Mo2C could be present in the most dispersed state (<2 nm). It was also demonstrated that during the reaction molybdenum-containing clusters with sizes of ∼1 nm were forming in the ZSM-5 channels.72
However, according to the XPS25,54,91 and EPR54,81,88,90 some molybdenum remained in higher oxidation state (Mo5+) even after prolonged reaction with methane. It was assumed that this molybdenum was located inside the zeolite channels.25,81
For solid-phase prepared 2–8% Mo/ZSM-5 catalysts before reaction it was shown92 that some molybdenum was located on the external zeolite surface as molybdenum oxide particles smaller than 3 nm, whereas the rest was in the zeolite channels in the form of [Mo5O12]6+ structures. It was supposed that the fraction of molybdenum located in the zeolite channels grew with molybdenum concentration.
It was shown that during calcination of MoO3 and H-ZSM-5 (Mo/Al < 0.4) in 20% O2/He at 700 °C molybdenum migrated into the zeolite channels and was stabilized in the form of Mo2O5 dimers in cation-exchange positions of the zeolite substituting two hydrogen atoms of the zeolite BAS.93 This conclusion was based on the measurements of H2O desorbed during synthesis and the results of D2 isotope exchange with the remaining OH groups of the zeolite. Predominant molybdenum location in the zeolite channels after the solid-phase synthesis was also supported by the results obtained by FTIR spectroscopy.57
On the basis of H/D exchange studies, 27Al MAS NMR and NH3 TPD methods it was determined94–96 that the anchoring mode of the molybdenum was strongly related to the Si/Al ratio of the parent ZSM-5 zeolite: monomeric bidentate species at low Si/Al ratio (15) and dimeric monodentate species at high Si/Al ratio (40).
Under reaction conditions the molybdenum oxide dimers formed initially are converted into 0.6–1 nm MoCx clusters containing ∼10 Mo atoms, which are located in the zeolite channels.76,93,97,98 This process is accompanied by partial regeneration of Brønsted acid sites participating in methane aromatization. It was noted76 that the formed molybdenum carbide clusters were stable with respect to agglomeration for a long time (10 h at 680 °C).
It was found92 that under reaction conditions molybdenum oxide species located on the external zeolite surface were converted to molybdenum carbide, whereas the ones located in the zeolite channels were transformed to MoOxCy. The authors of another paper77 paid attention to the presence of a metastable fcc α-Mo2C phase in addition to the stable hcp β-Mo2C in the samples prepared by solid-phase synthesis. Meanwhile, only the stable hcp β-Mo2C phase was observed by XRD in the catalysts prepared by impregnation.
3.2. State of the zeolite matrix in Mo/ZSM-5 catalysts
At it was noted by many researchers,54,58,72 molybdenum introduction in the H-ZSM-5 zeolite followed by calcination decreases the surface area and pore volume of the zeolite. This effect strengthens when the molybdenum concentration54,58,72,77 or the calcination temperature58 is increased. Large molybdenum oxide crystallites formed at high molybdenum concentrations block the zeolite channels.It was found that the crystallinity of the H-ZSM-5 zeolite decreased when the calcination temperature of Mo/ZSM-5 catalysts was increased from 500 to 700 °C.54,58 According to the 29Si and 27Al MAS NMR and XRD data,56 the zeolite matrix is subjected to substantial dealumination under these conditions with the formation of aluminium molybdate Al2(MoO4)3 and significant increase of the zeolite Si/Al ratio.
As it was shown by FTIR using adsorbed pyridine as a probe,61 the number of BAS in H-ZSM-5 zeolite did not change after introducing 3% Mo by impregnation followed by calcination at 500 °C for 2 h. However, the acid properties of the zeolite also depend on the composition of the gas phase, temperature and zeolite calcination time.25,54,74 For instance, pretreatment of 2% Mo/ZSM-5 catalyst in the flow of He or dry oxygen at 700 °C decreases the concentrations of all OH groups in the zeolite.25 After 0.5 h the concentration of BAS OH groups decreases by 30%, whereas that of terminal silanol groups, by 70%. The use of air leads to similar changes of the zeolite aid properties even at lower temperature −500 °C.25,71 This is caused by the effect of the zeolite treatment conditions on molybdenum diffusion.
Longer calcination of Mo/ZSM-5 catalysts leads to a decrease of the BAS concentration.74 For example, the concentration of such sites measured by 1H MAS NMR dropped to only 32% of their initial concentration after calcination at 500 °C for 3 h and to 15% of the initial value after calcination for 18 h.
The changes of the zeolite acid properties after introducing molybdenum also depend on the catalyst synthesis method.57 For instance, terminal silanol groups were predominantly affected in catalysts prepared by impregnation.25,57,71 This effect was more significant when the molybdenum concentration was increased.71 Thus, the concentrations of BAS and terminal OH groups in 2% Mo/ZSM-5 catalyst after heat treatment were 70% and 47% of the initial ones. Meanwhile, in 10% Mo/ZSM-5 catalyst the BAS concentration decreased to 23% whereas the concentration of remaining silanol groups was only 2% of the initial value. Meanwhile, it was shown57 that the zeolite BAS were preferentially destroyed for catalysts prepared by solid-phase synthesis.
In most cases,57,71 the observed decrease in the concentration of zeolite OH groups was associated with the H+ ion exchange for molybdenum ions. However, the problem of BAS regeneration under reaction conditions remains a subject of discussion. Some researchers76 observed the growth of the BAS concentration at the first stage of the reaction during carbidization of molybdenum oxide structures. However, another group reported99 that this process was accompanied by further decrease in the concentration on the zeolite acid sites. In addition, the concentration of Lewis acid sites (LAS) was observed to slightly increase.
Not so many papers are devoted to the investigation of the changes of the physicochemical properties of the zeolite matrix after reaction. It was found that the textural properties of the catalyst worsened, probably due to the formation of carbonaceous residues during reaction. The BAS concentration also decreases during reaction for the same reason according to the FTIR61 and ammonia TPD54 data.
3.3. The nature of carbonaceous deposits
The formation of carbonaceous deposits due reaction of Mo-substituted zeolite catalysts with methane was observed in many studies.54,57,67,69,72,81,100 They are formed with 20–40% selectivity.67 The concentration of the carbonaceous deposits increases with temperature and reaction time.54,100The formation rate of the carbonaceous deposits was found81 to grow linearly with the molybdenum concentration in the zeolite matrix, increasing from 0 to 2% and remain almost constant at molybdenum concentrations 2–10%. It is important that it remained constant during reaction for three hours.
Several types of the carbonaceous residues are distinguished. According to the XPS data,101 it is possible to distinguish three types of carbon. These include carbide carbon in Mo2C (C1s 282.7 eV), carbon in pre-graphite carbonaceous deposits (sp-type, C1s 283.2 eV) and carbon in carbonaceous deposits with graphite structure (C1s 284.6 eV). The pre-graphite deposits are formed on the external surface of the zeolite, whereas graphite is formed in the zeolite channels. It was noted101 that it is pre-graphite deposits that predominantly grow during reaction.
It was determined72,89,100 that during the DHA reaction the carbonaceous deposits were formed as graphite layers with the thickness of ∼2 nm on the surface of Mo2C nanoparticles that were >2 nm in size, and as friable layers with the thickness of up to 3 nm and a disordered structure on the external surface of the zeolite. According to the EDX, XRD, and DTA data, the content of the carbonaceous deposits and the extent of their condensation (the C/H ratio) increase with the time on stream, methane concentration in the feed, temperature and feed flow rate.72,100 For all the studied molybdenum contents (1%–10%) and reaction times (0.5–6 h), the carbonaceous deposits formed in the catalysts with Si/Al = 17 were characterized by one maximum of the exothermic burn-out effect in DTA, whereas in the catalysts with Si/Al = 30 and 45, they were characterized by two maxima.
According to 13C NMR data, some carbonaceous deposits are associated with Brønsted acid sites, whereas the others are associated with carbide or other molybdenum species with partially reduced molybdenum.69 Two types of carbonaceous deposits characterized by low (503 °C,102∼470 °C74,86) and high (592 °C,102 543 °C,74 557 °C86) oxidation temperatures were distinguished by TPO. These data agree with the results obtained by investigation of the carbonaceous deposits by thermogravimetric analysis (TGA).85 It was supposed74,103 that the deposits with lower oxidation temperature were located on the surface of molybdenum carbide, whereas the ones with higher oxidation temperature were associated with the zeolite BAS.
The existence of three TPO peaks at (459 °C, 511 °C and 558 °C) in the spectrum of 6% Mo/ZSM-5 catalyst with Si/Al = 25, after working for 1 h in the DHA of 90% CH4 at 700 °C, allowed the authors to distinguish three types of carbonaceous deposits: (1) associated with Mo sites located on the external zeolite surface; (2) associated with Mo sites in the zeolite channels; and (3) associated with Brønsted acid sites.88 The concentration of the carbonaceous deposits localized on BAS substantially decreases after the catalyst treatment in hydrogen. Meanwhile, the concentration of the deposits associated with Mo sites on the external surface increases.
Other researchers using TPO showed that, on the contrary, the carbonaceous deposits were uniform and associated only with the zeolite BAS.54 Based on the investigation of the carbonaceous deposits formed during reaction over a physical mixture of Mo2C/Al2O3 and H-ZSM-5, it was found that the carbonaceous deposits were predominantly formed on the Brønsted acid sites of the zeolite.104 Only one type of the carbonaceous residues was observed by TGA on 6% Mo/ZSM-5 catalyst (Si/Al = 25) after 6 h on stream in DHA of 100% CH4 at 700 °C.79 According to the HRTEM data, these deposits consisted of filamentous carbon.
The concentration and burn-out temperature of the carbonaceous deposits were found to depend on the type of molybdenum carbide phase formed on the zeolite surface.85 Using TGA, it was found that the carbonaceous deposits with the lower burn-out temperature preferentially formed over more active and stable α-MoC1-x/ZSM-5 catalyst compared to β-Mo2C/ZSM-5. However, the total concentration of the carbonaceous deposits was twice higher on the α-MoC1-x/ZSM-5 than on β-Mo2C/ZSM-5. It was supposed84 that this could be caused both by the different types of molybdenum carbides and their different dispersity or location.
According to the H2 TPR and CO2 TPR data, the treatment of the carbonized catalyst with CO2 decreases the concentration of deposits with both the low and high oxidation temperatures, whereas treatment with hydrogen decreases only the concentration of the deposits with the high oxidation temperature.102 However, it was not possible to remove the carbon deposits completely by carrying out successive H2 TPR and CO2 TPR experiments.
It was shown that the formation of carbonaceous deposits considerable decreased (up to two times) when the methane dehydroaromatization reaction over Mo/ZSM-5 catalyst was performed in a recycle condition with collection of benzene.105
4. Ways of regulating the activity of Mo/ZSM-5 catalysts in CH4 DHA
4.1. Effect of synthesis conditions on the catalytic activity of Mo/ZSM-5 catalysts
As noted above, different methods are used for introducing molybdenum during synthesis of Mo/ZSM-5 catalysts as well as different conditions of the following thermal treatment. It is the difference in the stabilization forms, location and dispersity of molybdenum that accounts for differences in the activity of Mo/ZSM-5 catalysts prepared by different methods.For example, it was found that the catalysts prepared by impregnation could be less active77 or more active57,106 in methane DHA than the samples prepared by solid-phase synthesis. Based on the XPS data,77 it was supposed that stronger molybdenum interaction with the zeolite matrix could be achieved in Mo/ZSM-5 catalysts prepared by impregnation rather than solid-phase synthesis. Such strong interaction primarily inhibits the formation of molybdenum carbide species necessary for the reaction and results in lower activity of such catalysts.
However, a Mo/ZSM-5 catalyst prepared by Mo2C deposition on H-ZSM-5 showed low activity in the studied reaction in comparison with the catalysts prepared by impregnation, and its main products were hydrogen and carbon rather than aromatic hydrocarbons. It was supposed55 that this was caused by low dispersity of molybdenum carbide. The molybdenum-zeolite catalyst with Mo in zero oxidation state also proved to be inactive in methane aromatization.55
When the catalysts are prepared by impregnation, it is possible to regulate the state of molybdenum already during preparation of the impregnation solutions. This is related to the known tendency of molybdenum to form monomeric or polymeric species in solution depending on the concentration and pH. The effects of acidification and basification of ammonium heptamolybdate solution during impregnation of HZSM-5 on the catalytic performance of Mo/HZSM-5 in methane dehydroaromatization were investigated.106,107 It was demonstrated107 that 2 wt.% Mo/HZSM-5 prepared with NH3-basified solution showed higher activity (7.1% aromatics yield) compared to that over a similar catalyst prepared without pH adjustment of the impregnation solution (4.9% aromatics yield). The authors attributed the improvement in the catalytic activity of this Mo/HZSM-5 catalyst to an increase in the concentration of the sites active in methane activation due to better Mo dispersion.
It was shown106,108 that the activity of both 2% Mo/ZSM-5 and 10% Mo/ZSM-5 catalysts in CH4 DHA slightly increased with an increase of pH in the ammonium heptamolybdate solution used for impregnation from 4 to 11.
The methane conversion and selectivity to aromatic hydrocarbons were found to decrease with an increase of the Mo/ZSM-5 calcination temperature from 500 to 700 °C.57,58,70 Meanwhile, the selectivity to C2 hydrocarbons, on the contrary, increased. This effect tended to become stronger as the molybdenum concentration increased. It was supposed58 that the high calcination temperature led to molybdenum aggregation into large particle blocking the zeolite channels. Later the same authors showed by FTIR that calcination at 700 °C resulted in molybdenum stabilization in the form of MoO42− reflecting strong interaction between molybdenum and oxygen atoms of the zeolite matrix. However, the thermal treatment at high temperature also destroys the crystalline lattice of the zeolite and leads to the formation of aluminium molybdate Al2(MoO4)3 This process is likely to be the cause of the poorer catalytic properties of the catalysts calcined at high temperature.
An increase of the 6% Mo/ZSM-5 catalyst calcination time at 500 °C from 3 to 18 h improved its catalytic activity.74 In addition it improved its stability as well. However, further increase of the calcination time to 30 h did not affect the catalytic properties. 1H MAS NMR showed that longer calcination time favored more complete molybdenum migration into the zeolite channels and its interaction with BAS. It was this additional migration that led to the catalytic activity increase.
4.2. Effect of molybdenum concentration on the activity of Mo/ZSM-5 catalysts
Molybdenum concentration in Mo/ZSM-5 catalysts was varied in wide range from 1 to 20%.54,56,58,70,77 It was shown that the catalytic activity of Mo/ZSM-5 catalysts prepared by impregnation grew when the molybdenum concentration was increased to 2–3%,54,58,87,109 2–5%,72 4%,57 6%71,77,81 or 10%.110 The activity of Mo/ZSM-5 catalysts prepared by solid-phase synthesis increased until Mo concentration of 16%.77 Further increase of the molybdenum concentration decreased the both total methane conversion and the selectivity to benzene. It was supposed54,58,77 that this decrease was mostly caused by the formation of large molybdenum particles blocking the zeolite channels. Another explanation of the observed dependence of the catalytic activity on the molybdenum concentration was based on the results of an XPS study of Mo/ZSM-5 catalysts with 1–15% Mo.87 It showed that the Mo2C/MoO3 ratio in the most active 3% Mo/ZSM-5 catalyst was about 0.4. An increase of the molybdenum concentration in the sample leads to increase of the fraction of molybdenum carbide. As far as it was supposed that methane activation requires the presence of both molybdenum oxide and carbide in the catalyst, this increase results in alteration of the composition from the optimal one.The specific benzene formation rate was much higher over samples with low molybdenum concentration (0.6–2%) compared to the samples with higher Mo concentrations (5–10%): 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–14000 nmol C6H6 gMo−1 s−1 and 1000–4000 nmol C6H gMo−1 s−1, respectively.72,106
000–14000 nmol C6H6 gMo−1 s−1 and 1000–4000 nmol C6H gMo−1 s−1, respectively.72,106
It was shown that the difference in the activity of catalysts with molybdenum concentrations of 2–10% increased when the calcination temperature was increased from 500 to 700 °C.70 A sharp decrease of the catalytic activity observed when the molybdenum concentration exceeded 2% was apparently caused by molybdenum interaction with the zeolite lattice resulting in the formation of aluminium molybdate.56
It was also found that an increase of molybdenum concentration shortened the induction period.57 This effect was supposed to be related to an increased fraction of more easily reduced polymolybdate compounds on the external surface of the zeolite.
4.3. Effect of zeolite physicochemical properties on the activity of Mo-zeolite catalysts
Different zeolites were used for synthesis of Mo-zeolite catalysts for methane dehydroaromatization: ZSM-5,54–58,67–70,75,76,80,111 ZSM-8,73 ZSM-11,73 MCM-41,73 MCM-22,112–114 FSM-16,67,115etc. The effect of the zeolite type on the catalytic properties was studied for 3% Mo-zeolite catalysts with Si/Al = 25.73 The catalysts prepared using ZSM-5, ZSM-8 and ZSM-11 zeolites were shown to be the most efficient in the studied reaction. Their activity was 5–10 times higher than that of the catalysts prepared using MCM-41 or SAPO-34. The catalysts prepared on such zeolites as MOR, X and Y were characterized by low methane conversion (< 1%) with the formation of only C2 hydrocarbons and CO. It is important that over such zeolites as MOR, USY, FSM-16 the selectivity to carbonaceous deposits was exceptionally high, above 80%.61 All the authors agree in the opinion61,73,116 that the structure of the ZSM-5 zeolite with a system consisting of two types of intersecting straight (d = 5.3 × 5.6 Å) and zig-zag (d = 5.1 × 5.6 Å) channels without large cavities favors the synthesis of small aromatic molecules but prevents accumulation of polycondensed aromatic substances.Indeed, the molecular diameter of benzene is ∼5 Å, while that of phenantrene is ∼8 Å.117,118 Therefore, narrow pores like those in SAPO-34 zeolite (d = 4.3 Å) hamper diffusion of aromatic molecules.116 Meanwhile, heavy aromatic products that are precursors of the carbonaceous deposits can be expected120 to form and accumulate over large-pore zeolites, such X or Y (d = 7.4 Å) with a system of cavities as large as 12 Å.119 However, there are several papers112–114 showing that the presence of large cavities in the zeolite structure does not necessarily worsen the catalyst performance in CH4 DHA. For instance, it was shown that large cavities (7.1 × 18.2 Å) with narrows entrance windows (4.0 × 5.4 Å) present in MCM-22 provide for high benzene selectivity and good stability to deactivation of Mo/MCM-22 catalysts.
A comparison of the catalytic activity of Mo/ZSM-5 catalysts prepared by impregnation of ZSM-5 in the H-form116 and NH4-form58,70 did not show any significant differences in their performance in CH4 DHA. However, catalysts prepared by impregnation of Na-ZSM-5 were inactive in methane dehydroaromatization.57,58 This is related to the lack of Brønsted acid sites in Mo/Na-ZSM-5 catalysts, which are required for oligomerization of the carbon fragments during reaction.57 The authors concluded that both the catalytic activity of Mo/ZSM-5 catalysts and the concentration of acid sites increased with the increase of the H+/Na+ ratio in the parent zeolite.
The Si/Al atomic ratio in the zeolite that can be varied in a wide range (Si/Al = 14,69,76 25,25,56–58,70,71,77,81 30,77 55,55 17–45,72,100 10–95061,67) can also affect the activity of Mo/ZSM-5 catalysts in CH4 DHA. For instance the highest activity was observed for the sample with Si/Al ≈ 2061,67 when Si/Al ratio was varied from 10 to 950. Similarly,121 the formation rate of aromatic hydrocarbons increased when Si/Al was increased from 14 to 28 but decreased after further Si/Al increase to 54. However, in some studies,2,114 the activity was observed to increase with the Si/Al decrease from 130 to 15114 and from 45 to 17.72
When the Si/Al ratio was increased from 17 to 45, the maximum methane conversion to benzene was achieved at lower molybdenum concentration 2% Mo for Si/Al = 17 and 1% Mo for Si/Al = 30 or 45.122 However, the maximum activity of Mo/ZSM-5 catalysts with Si/Al = 17 exceeded those of the catalysts with higher Si/Al ratios and was observed in a wider range of Mo concentrations (2–5%).
Dealumination of the initial zeolite is one of the known methods for increasing the Si/Al ratio. Such dealumination can be carried out under different conditions: zeolite treatment with hydrochloric acid,123 in steam at 500–550 °C for 6 h,80 in dry N2 at 600 °C for 6 h124 or in oxalic acid at 70 °C.125 The effect of the zeolite dealumination on the activity of Mo/ZSM-5 catalysts was controversial. In different studies it was observed to decrease123 or increase80,124,125 even when the dealumination degree was about the same (Si/Al growth from 20 to 50123 and from 27.2 to 38.8124). Interestingly, the selectivity to carbonaceous deposits was found to decrease from 20 to 8% for Mo/ZSM-5 catalyst prepared using a dealuminated zeolite.80
It is well known that variation of the Si/Al ratio is accompanied by changes in the concentration and strength of Brønsted acid sites.126 So, in the papers cited above the discussion of the observed results on the effect Si/Al ratio on the activity of Mo/ZSM-5 catalysts was based on this phenomenon. For example, the observed maximum of the catalytic activity at Si/Al ≈ 20 was found61,67 to correspond to the maximum BAS concentration determined from the FTIR spectra of adsorbed pyridine. The increase of the BAS concentration was supposed to favor oligomerization of CHx to benzene. The same catalysts also featured the lowest selectivity to coke formation of ∼35%.67
An alternative hypothesis on the growth of the activity of Mo/ZSM-5 catalysts with the Si/Al decrease is based on the growth of molybdenum dispersion in the zeolite matrix.113 BAS were assumed to act as molybdenum stabilization sites. So, their increase with the decrease of the Si/Al ratio favors the formation of small Mo species. An increase of Si/Al in Mo/ZSM-5 catalyst due to dealumination of the parent zeolite was found124 to decrease the concentration of Brønsted acid sites and increase that of Lewis acid sites. This led to an increase of both total methane conversion and its conversion to benzene. So, dealumination can be used to obtain an optimal BAS concentration.80
One should take into account that besides the Si/Al increase in the studied zeolites, dealumination of some zeolites (Y,127 BEA128) in the water vapor flow can result in the formation of secondary porosity (mesoporosity) of the zeolite matrix. Such mesoporosity can facilitate diffusion of aromatic molecules.129 Indeed, it was shown130 that the formation of a mesoporous structure in the H-ZSM-5 zeolite pretreated in NaOH at 80 °C improved the activity and stability of the 6% Mo/ZSM-5 catalyst.
Also it should be noted that the decrease of the Si/Al ratio in the zeolite is accompanied by the growth of the fraction of extraframework (octahedral) aluminium.131 Strong interaction with the latter is known for several metal ions (Cu, Co).132 Obviously, the presence of extraframework aluminium can affect the state of molybdenum in Mo/ZSM-5 catalysts and, therefore, their catalytic activity.
Preliminary hydrothermal treatment of H-ZSM-5 (Si/Al = 28.6) in 0.5 M solution of aluminium nitrate133 or 0.04 M NaOH solution134 was shown to improve the activity and stability of the 6% Mo/ZSM-5 catalyst. According to the data obtained by 27Al NMR and NH3 desorption, the concentration of extraframework aluminium decreases after such treatment. Additionally, the concentration of strong BAS goes down whereas that of BAS with medium strength goes up. Also this treatment slightly increases the zeolite microporosity.133,134 On the contrary, the hydrothermal treatment in water was shown to decrease the H-ZSM-5 crystallinity and increase the mesopore volume. Meanwhile, the activity of the Mo/ZSM-5 catalyst prepared using this zeolite was lower.
The zeolite crystallite size was found to have a significant effect on the activity of Mo/ZSM-5 catalysts.135 Methane conversion was 1.5 higher over the catalyst with the zeolite crystal size 100 nm compared to that over a zeolite with 70 nm crystals. The benzene selectivity was also higher over the sample with large crystals.135 According to the 1H MAS NMR data, the growth of the zeolite crystals led to migration of more molybdenum into the zeolite channels. In the sample with small zeolite crystals, molybdenum was predominantly located on the external zeolite surface. This was caused by an increase of the fraction of the external surface in the sample with small crystals.135
4.4. Effect of pretreatment and reaction conditions on the activity of Mo/ZSM-5 catalysts
The effect of the Mo/ZSM-5 catalyst pretreatment with a mixture of methane with one of the gases H2, C2H4, H2O or CO2 on the induction period time and the initial catalytic activity was studied for CH4 DHA reaction.136 The presence of ethylene in the mixture with methane (C2H4/CH4 = 0.005) was shown to shorten the induction period but have no effect on the benzene formation rate. An increase of the ethylene concentration (C2H4/CH4 = 0.1) shortened the induction period even further but decreased the activity of the catalyst. The catalyst pretreatment with H2/CH4 = 0.1 mixture had practically no effect on the induction period. Meanwhile, the presence of H2O or CO2 made the induction period much longer. So, to shorten the induction period, it was suggested to treat Mo/ZSM-5 catalysts with C2H4 or H2,69 or 1 : 4 CH4/H2 mixture.25,55,86The catalyst pretreatment in oxygen was also shown to shorten the induction period.87In situ FTIR studies showed that such treatment led to the formation of O22− (888 cm−1) and O–Al (670 cm−1) species on the catalyst surface that may take part in the reaction during the starting period of time.
Besides the induction period,84,88 the composition of the gas mixture used for pretreatment of the Mo/ZSM-5 sample also affected the phase composition and activity of the catalysts.84,88,137 For instance, it was shown that treatment in oxygen increased the methane conversion compared to the treatment in helium.137
After pretreatment with a hydrogen/butane mixture, the activity of Mo/ZSM-5 catalysts and its resistance to coking were better than after pretreatment with a hydrogen/methane mixture.84 Molybdenum carbide α-MoC1-x with face-centered cubic (fcc) structure was found in the catalyst in the former case. Meanwhile, β-Mo2C phase with hexagonal close-packed (hcp) structure was observed in the latter case. The authors supposed84 that the type of molybdenum carbide phase accounted for the differences in the activity of the catalysts. Similar conclusions were made by Liu et al.88 They found that hydrogen pretreatment at 350 °C of 6% Mo/ZSM-5 catalyst prepared by impregnation led to higher activity and better stability of the catalyst. They supposed that calcination in hydrogen led to the formation of MoCxOy phase with fcc rather than hcp. The former is more resistant to further reduction to the MoCx phase that is less active in CH4 DHA.
The feed composition also affects the activity of Mo/ZSM-5 catalysts in CH4 DHA reaction. The effect of ethane was studied most. This is realted to fact that C2–C4 hydrocarbons are present in the main sources of methane—natural gas and oil-associated gas. Ethane is the major component among them. For example, the average natural gas contains ∼4.7% ethane, ∼1.7% propane and ∼0.8% butane.15
Mo/ZSM-5 catalysts are known to have high activity in ethane conversion to aromatic hydrocarbons.138,139,140 For instance, benzene yield of 30% was observed using a mixture containing 1.1% C2H6/N2.138 If the feed consisted of only ethane the yield of aromatic hydrocarbons reached 65%.140 The main by-product was methane formed with the selectivity as high as 40%. The methane formation was supposed to result from demethylation of aromatic hydrocarbons, e.g. xylene.
The activity of 6% Mo/ZSM-5 catalyst was shown to grow linearly when the ethane concentration in the feed containing also CH4 and Ar was increased from 2.2 to 16.8 vol.%.141 The benzene formation rate when the feed consisted of two hydrocarbons (CH4 + C2H6 + Ar) was observed to be about two times higher than a sum of the reaction rates observed for one-component feeds (CH4 + Ar; C2H6 + Ar). However, it was noted that the increased activity was accompanied by faster catalyst deactivation.
It was found that addition of just 1% ethane to the feed increased the benzene yield by 7 to 10%.138 When 2.8% ethane was added to the feed containing N2/CH4/CO2 = 25/25/1, the increased formation rate of aromatic hydrocarbons was accompanied by negative methane conversion due to its formation in the reaction in agreement with the results of thermodynamic calculations.142 The ethane conversion that was 100% in the first minutes went down to 50% after 25 h on stream. The TPO study of Mo/ZSM-5 catalysts after reaction showed that the amount of carbonaceous deposits was about two times greater in the presence of ethane than without it.142
The total methane conversion and selectivity to aromatic were found to decrease when the feed flow rate was increased.3,143
Both the methane conversion and selectivity to benzene were found to increase when the reaction temperature was increased from 600 to 840 °C.58 However, this was typical only for the first ∼100 min on stream. Generally, higher reaction temperature leads to faster deactivation of Mo/ZSM-5 catalysts.70,106,144
5. Mechanism of methane dehydroaromatization in the presence Mo/ZSM-5 catalysts
5.1. The nature of active sites
A bifunctional mechanism of methane dehydroaromatization over Mo/ZSM-5 catalysts with participation of Mo sites and Brønsted acid sites is most widely accepted in recent papers.25,55,57,61,67,76 In this mechanism methane is activated on Mo sites with hydrogen release to the gas phase and formation of surface CHx species (Scheme 2a). Then the products of their dimerization C2Hy are subjected to oligomerization on the zeolite BAS to form benzene and naphthalene (Scheme 2b).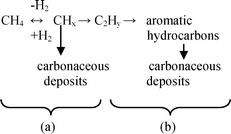
Using the infrared spectroscopy, XPS, and chemical reactivity measurements it was demonstrated that thermally stable alkylidenes could be formed on the surface of α-Mo2C at or above 900 K. It was proposed that carbenes on the molybdenum carbide component of the bifunctional Mo/ZSM-5 catalyst participated in the aromatization reaction.145
Usually3,25,55 ethylene is considered to be a reaction intermediate. For example, based on the data obtained by H2 TPR of Mo/ZSM-5 catalyst subjected to temperature-programmed reaction with CH4, both CH4 activation to surface [CH3] species and dehydrogenation of the latter to [CH2] followed by condensation to C2H4 were supposed to take place on Mo sites.103 The C2H4 formation was also observed during investigation of the CH3Cl and CH2I2 adsorption on the Mo2C surface.146,147 Note that [CH3] decomposition reaction predominates over Pt catalysts.
Alternatively, acetylene rather than ethylene was supposed to be the primary product formed from methane.113,148 Then, it can undergo either hydrogenation to C2H4 or oligomerization over the zeolite acid sites to form aromatic compounds. The main argument in favor of this mechanism is based on the dependence of the partial pressures of acetylene, ethylene and benzene on the feed contact time with the catalyst sample. The acetylene pressure decreased when the contact time was maid longer. Meanwhile, the ethylene and benzene pressures, on the contrary, increased. However, the acetylene pressure was only 0.01–0.04 Torr that was almost two orders lower than those of ethylene (0.2–1 Torr) and benzene (1–5 Torr).148
Heterolytic splitting of the C–H bond in CH4 was one of the first suggested mechanisms for this reaction.58 The authors believed that the C–H bond was split on the zeolite acid sites followed by the formation of MoO3CH2 intermediates, which were then dimerized to form ethylene.
Methane activation through the formation of free CH3˙ radicals was also one of the early hypotheses.54 MoOx groups located close to BAS were considered to be the active sites.
It was reported25,55,114 that Mo2C structures were the methane activation sites, and their partially oxidation decreased the activity of the Mo/ZSM-5 catalysts.114 Alternatively, the active sites were supposed88,92,149 to have oxycarbide structure MoCxOy.
It was established that the [Mo5OxCy]n+ units inside the channels of ZSM-5, which interacted with the framework of ZSM-5 and could hardly aggregate and sublime during the reaction, maintained the catalytic activity of the catalysts.149 The synergetic effect of the catalytic activity of [Mo5OxCy]n+ units and the shape selectivity of ZSM-5 was beneficial for the selectivity to benzene.
A good correlation between the amount of the exchanged molybdenum carbide species and the formation rate of aromatics compound was obtained.82 It was concluded that the molybdenum species originating from the exchanged Mo species were the active centers for the methane dehydroaromatization reaction.
It was supposed61,74,76,92,97,149,150 that molybdenum location in the zeolite channels was the main reason for the long-term activity of the catalysts. However, there is an alternative opinion84,115 that the activity of Mo/ZSM-5 catalysts is related to molybdenum carbide located on the external surface of the zeolite. The activity of the catalysts was found to correlate with the type of the molybdenum carbide phase.
The carbonaceous deposits may also participate in the formation of the catalyst active sites.25,69,115 It is possibly that the Mo-associated carbonaceous deposits decrease the Mo2C activity in side reaction of methane decomposition to hydrogen and carbon.
5.2. Deactivation of Mo/ZSM-5 catalysts and methods of their regeneration
The deactivation of Mo/ZSM-5 catalysts due to their substantial carbonization is a generally accepted interpretation.54,67,101,103,151 As noted above, the concentration of the carbonaceous deposits grows with time on stream72,101,103 and reaction temperature.54 The amount of the carbonaceous deposits located on the external surface of the zeolite was shown to grow during reaction.102 The time required for the catalyst to loose almost all its activity can be different and equal, e.g. 4 h54 or 16 h.25The results obtained by TPO of Mo/ZSM-5 catalyst after reaction suggested103 that the main reason for the catalyst deactivation was related to the formation of the carbonaceous deposits associated with the zeolite BAS and their blocking of the zeolite channels.
Note that the catalyst deactivation is not always directly related to the formation rate of the carbonaceous deposits. For example, the morphology of the formed carbonaceous deposits was found to be an important parameter affecting the activity of H-ZSM-5 catalysts for methanol conversion.152 The H-ZSM-5 channels remain accessible to reagents until the deposits have amorphous structure. So, no catalyst deactivation is observed in this case. However, when three-dimensional networks with aromatic structure parallel to the zeolite surface area are formed, the zeolite channels are blocked, and the catalyst loses its activity.152
It was reported115 that deactivation of Mo/FSM-16 sample in CH4 DHA could result from the change of molybdenum carbide phase in the catalyst. It was found that the interaction of η-Mo3C2 carbide with the carbonaceous deposits resulted in the formation of less active molybdenum carbide phases α-MoC1-x and β-Mo2C.
The catalytic activity of carbonized Mo/ZSM-5 samples can be regenerated by the oxidative treatment. Several methods were suggested for regeneration of deactivated Mo/ZSM-5 catalysts.93,105,153,154 For example, the Mo/ZSM-5 catalyst was regenerated by treatment in a NO/air mixture (1/50 vol/vol) at 450 °C.153 A small addition of nitric oxide decreased the temperature required for complete removal of the carbonaceous deposits by 100 °C compared to regeneration in pure air. The following mechanism for regeneration of the carbonized catalyst based on the results of the TPO experiments was suggested:
| 2NO + O2→ 2NO2 | (14) |
| NO2 + [carbonaceous deposits] → NO + N2O + N2 + COx + H2O | (15) |
The low temperature used for the oxidative treatment made it possible to avoid sublimation and migration of MoOx. This preserved the initial molybdenum distribution in the zeolite matrix and provided for more stable functioning of the catalyst in the mode alternating reaction with regeneration.153
Oxidative regeneration of 2% Mo/ZSM-5 catalysts after ∼6 and ∼20 h on stream was carried out at 520 and 600 °C, respectively.100 It was shown that after 6 h on stream the catalytic activity remained practically constant after 5 reaction-oxidative regeneration cycles. Meanwhile, an increase of the reaction time to ∼20 h led to faster catalyst deactivation in the third cycle: methane conversion to benzene decreased from 9% to 2% for ∼15 h, whereas after the first reaction cycle such decrease occurred only after ∼20 h on stream.
If the process is carried our in a periodic mode (alternating feed of CH4 and H2), the yield of aromatic compounds can be increased and the catalyst carburization can be lowered.155,156 However, the stable functioning of the catalyst in such mode required the cycles to be as short as 5–10 min.155
The treatment of carbonized catalysts with a 20% H2/He mixture at 680 °C regained the initial catalyst activity and selectivity to benzene and eliminated the induction period.93,154 However, this method proved to be not effective when the catalyst deactivation was very significant. After the treatment of the Mo/ZSM-5 catalyst under TPO conditions (130–680 °C, 5 °C min−1 heating rate) the induction period appeared again whereas the reaction rate in the steady state was equal to that observed over the fresh catalyst.93
CO2 TPR followed by H2 TPR was shown to be the most acceptable method for regeneration of Mo/ZSM-5 catalysts because it removed all types of the carbonaceous deposits.103 About 90% of the carbonaceous deposits related to BAS and 60% of those related to Mo sites were removed when the catalyst was regenerated only by H2 TPR.103
6. Ways of improving the methane dehydroaromatization process
In several papers54,62,68,75,137,143,157 it was shown that the presence of a second metal in the zeolite matrix in addition to molybdenum could have a significant effect and the activity, selectivity and coking resistance of Mo/ZSM-5 catalysts.For instance, the copper introduction to H-ZSM-5 zeolite by ion exchange followed by solid-phase synthesis of Mo/Cu/ZSM-5 (Cu/ZSM-5 + MoO3) system gave a more active and stable catalyst.75 The catalyst after reaction was studied by XPS and EPR. It was found that the presence of copper increased the concentration of Mo5+ ions in the catalyst. In addition, it was found by XRD and 27Al NMR that the copper introduction decreased the zeolite dealumination rate and its carbonization, thus, increasing the catalyst life time. The character of the carbonaceous deposits also changed: their oxidation temperature decreased, and there were more carbon radicals in the sample.
The addition of Co,67,158 W, Zr137 or Ru143,159 to Mo/ZSM-5 catalysts was shown to increase their activity and selectivity. The improvement of the characteristics of Mo/ZSM-5 catalysts modified with Ru was related to the decreased concentration of strong BAS and increased concentration of the weak and medium-strength ones as well as easier reduction of the initially formed molybdenum oxide.143 The use of platinum as the modifying additive increased the stability of Mo/ZSM-5 catalyst due to the lower concentration of carbonaceous deposits formed during reaction over Pt-doped catalyst.157 The addition of Zn or La was also found to decrease the carbonization rate of Mo-zeolite catalysts.68
The activity and stability of the catalyst also improved after the introduction of Fe (Fe/Mo = 0.25 mol mol−1).158 However, in this case the elemental analysis showed that the concentration of the carbonaceous deposits increased as well. Taking into the fact that the experiment in this case lasted only for 6 h, one can expect that the observed improvement of the catalyst will not be long-term. No positive effect of Co on the activity of Mo/ZSM-5 catalyst was observed in that study.
Lithium,54 phosphorus54 or vanadium137 present in the Mo/ZSM-5 catalysts were found to decrease their catalytic activity due to the decrease of the concentration of Brønsted acid sites participating in the methane aromatization.
Dealumination of the parent zeolite is an alternative way to reduce the formation of the carbonaceous residues during reaction.80,114,124 According to the TPO data, the zeolite dealumination decreases the concentration of the carbonaceous deposits with high oxidation temperature related to Brønsted acid sites.80,124 The selectivity to the carbonaceous deposits was observed to decrease from 37.9% to 18.2% over dealuminated Mo/ZSM-5 catalyst at the same methane conversion (9.5%) whereas the yield of aromatic hydrocarbons increased by 32%.80
The effect of silanation of the parent H-ZSM-5 zeolite on the activity of Mo/ZSM-5 catalysts in CH4 DHA was also studied.98,160 To preserve the acidic OH groups located in the zeolite channels, 3-aminopropyl-trietoxysilane was used as the source of silicon. Due to steric limitations it reacted only with the surface OH groups of the zeolite. The silanation was found to remove 24% of the zeolite OH groups. The optimum concentration of the additive was found to be 0.5% (calculated as SiO2).160 In Mo/SiO2/ZSM-5 catalyst, molybdenum reacted predominantly with the acidic groups inside the zeolite channels. In this case the CH4 DHA took place only inside the channels. As a result, the benzene selectivity of silanated Mo/ZSM-5 catalyst increased, whereas the selectivity to C12+ products decreased. Similar results were obtained by modifying the external surface of the initial zeolite with an organometallic tin compound (Sn(Bu)4).161
Adding small amounts of CO,62,67,138 CO2,62,67,162 O2163 or H2O164 to the feed, i.e. to methane, is another approach to improve the stability of Mo/ZSM-5 catalysts. Interestingly, the positive effect was observed only in a narrow concentration range of the added reagent.
When 1.6–12% CO was added to the feed, the methane conversion after 30 h on stream was 8–9% with benzene formation rate ∼500 nmol gcat−1 s−1.62 Both values were approximately twice higher that in the absence of CO. If CO2 is added to methane, its concentration should not exceed 1.6%. The increase of the CO2 concentration to 12% led to the loss of the 3% Mo/ZSM-5 catalyst activity.62,138 When CO2 is added to methane, it is first converted to CO:165
| CO2 + CH4→ 2CO + H2 | (16) |
| CO2 + [C] → 2CO | (17) |
| 2CO ↔ CO2 + [C], where [C] is the “active” carbon | (18) |
| CO2 + CHx (x = 0–4) ↔ 2CO + x/2H2 | (19) |
| [C] + H2↔ CHx↔ C2Hy→ C6H6, C10H8 | (20) |
There is also an alternative mechanism explaining the effect of CO2 addition:162
| CO2 + * → CO + O* | (21) |
| CH4 + 2* → CH3* + H* → C* + 4H* | (22) |
| C* + O* → CO + 2* | (23) |
| H* + H* → H2 + 2* | (24) |
| CHx* + CHy* → hydrocarbons | (25) |
The oxygen presence in the feed was also reported to have a positive effect.163 The critical oxygen concentration in the feed, which should not be exceeded to avoid deep oxidation of methane, grows with reaction temperature. For instance, at 700 °C the O2/CH4 molar ratio should not exceed 6.5 × 10−3 whereas at 800 °C this critical value is equal to 2.4 × 10−2. If no oxygen is present in the feed, after contact with methane molybdenum oxide is first converted to oxycarbide (MoOxCy), then, to less active carbide (Mo2C). This effect contributes to the decrease of the Mo/ZSM-5 activity during reaction.163 The presence of oxygen in the feed in small amounts was supposed163 to decrease the concentration of the carbonaceous deposits and prevent reduction of molybdenum oxycarbide (MoOxCy) for an extended period time.
The addition of small amounts of water (1.7–2.2%) was shown164 to improve the activity and stability of 6% Mo/ZSM-5 catalyst. The positive effect of water is related to the following reactions:
| CH4→ C + 2H2 | (26) |
| H2O + C → CO + H2 | (27) |
In addition, the concentration of the carbonaceous residues and their oxidation temperature also decrease. However, when the water concentration exceeds the optimal value, the catalytic activity quickly goes down. The results of the 27Al NMR studies suggest that this is related to the zeolite dealumination.164
Another general approach to improving the efficiency of CH4 DHA process is based on the improvement of process technology and engineering.138,145,166–173 An interesting example of such scheme is the combination of two reactions—oxidative dimerization of methane (ODM) and DHA of CH4 and C2 hydrocarbons formed in the ODM reaction—in one reactor or two successive reactors.138,166,167 However, the results of these experiments proved to be contradictory. In some studies,166,167 the combination of these two processes was shown to be very effective. After 20 h on stream the yield of aromatic hydrocarbons was 8.8% with a methane conversion of 13%. For comparison, these values were equal to 0.1% and 1%, respectively, if CH4 DHA was carried out in a single reactor. The improvement of the catalyst stability was supposed to be caused by the reaction of CO2 produced in ODM with the carbonaceous deposits formed on Mo/ZSM-5. However, in another study138 the combination of these two processes was shown to result in a considerable decrease of the amount of aromatic products formed in the reaction. This effect was ascribed to the deactivating effect of CO2 and H2O formed in the ODM process on the performance of the Mo/ZSM-5 catalyst. Most likely, the opposite results obtained in these studies are related to some important differences in the reaction conditions.138,166
The calculations carried out using a kinetic model describing the methane DHA with participation of a catalyst suggested that elimination of hydrogen formed in this reaction would make it possible to lift the thermodynamic limitations for methane conversion to aromatic hydrocarbons.168 This goal can be achieved by selecting a proper membrane that should provide for the hydrogen penetration rate comparable with the rate of its formation in the catalytic reaction. Until now, only few papers devoted to the use of membranes in CH4 DHA process have been published. CH4 DHA was carried out in a reactor with a Pd- or Pd-Ag membrane.169–171 However, in this case the about two times faster methane conversion to benzene was accompanied by faster catalyst deactivation due to the formation of carbonaceous deposits with high C/H ratio. The application of an oxide membrane with SrCe0.95Yb0.05O3-α proved to be inefficient due to slow hydrogen elimination from the reactor volume.172
Multiple recycling of CH4 with collection of the aromatic product was performed to increase the overall methane conversion.105 It was shown that if the reaction was carried out under flow recycling conditions the benzene formation rate (mgC6H6 gcat.−1 h−1) was significantly improved (up to four times). The benzene yield increased when the circulating factor was increased.105
Conclusions
The following conclusions can be made on the basis of the discussed experimental data. Methane dehydroaromatization reaction takes place at atmospheric pressure and high temperatures (T > 700 °C) typical for methane processing reactions. The fact that aromatic products can be formed from methane with high selectivity makes it possible to consider CH4 DHA to be an alternative method for rational utilization of the natural gas and oil-associated gas. Today CH4 DHA can be carried out over Mo/ZSM-5 catalysts with benzene selectivity as high as 80% and total methane conversion up to 14%.The synthesis conditions and composition of Mo/ZSM-5 catalysts have a significant effect on the state and location of molybdenum in the zeolite, which affect the catalytic activity in CH4 DHA. The tendency to sublimation of molybdenum oxides that have relatively high vapor pressures makes possible molybdenum redistribution in the zeolite matrix through the gas phase during the thermal treatment. This results in the molybdenum stabilization both on the external zeolite surface and inside its channels. During the CH4 DHA reaction the oxide forms of molybdenum are converted to carbides.
The physiochemical properties of the parent zeolite (structure, Si/Al atomic ratio, etc.) also have a substantial effect on the activity of Mo-zeolite catalysts. The zeolite pore structure with the diameters of entrance windows close to the diameter of benzene molecules provide for high methane conversion and benzene selectivity. On the one hand, the catalytic activity was found to grow with the number of Brønsted acid sites. On the other hand, dealumination of the zeolite matrix aimed at decreasing the BAS concentration improves some process conditions. Apparently, such differences are related to the participation of BAS both in oligomerization of C2 intermediates to benzene and in the formation of the carbonaceous deposits. Also, BAS participate in molybdenum stabilization in the zeolite matrix.
Molybdenum introduction in the zeolite substantially alters its textural and acidic properties. The changes in the physicochemical properties of the zeolite depend on the concentration and location of molybdenum in the zeolite, which are determined with the synthesis conditions of Mo/ZSM-5 catalysts. Subsequent changes of the zeolite physicochemical properties during the CH4 DHA reaction are mostly related to the formation of the carbonaceous deposits.
Carbonization of Mo/ZSM-5 catalysts is a side process in the CH4 DHA reaction, gradually leading to the catalyst’s deactivation. The available data on the type of the carbonaceous deposits indicate that their formation rate and selectivity can be regulated by varying the conditions used for the reaction and for synthesis of Mo/ZSM-5 catalysts. The treatment of carbonized Mo/ZSM-5 with a gas mixture containing oxygen or hydrogen allows for full or partial regeneration of the catalyst activity. However, the data on the activity of Mo/ZSM-5 catalysts, the states of molybdenum and zeolite after multiple regeneration cycles are very limited.
Although the concept of bifunctional action of Mo/ZSM-5 catalysts is generally accepted, there is no information about the detailed reaction mechanism. The questions about the nature and distribution of Mo sites in the zeolite matrix are still in the focus of discussion. The role of carbide or oxycarbide Mo clusters located in the zeolite channels in activating methane was emphasized in many published papers.
Based on the information available in the literature, three main approaches for possible improvement of the methane dehydroaromatization process can be distinguished:
—Optimization of the Mo/ZSM-5 catalyst composition by adding various dopes;
—Variation of the feed composition by adding low-molecular oxygenated reagents to methane;
—Improvement of the process technology and engineering.
The problems that should be addressed later include development of a synthesis method for preparation of Mo/ZSM-5 catalysts with certain compositions and distributions of Mo clusters in the zeolite matrix. The detailed mechanism of methane activation has to be studied. For practical implementation of the process it is important to study the activity and selectivity of Mo/ZSM-5 catalysts using a feed composition close that of the natural/oil-associated gas. Of special interest are the problems of increasing the catalyst life time and optimizing the conditions of its regeneration. One can expect that proper technological implementation will make the synthesis of benzene and hydrogen by CH4 DHA economically profitable.
References
- T. V. Choudhary, E. Aksoylu and D. W. Goodman, Catal. Rev., 2003, 45, 151–203 CrossRef CAS.
- E. F. Sousa-Aguiar, L. G. Appel and C. Mota, Catal. Today, 2005, 101, 3–7 CrossRef CAS.
- Y. Xu, X. Bao and L. Lin, J. Catal., 2003, 216, 386–395 CrossRef CAS.
- O. V. Krylov, Kinet. Catal., 1999, 40, 138–143 CAS.
- A. Ya. Rozovskii, Kinet. Catal., 1999, 40, 322–333 CAS.
- J. H. Lunsford, Catal. Today, 2000, 63, 165–174 CrossRef CAS.
- T. V. Vasina, A. V. Preobrazhenskii, S. A. Isaev, O. V. Chetina, O. V. Masloboishchikova and O. V. Bragin, Kinet. Catal., 1994, 35, 106–109 CAS.
- G. R. Meima, B. R. Maughon, A. E. Schweizer and M. E. Jones, Book of abstracts, Seventh European Congress of Catalysis, Sofia, Bulgaria, 2005, pp. 157–158 Search PubMed.
- J. R. H. Ross, Catal. Today, 2005, 100, 151–158 CrossRef CAS.
- J. R. Rostrup-Nielsen, J. Sehested and J. K. Norskov, Adv. Catal., 2002, 47, 65–139 CAS.
- K. Otsuka and Y. Wang, Appl. Catal., A, 2001, 222, 145–161 CrossRef CAS.
- M. C. Bahome, L. L. Jewell, D. Hildebrandt, D. Glasser and N. J. Coville, Appl. Catal., A, 2005, 287, 60–67 CrossRef.
- Q. Zhang, X. Li, K. Asami, S. Asaoka and K. Fujimoto, Fuel Process. Technol., 2004, 85, 1139–1150 CrossRef CAS.
- G. V. Echevsky, E. G. Kodenev, O. V. Kikhtyanin and V. N. Parmon, Appl. Catal., A, 2004, 258, 159–171 CrossRef CAS.
- V. S. Arutyunov and O. V. Krylov, Oxidative Conversion of Methane, Nauka, Moscow, 1998 Search PubMed.
- J. S. Lee and S. T. Oyama, Catal. Rev. Sci. Eng., 1988, 30, 249–280 CAS.
- J. Wang, L. Chou, B. Zhang, H. Song, J. Zhao, J. Yang and S. Li, J. Mol. Catal. A: Chem., 2006, 245, 272–277 CrossRef CAS.
- H. Wang, Y. Cong and W. Yang, Catal. Today, 2003, 82, 157–166 CrossRef CAS.
- K. Tabata, Y. Teng, T. Takemoto, E. Suzuki, M. A. Banares, M. A. Pena and J. L. G. Fierro, Catal. Rev., 2002, 44, 1–58 CrossRef CAS.
- J. H. Lunsford, Catal. Today, 1990, 6, 235–259 CrossRef CAS.
- M. J. Brown and N. D. Parkyns, Catal. Today, 1991, 8, 305–335 CrossRef CAS.
- J. R. Anderson, Appl. Catal., 1989, 47, 177–196 Search PubMed.
- O. V. Krylov, Catal. Today, 1993, 18, 209–302 CrossRef CAS.
- L. Wang, L. Tao, M. Xie, G. Xu, J. Huang and Y. Xu, Catal. Lett., 1993, 21, 35–41 CrossRef CAS.
- D. Wang, J. H. Lunsford and M. P. Rosynek, J. Catal., 1997, 169, 347–358 CrossRef CAS.
- J. R. Rostrup-Nielsen, J. Catal., 1973, 31, 173–199 CrossRef CAS.
- N. Laosiripojana and S. Assabumrungrat, Appl. Catal., A, 2005, 290, 200–211 CrossRef CAS.
- S. C. Tsang, J. B. Clarige and M. L. H. Green, Catal. Today, 1995, 23, 3–15 CrossRef CAS.
- F. Pompeo, N. N. Nichio, M. M. V. N. Souza, D. V. Cesar, O. A. Ferretti and M. Schmal, Appl. Catal., A, 2007, 316, 175–183 CrossRef CAS.
- M. Yang and H. Papp, Catal. Today, 2006, 115, 199–204 CrossRef CAS.
- F. B. Noronha, E. C. Fendley, R. R. Soares, W. E. Alvarez and D. E. Resasco, Chem. Eng. J., 2001, 82, 21–31 CrossRef CAS.
- N. Sahli, C. Petit, A. C. Roger, A. Kiennemann, S. Libs and M. M. Bettahar, Catal. Today, 2006, 113, 187–193 CrossRef CAS.
- A. Olafsen, C. Daniel, Y. Schuurman, L. B. Råberg, U. Olsbye and C. Mirodatos, Catal. Today, 2006, 115, 179–185 CrossRef CAS.
- K. C. Mondal, V. R. Choudhary and U. A. Joshi, Appl. Catal., A, 2007, 316, 47–52 CrossRef CAS.
- D. Dissanayake, M. P. Rosynek, K. C. C. Kharas and J. H. Lunsford, J. Catal., 1991, 132, 117–127 CrossRef CAS.
- L. Majocchi, G. Groppi, C. Cristiani, P. Forzatti, L. Basini and A. Guarinoni, Catal. Lett., 2000, 65, 49–56 CrossRef CAS.
- T. Utaka, S. A. Al-Drees, J. Ueda, Y. Iwasa, T. Takeguchi, R. Kikuchi and K. Eguchi, Appl. Catal., A, 2003, 247, 125–131 CrossRef CAS.
- E. V. Slivinskii, G. A. Kliger, A. E. Kuz'min, A. V. Abramova, A. N. Shuikin, V. I. Kurkin, E. I. Bogolepova and L. A. Vytnova, Kinet. Catal., 1999, 40, 338–344 CAS.
- B. Michalkiewicz, Appl. Catal., A, 2004, 277, 147–153 CrossRef CAS.
- J. W. M. H. Geerts, J. H. B. J. Hoebink and К. Wiele, Catal. Today, 1990, 6, 613–620 CrossRef CAS.
- Q. Zhang, D. He, J. Li, B. Xu, Y. Liang and Q. Zhu, Appl. Catal., A, 2002, 224, 201–207 CrossRef CAS.
- L. D. Nguyen, S. Loridant, H. Launay, A. Pigamo, J. L. Dubois and J. M. M. Millet, J. Catal., 2006, 237, 38–48 CrossRef CAS.
- M. R. Smith and U. S. Ozkan, J. Catal., 1993, 142, 226–236 CrossRef.
- F. Arena, G. Gatti, G. Martra, S. Coluccia, L. Stievano, L. Spadaro, P. Famulari and A. Parmaliana, J. Catal., 2005, 231, 265–380.
- Y. Zeng, F. T. Akin and Y. S. Lin, Appl. Catal., A, 2001, 213, 33–45 CrossRef CAS.
- S. Kus, M. Otremba and M. Taniewski, Fuel, 2003, 82, 1331–1338 CrossRef CAS.
- H. Wang, Y. Cong and W. Yang, Catal. Today, 2005, 104, 160–167 CrossRef CAS.
- J. A. S. P. Carreiro and M. Baerns, J. Catal., 1989, 117, 258–265 CAS.
- J. Da, X. Ding and S. Shen, Appl. Catal., A, 1994, 116, 81–94 CrossRef CAS.
- A. Lucas, J. L. Valverde, L. Rodriguez, P. Sanchez and M. T. Garcia, Appl. Catal., A, 2000, 203, 81–90 CrossRef.
- H. Kim, H. M. Suh and H. Paik, Appl. Catal., A, 1992, 87, 115–127 CrossRef CAS.
- K. Otsuka, M. Hatano and T. Amaya, J. Catal., 1992, 137, 487–496 CrossRef CAS.
- S. Han, D. J. Martenak, R. E. Palermo, J. A. Pearson and D. E. Walsh, J. Catal., 1992, 136, 578–583 CAS.
- L. Chen, L. Lin, Z. S. Xu, X. S. Li and T. Zhang, J. Catal., 1995, 157, 190–200 CrossRef CAS.
- F. Solymosi, A. Cserenyi, A. Szoke, T. Bansagi and A. Oszko, J. Catal., 1997, 165, 150–161 CrossRef CAS.
- W. Liu, Y. Xu, S.-T. Wong, L. Wang, J. Qui and N. Yang, J. Mol. Catal. A: Chem., 1997, 120, 257–265 CrossRef CAS.
- B. M. Weckhuysen, D. Wang, M. P. Rosynek and J. H. Lunsford, J. Catal., 1998, 175, 338–346 CrossRef CAS.
- Y. Xu, S. Liu, X. Guo, L. Wang and M. Xie, Catal. Lett., 1995, 30, 135–149 CrossRef.
- O. V. Bragin, T. V. Vasina, A. V. Preobrazhenskii and Kh. M. Minachev, Izv. Akad. Nauk. SSSR, Ser. Khim., 1989, 3, 750–751 (Russian).
- M. S. Scurrell, Appl. Catal., 1987, 32, 1–22 Search PubMed.
- S. Liu, L. Wang, R. Ohnishi and M. Ichikawa, J. Catal., 1999, 181, 175–188 CrossRef.
- R. Ohnishi, S. Liu, Q. Dong, L. Wang and M. Ichikawa, J. Catal., 1999, 182, 92–103 CrossRef CAS.
- G. Bonura, O. D. Blasi, L. Spadaro, F. Arena and F. Frusteri, Catal. Today, 2006, 116, 298–303 CrossRef CAS.
- L. B. Avdeeva, T. V. Reshetenko, Z. R. Ismagilov and V. A. Likholobov, Appl. Catal., A, 2002, 228, 53–63 CrossRef CAS.
- M. A. Ermakova, D. Yu. Ermakov, A. L. Chuvilin and G. G. Kuvshinov, J. Catal., 2001, 201, 183–197 CrossRef CAS.
- T. V. Reshetenko, L. B. Avdeeva, Z. R. Ismagilov, A. L. Chuvilin and V. F. Fenelonov, Catal. Today, 2005, 102–103, 115–120 CrossRef CAS.
- S. Liu, L. Wang, R. Ohnishi and M. Ichikawa, Kinet. Catal., 2000, 41, 132–144 CrossRef CAS.
- J.-L. Zeng, Z.-T. Xiong, H.-B. Zhang, G.-D. Lin and K. R. Tsai, Catal. Lett., 1998, 53, 119–124 CrossRef CAS.
- H. Jiang, L. Wang, W. Cui and Y. Xu, Catal. Lett., 1999, 57, 95–102 CrossRef CAS.
- Y. Xu, Y. Shu, S. Liu, J. Huang and X. Guo, Catal. Lett., 1995, 35, 233–243 CrossRef CAS.
- D. Ma, W. Zhang, Y. Shu, Y. Xu and X. Bao, Catal. Lett., 2000, 66, 155–160 CrossRef CAS.
- E. V. Matus, I. Z. Ismagilov, O. B. Sukhova, V. I. Zaikovskii, L. T. Tsikoza, Z. R. Ismagilov and J. A. Moulijn, Ind. Eng. Chem. Res., 2007, 46, 4063–4074 CrossRef CAS.
- C.-L. Zhang, S. Li, Y. Yuan, W.-X. Zhang, T.-H. Wu and L.-W. Lin, Catal. Lett., 1998, 56, 207–213 CrossRef CAS.
- H. Liu, W. Shen, X. Bao and Y. Xu, Appl. Catal., A, 2005, 295, 79–88 CrossRef CAS.
- S. Li, C. Zhang, Q. Kan, D. Wang, T. Wu and L. Lin, Appl. Catal., A, 1999, 187, 199–206 CrossRef CAS.
- W. Ding, S. Li, G. D. Meitzner and E. Iglesia, J. Phys. Chem. B, 2001, 105, 506–513 CrossRef CAS.
- L. Chen, J. Lin, H. S. Zeng and K. L. Tan, Catal. Commun., 2001, 2, 201–206 CrossRef CAS.
- G. Dantsin and K. S. Suslick, J. Am. Chem. Soc., 2000, 122, 5214–5212 CrossRef CAS.
- S. Qi and B. Yang, Catal. Today, 2004, 98, 639–645 CrossRef CAS.
- Y. Lu, D. Ma, Z. Xu, Z. Tian, X. Bao and L. Lin, Chem. Commun., 2001, 2048–2049 RSC.
- D. Ma, Y. Shu, X. Bao and Y. Xu, J. Catal., 2000, 189, 314–325 CrossRef CAS.
- H. Zheng, D. Ma, X. Bao, J. Z. Hu, J. H. Kwak, Y. Wang and C. H. F. Peden, J. Am. Chem. Soc., 2008, 130, 3722–3723 CrossRef CAS.
- D. Zhou, D. Ma, X. Liu and X. Bao, J. Mol. Catal. A: Chem., 2001, 168, 225–232 CrossRef CAS.
- C. Bouchy, I. Schmidt, J. R. Anderson, C. J. H. Jacobsen, E. G. Derouane and S. B. Derouane-Abd Hamid, J. Mol. Catal. A: Chem., 2000, 163, 283–296 CrossRef CAS.
- S. B. Derouane-Abd Hamid, J. R. Anderson, I. Schmidt, C. Bouchy, C. J. H. Jacobsen and E. G. Derouane, Catal. Today, 2000, 63, 461–469 CrossRef CAS.
- D. Ma, Y. Shu, M. Cheng, Y. Xu and X. Bao, J. Catal., 2000, 194, 105–114 CrossRef CAS.
- H. Y. Chen, S. Tang, Z. Y. Zhong, J. Lin and K. L. Tan, Surf. Rev. Lett., 2001, 8, 627–632 CAS.
- H. Liu, X. Bao and Y. Xu, J. Catal., 2006, 239, 441–450 CrossRef CAS.
- V. I. Zaikovskii, A. V. Vosmerikov, V. F. Anufrienko, L. L. Korobitsyna, E. G. Kodenev, G. V. Echevskii, N. T. Vasenin, S. P. Zhuravkov, E. V. Matus, Z. R. Ismagilov and V. N. Parmon, Kinet. Catal., 2006, 47, 389–394 CrossRef CAS.
- N. T. Vasenin, V. F. Anufrienko, I. Z. Ismagilov, T. V. Larina, E. A. Paukshtis, E. V. Matus, L. T. Tsikoza, M. A. Kerzhentsev and Z. R. Ismagilov, Top. Catal., 2005, 32, 61–70 CrossRef CAS.
- I. Z. Ismagilov, V. Keller, M. A. Kerzhentsev, E. V. Matus, O. B. Sukhova, L. T. Tsikoza, T. V. Larina, E. A. Paukshtis, Z. R. Ismagilov, S. Libs, P. Bernhardt, P. Ligari and F. Garin, Proceedings of the Eighth European Congress of Catalysis, Turku, Finland, 2007 Search PubMed.
- B. Li, S. Li, N. Li, H. Chen, W. Zhang, X. Bao and B. Lin, Microporous Mesoporous Mater., 2006, 88, 244–253 CrossRef CAS.
- Y.-H. Kim, R. W. Borry III and E. Iglesia, Microporous Mesoporous Mater., 2000, 35–36, 495–509 CrossRef CAS.
- J.-P. Tessonnier, B. Louis, S. Walspurger, J. Sommer, M.-J. Ledoux and C. Pham-Huu, J. Phys. Chem. B, 2006, 110, 10390–10395 CrossRef CAS.
- J.-P. Tessonnier, B. Louis, S. Rigolet, M.-J. Ledoux and C. Pham-Huu, Appl. Catal., A, 2008, 336, 79–88 CrossRef CAS.
- J.-P. Tessonnier, B. Louis, M.-J. Ledoux and C. Pham-Huu, Catal. Commun., 2007, 8, 1787–1792 CrossRef CAS.
- H. S. Lacheen and E. Iglesia, J. Catal., 2005, 230, 173–185 CrossRef CAS.
- W. Ding, G. D. Meitzner and E. Iglesia, J. Catal., 2002, 206, 14–22 CrossRef CAS.
- L. Ovari and F. Solymosi, J. Mol. Catal. A: Chem., 2004, 207, 35–40 CrossRef CAS.
- Z. R. Ismagilov, L. T. Tsikoza, E. V. Matus, G. S. Litvak, I. Z. Ismagilov and O. B. Sukhova, Eurasian Chem.-Technol. J., 2005, 7, 115–121 Search PubMed.
- B. M. Weckhuysen, M. P. Rosynek and J. H. Lunford, Catal. Lett., 1998, 52, 31–36 CrossRef CAS.
- H. Liu, T. Li, B. Tian and Y. Xu, Appl. Catal., A, 2001, 213, 103–112 CrossRef CAS.
- D. Ma, D. Wang, L. Su, Y. Shu, Y. Xu and X. Bao, J. Catal., 2002, 208, 260–269 CrossRef CAS.
- K. Honda, X. Chen and Z.-G. Zhang, Catal. Commun., 2004, 5, 557–561 CrossRef CAS.
- E. V. Matus, I. Z. Ismagilov, O. B. Sukhova, L. T. Tsikoza, M. A. Kerzhentsev and Z. R. Ismagilov, Proceedings of III International conference «Catalysis: theory and practice», Novosibirsk, Russia, 2007 Search PubMed.
- E. V. Matus, PhD thesis, Boreskov Insitute of Catalysis, Novosibirsk, Russia, 2007.
- P. L. Tan, C. T. Au and S. Y. Lai, Appl. Catal., A, 2007, 324, 36–41 CrossRef CAS.
- E. V. Matus, L. T. Tsykoza, Z. R. Ismagilov and V. V. Kuznetsov, Chem. Sustainable Dev., 2003, 11, 167–171 Search PubMed.
- Y. Xu, W. Liu, S.-T. Wong, L. Wang and X. Guo, Catal. Lett., 1996, 40, 207–214 CrossRef CAS.
- H. Liu and Y. Xu, Chin. J. Catal., 2006, 27, 319–323 Search PubMed.
- B. M. Weckhuysen, D. Wang, M. P. Rosynek and J. H. Lunsford, J. Catal., 1998, 175, 347–351 CrossRef CAS.
- Y. Shu, D. Ma, L. Xu, Y. Xu and X. Bao, Catal. Lett., 2000, 70, 67–73 CrossRef CAS.
- V. T. T. Ha, L. T. Tiep, P. Meriaudeau and C. Naccache, J. Mol. Catal. A: Chem., 2002, 181, 283–290 CrossRef CAS.
- Y. Shu, R. Ohnishi and M. Ichikawa, Appl. Catal., A, 2003, 252, 315–329 CrossRef CAS.
- M. Nagai, T. Nishibayashi and S. Omi, Appl. Catal., A, 2003, 253, 101–112 CrossRef CAS.
- S.-T. Wong, Y. Xu, W. Liu, L. Wang and X. Guo, Appl. Catal., A, 1996, 136, 7–17 CrossRef CAS.
- S. Villar-Rodil, A. Martinez-Alonso, J. A. Pajares, J. M. D. Tascon, M. Jasienko-Hajat, E. Broniek, J. Kaczmarczyk, A. Jancowska, A. Albiniak and T. Siemieniewska, Microporous Mesoporous Mater., 2003, 64, 11–19 CrossRef CAS.
- M. Nomura, K. Akagi, S. Murata and H. Matsui, Catal. Today, 1996, 29, 235–240 CrossRef CAS.
- J.-H. Chen, J.-N. Lin, Y.-M. Kang, W.-Y. Yu, C.-N. Kuo and B.-Z. Wan, Appl. Catal., A, 2005, 291, 162–169 CrossRef CAS.
- W. Zhang and P. G. Smirniotis, Appl. Catal., A, 1998, 168, 113–130 CrossRef CAS.
- A. Sarıoğlan, Ö. T. Savaşçı, A. Erdem-Şenatalar, A. Tuel, G. Sapaly and Y. B. Taârit, J. Catal., 2007, 246, 35–39 CrossRef.
- E. V. Matus, I. Z. Ismagilov, O. B. Sukhova, V. I. Zaikovskii, L. T. Tsikoza, Z. R. Ismagilov and J. A. Moulijn, Proceedings of the International Symposium on Catalysis Engineering, Delft, The Netherlands, 2007 Search PubMed.
- S. Tang, Y. Chen, J. Lin and K. L. Tan, Catal. Commun., 2001, 2, 31–35 CrossRef CAS.
- X. Dong, Y. Song and W. Lin, Catal. Commun., 2007, 8, 539–542 CrossRef CAS.
- A. Sarioglan, A. Erdem-Senatalar, Ö. T. Savasci and Y. B. Taârit, J. Catal., 2004, 226, 210–214 CrossRef CAS.
- E. A. Paukshtis, IR Spectroscopy for Henerogeneous Acid–Base Catalysis, Nauka, Novosibirsk, 1992 Search PubMed.
- Y. Sasaki, T. Suzuki, Y. Takamura, A. Saji and H. Saka, J. Catal., 1998, 178, 94–100 CrossRef CAS.
- S. Bernasconi, J. A. Bokhoven, F. Krumeich, G. D. Pirngruber and R. Prins, Microporous Mesoporous Mater., 2003, 66, 21–26 CrossRef.
- M. Ogura, S. Shinomiya, J. Tateno, Y. Nara, N. Nomura, E. Kikuchi and M. Matsukata, Appl. Catal., A, 2001, 219, 33–43 CrossRef CAS.
- L. Su, L. Lin, J. Zhuang, H. Wang, Y. Li, W. Shen, Y. Xu and X. Bao, Catal. Lett., 2003, 91, 155–167 CrossRef CAS.
- T. Hibino, M. Niwa and Y. Murakami, Zeolites, 1993, 13, 518–523 CrossRef CAS.
- O. P. Krivoruchko, V. F. Anufrienko, E. A. Paukshtis, T. V. Larina, E. B. Burgina, S. A. Yashnik, Z. R. Ismagilov and V. N. Parmon, Dokl. Phys. Chem., 2004, 398, 226–230 CrossRef CAS.
- Y. Song, C. Sun, W. Shen and L. Lin, Appl. Catal., A, 2007, 317, 266–274 CrossRef CAS.
- Y. Song, C. Sun, W. Shen and L. Lin, Catal. Lett., 2006, 109, 21–24 CrossRef CAS.
- W. Zhang, D. Ma, X. Han, X. Liu, X. Bao, X. Guo and X. Wang, J. Catal., 1999, 188, 393–402 CrossRef CAS.
- H. S. Lacheen and E. Iglesia, Phys. Chem. Chem. Phys., 2005, 7, 538–547 RSC.
- L. Wang, Y. Xu, W. Wong, W. Cui and X. Guo, Appl. Catal., A, 1997, 152, 173–182 CrossRef CAS.
- K. Skutil and M. Taniewski, Fuel Process. Technol., 2006, 87, 511–521 CrossRef CAS.
- F. Solymosi and A. Szoke, Appl. Catal., A, 1998, 166, 225–235 CrossRef CAS.
- O. A. Anunziata, G. A. Eimer and L. B. Pierella, Appl. Catal., A, 1999, 182, 267–274 CrossRef CAS.
- W. Chu and F. Qiu, Top. Catal., 2003, 22, 131–134 CrossRef CAS.
- M. C. J. Bradford, M. Te, M. Konduru and D. X. Fuentes, Appl. Catal., A, 2004, 266, 55–66 CrossRef CAS.
- Y. Shu, Y. Xu, S.-T. Wong, L. Wang and X. Guo, J. Catal., 1997, 170, 11–19 CrossRef CAS.
- P. L. Tan, K. W. Wong, C. T. Au and S. Y. Lai, Appl. Catal., A, 2003, 253, 305–316 CrossRef CAS.
- M. Siaj, H. Oudghiri-Hassani, C. Maltais and P. H. McBreen, J. Phys. Chem. C, 2007, 111, 1725–1732 CrossRef CAS.
- J. Cserenyi, L. Ovari, T. Bansagi and F. Solymosi, J. Mol. Catal. A: Chem., 2000, 162, 335–352 CrossRef CAS.
- F. Solymosi, L. Bugyi, A. Oszko and I. Horvath, J. Catal., 1999, 185, 160–169 CrossRef CAS.
- P. Mériaudeau, L. V. Tiep, V. T. T. Ha and C. G. NaccacheSzabo, J. Mol. Catal. A: Chem., 1999, 144, 469–471 CrossRef CAS.
- B. Li, S. Li, N. Li, H. Chen, W. Zhang, X. Bao and B. Lin, Microporous Mesoporous Mater., 2006, 88, 244–253 CrossRef CAS.
- H. Liu, W. Shen, X. Bao and Y. Xu, J. Mol. Catal. A: Chem., 2006, 244, 229–236 CrossRef CAS.
- C. Descorme, P. Gelin, C. Lecuyer and A. Primet, Appl. Catal., B, 1997, 13, 185–195 CrossRef CAS.
- T. Behrsing, H. Jaeger and J. V. Sanders, Appl. Catal., 1989, 54, 289–302 Search PubMed.
- H. Ma, R. Kojima, R. Ohnishi and M. Ichikawa, Appl. Catal., A, 2004, 275, 183–187 CrossRef CAS.
- R. W. Borry III, E. C. Lu, Y.-H. Kim and E. Iglesia, Stud. Surf. Sci. Catal., 1998, 119, 403–410.
- K. Honda, T. Yoshida and Z.-H. Zhang, Catal. Commun., 2003, 4, 21–26 CrossRef CAS.
- A. C. C. Rodrigues and J. L. F. Monteiro, Catal. Commun., 2008, 9, 1060–1065 CrossRef CAS.
- L. Chen, L. Lin, Z. Xu, T. Zhang and X. Li, Catal. Lett., 1996, 39, 169–172 CrossRef CAS.
- S. Burns, J. S. J. Hargreaves, P. Pal, K. M. Parida and S. Parija, Catal. Today, 2006, 114, 383–387 CrossRef CAS.
- P. D. Sily, F. B. Noronha and F. B. Passos, J. Nat. Gas Chem., 2006, 15, 82–86 Search PubMed.
- S. Kikuchi, R. Kojima, H. Ma, J. Bai and M. Ichikawa, J. Catal., 2006, 242, 349–356 CrossRef CAS.
- P. Wu, Q. Kan, X. Wang, D. Wang, H. Xing, P. Yang and T. Wu, Appl. Catal., A, 2005, 282, 39–44 CrossRef CAS.
- Z. Liu, M. A. Nutt and E. Iglesia, Catal. Lett., 2002, 81, 271–279 CrossRef CAS.
- S. Yuan, J. Li, Z. Hao, Z. Feng, Q. Xin, P. Ying and C. Li, Catal. Lett., 1999, 63, 73–77 CrossRef CAS.
- S. Liu, R. Ohnishi and M. Ichikawa, J. Catal., 2003, 220, 57–65 CrossRef CAS.
- Y. Shu and M. Ichikawa, Catal. Today, 2001, 71, 55–67 CrossRef CAS.
- Y. Li, L. Su, H. Wang, H. Liu, W. Shen, X. Bao and Y. Xu, Catal. Lett., 2003, 89, 275–279 CrossRef CAS.
- P. Qiu, J. H. Lunsford and M. P. Rosynek, Catal. Lett., 1997, 48, 11–15 CrossRef CAS.
- L. Li, R. W. Borry and E. Iglesia, Chem. Eng. Sci., 2002, 57, 4595–4604 CrossRef CAS.
- O. Rival, B. P. A. Grandjean, C. Guy, A. Sayari and F. Larachi, Ind. Eng. Chem. Res., 2001, 40, 2212–2219 CrossRef CAS.
- M. C. Iliuta, F. Larachi, B. P. A. Grandjean, I. Iliuta and A. Sayari, Ind. Eng. Chem. Res., 2002, 41, 2371–2378 CrossRef CAS.
- M. C. Iliuta, B. P. A. Grandjean and F. Larachi, Ind. Eng. Chem. Res., 2003, 42, 323–330 CrossRef CAS.
- Z. Liu, L. Li and E. Iglesia, Catal. Lett., 2002, 82, 175–180 CrossRef CAS.
- K. Skutil and M. Taniewski, Fuel Process. Technol., 2007, 88, 877–882 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2008 |