A high-power glucose/oxygen biofuel cell operating under quiescent conditions†
Hideki
Sakai
a,
Takaaki
Nakagawa
a,
Yuichi
Tokita
a,
Tsuyonobu
Hatazawa
*a,
Tokuji
Ikeda
b,
Seiya
Tsujimura
c and
Kenji
Kano
*c
aAdvanced Materials Laboratories, Sony Corporation, Okata, Atsugi-shi, Kanagawa 243-0021, Japan. E-mail: Tsuyonobu.Hatazawa@jp.sony.com
bDepartment of Biological Resources, Graduate School of Bioscience and Biotechnology, Fukui Prefectural University, Yoshida-gun, Fukui 910-1142, Japan
cDivision of Applied Life Sciences, Graduate School of Agriculture, Kyoto University, Sakyo-ku, Kyoto 606-8502, Japan. E-mail: kkano@kais.kyoto-u.ac.jp
First published on 29th September 2008
Abstract
Biofuel cells are a next-generation energy device because they use renewable fuels with high energy density and safety. We have developed passive-type biofuel cell units, which generate a power over 100 mW (80 cm3, 39.7 g). Our biofuel cell, in which two-electron oxidation of glucose and four-electron reduction of O2 occurs at pH 7 in mediated bioelectrochemical processes under quiescent conditions, accomplished the maximum power density of 1.45 ± 0.24 mW cm−2 at 0.3 V. This performance was achieved by introducing three technologies: (1) Enzymes and mediator are densely entrapped on carbon-fiber electrodes with the enzymatic activity retained, (2) the concentration of buffer in electrolyte solution was optimized for the immobilized enzymes, and (3) the cathode structure was designed to supply O2 efficiently. The cell units with a multi-stacked structure successfully operate a radio-controlled car (16.5 g), which demonstrates the potential of biofuel cells in practical applications.
Introduction
Biofuel cells can generate electricity under mild conditions (ambient temperature, neutral pH) through the oxidation of renewable energy sources such as sugars or organic acids, coupled with the reduction of O2, by using enzymes or microorganisms as catalysts1–14 and are expected to be next-generation energy devices with higher energy density and more safety, compared to the present devices such as a Li-ion battery and a direct methanol fuel cell. The power density of miniature biofuel cells proposed as an implantable power supply for medical devices1,10 is in the order of sub-mW cm−2 even with fuels- and O2-convective system such as mechanical stirring or a pump, resulting in a few µW per single biofuel cell. The power density without a convective system should significantly decrease by some orders of magnitude, because of the limit of mass transfer rate of fuels and O2, and the permeability barrier in the layer of immobilized bioelectrocatalysts on an electrode.16The required power for portable electronic devices should be at least 100 mW, sufficient to operate a memory-type Walkman (Sony, NW-E407) for instance. Attempting to realize such a power, we adopted a mediated electron-transfer (MET) type biofuel cell system in which electron-transfer mediators shuttle electrons between electrodes and enzymes (Fig. 1). This system has an advantage in enhancing current density as long as the mediator concentration is sufficiently high, compared with direct electron-transfer (DET) type systems or systems using microorganisms.6,12,13 We also must design a cathode structure having an efficient O2-supplying system, because a minimal concentration of O2 in the electrolyte solution is particularly attributed to a lower current density on the cathode.29 Furthermore, we must make not only a biofuel cell with a power density of mW cm−2 per single biofuel cell (Fig. 2) but also a multi-stacked cell unit (Fig. 3) composed of some cells.
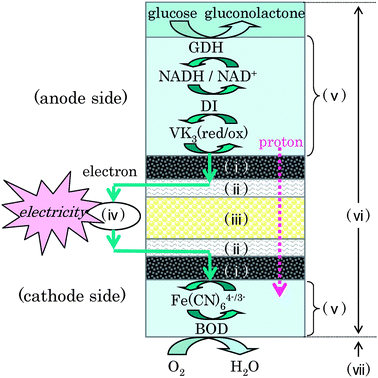 | ||
| Fig. 1 Reaction scheme of the biofuel cell. (i) Carbon-fiber electrode, (ii) Ti-mesh collector, (iii) cellophane separator, (iv) external circuit, (v) enzyme/mediator immobilized layer, (vi) electrolyte solution including the phosphate buffer, (vii) air. | ||
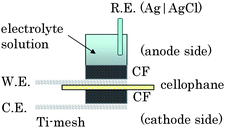 | ||
| Fig. 2 Schematic structure of an electrochemical cell for the single biofuel cell with a solution volume of 3 ml. | ||
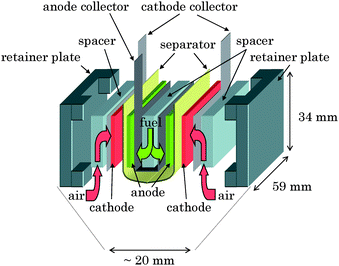 | ||
| Fig. 3 Schematic view of a multi-stacked biofuel cell unit composed of two biofuel cells (Fig. 2) connected in parallel. | ||
Experimental
Materials
The following six reagents were used to make the bioanodes. Glucose dehydrogenase (GDH, EC 1.1.1.47) from Bacillus sp. and diaphorase (DI, EC.1.6.99.-) from Bacillus stearothermophilus were purchased from Amano Enzyme Inc. (Japan) and used without further purification. β-Nicotinamide adenine dinucleotide disodium salt (reduced form) (NADH), poly-L-lysine (PLL, Mw = ca. 93![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 800) and polyacrylic acid sodium salt (PAANa, Mw = ca. 240000) were purchased from Sigma-Aldrich Co. 2-Methyl-1,4-naphthoquinone (vitamin K3, VK3) was purchased from Nacalai Tesque, Inc. (Japan). The following three reagents were used to make the biocathodes. Bilirubin oxidase (BOD, EC 1.3.3.5) from Myrothecium sp. was purchased from Amano Enzyme Inc. (Japan) and used without further purification. K3[Fe(CN)6] was purchased from Wako Pure Chemical Industries, Ltd. (Japan). Poly-L-lysine (PLL, Mw = ca. 8000) was purchased from Peptide Institute Inc., Ltd. (Japan). All other chemicals used in this study were of analytical reagent grade.
800) and polyacrylic acid sodium salt (PAANa, Mw = ca. 240000) were purchased from Sigma-Aldrich Co. 2-Methyl-1,4-naphthoquinone (vitamin K3, VK3) was purchased from Nacalai Tesque, Inc. (Japan). The following three reagents were used to make the biocathodes. Bilirubin oxidase (BOD, EC 1.3.3.5) from Myrothecium sp. was purchased from Amano Enzyme Inc. (Japan) and used without further purification. K3[Fe(CN)6] was purchased from Wako Pure Chemical Industries, Ltd. (Japan). Poly-L-lysine (PLL, Mw = ca. 8000) was purchased from Peptide Institute Inc., Ltd. (Japan). All other chemicals used in this study were of analytical reagent grade.
Glassy carbon (GC) plate electrodes of 3 mm ϕ were purchased from BAS Inc. and used as the working electrodes to characterize the bioelectrochemical oxidation of glucose. The GC-electrode was polished with 0.05 µm alumina powder and washed with distilled water before immobilizing the enzymes and mediator on the electrode.
Carbon-fiber (CF) sheets (BO050, 750 µm thickness) were purchased from Toray Industries, Inc. (Japan), cut into 10 mm squares, and used to fabricate the biocathodes and the bioanodes. The BET specific surface area of the CF-sheet, which was degassed for 2 h (200 °C), was evaluated to be 0.17 m2 g−1 by nitrogen adsorption-desorption isotherm analysis at 77 K using a Belsorp-Max equipment (BEL Japan Inc., Japan). The total specific surface area of the 10 mm squares of the four piled-up CF-sheets for the anode was evaluated to be 20.4 cm2. The ozone treatment was performed on the CF-sheet using a UV. TC. 110 (BIOFORCE Nanosciences. Inc.) before immobilizing the enzymes and mediator, in order to improve adhesion of the immobilized layer on the CF-sheets.
Cellophane membranes were purchased from Futamura Chemical Co., Ltd. (Japan) and were used as separators. Ti-meshes were purchased from Thank-Metal Co. (Japan).
Preparation of GC-bioanodes
On the GC-electrode, 10 µl of a PLL aqueous solution (2% (w/v)), 8 µl of a GDH solution (1 U µl−1 in a 0.1 M PBS (pH 7.0)), 8 µl of a DI solution (10 U µl−1 in the PBS), 2 µl of a NADH solution (0.9 µmol µl−1 in the PBS), 4 µl of a VK3 solution (0.29 M in acetone) and 4 µl of a PAAcNa aqueous solution (0.066% (w/v)) were added in the order mentioned. Drying at 40 °C for 10 min followed after each addition step. The modified electrode was rinsed in distilled water for 5 min before electrochemical measurements. The thickness of the immobilized layer was estimated by using a confocal microscope (FV1000-BX6, Olympus Co., Japan). The immobilized layer has 53 µm thickness in the dry state (Fig. S1†).Preparation of CF-bioanodes
A similar procedure to the GC-bioanodes was adapted to fabricate the CF-bioanodes. In the immobilization process, a 14-times volume of each solution used for the preparation of GC-bioanode was applied to a pile of four CF-sheets, while the drying condition was identical with that for GC-bioanodes. The thickness of the CF-bioanode before electrochemical measurements was 2.11 mm, which was determined with a coolant-proof micrometer (Series No. 293, Mitutoyo Corporation, Japan) under pressure at 26.5 N cm−2.Preparation of CF-biocathodes
On two piled-up CF-sheets, 80 µl of a K3[Fe(CN)6] aqueous solution (0.1 M), 80 µl of a PLL aqueous solution (1% (w/v)) and 80 µl of a BOD solution (50 mg ml−1 in 50 mM PBS (pH 7.0)) were added in the order mentioned. Drying at 30 °C for 10 min followed after each addition step. The thickness of the CF-biocathode before electrochemical measurements was 0.905 mm using the same method as for the CF-bioanode.Electrochemical measurements for bioanode and biocathode
Cyclic voltammetry (CV) and chronoamperometry (CA) were performed by using a 1480 Multi-Stat (Solartron Analytical) in a three-electrode system without forced convection at room temperature (25 ± 2 °C), where a GC-bioanode, a CF-bioanode, and a CF-biocathode were used as a working electrode (W.E.), a Pt wire was used as a counter electrode (C.E.) and an Ag|AgCl|sat. KCl electrode was used as reference electrode (R.E.), to which all potentials are referred in this paper.GC-bioanodes were characterized using a conventional one-compartment cell (Fig. S2a†). The measurement solutions used were from 0.05 to 2.0 M PBS (pH 7.0) containing glucose in the range from 0 to 0.4 M. O2 in the buffer solutions was removed by bubbling with H2O-saturated Ar gas before measurements.
Confocal microscopic imaging of the GC-bioanode suggested that a large amount of VK3 was immobilized uniformly (Fig. S1†). In the absence of glucose, the redox wave can be ascribed to the redox reaction of VK3 entrapped on the GC-bioanode (Fig. S3†). The linear dependence of the peak current on the square root of the scan rate (v) (inset in Fig. S3†) indicates that VK3 can diffuse within the immobilized layer, having 53 µm thickness in a dry state (Fig. S1†). The concentration of VK3 in the immobilized layer was 2.8 mM determined by the faradaic electricity calculated from the oxidation of reduced VK3 (2.8 mC cm−2).
When the CF-bioanode was used as the working electrode (W.E.), the electrode was set at the bottom of an electrochemical cell and a Ti-mesh was used as the collector for electric contact with the CF-electrode (Fig. S2b†).
In order to characterize the efficiency of mass transfer of O2 into the CF-biocathode, two kinds of electrochemical cells were constructed. In both cells, the CF-biocathode was sandwiched between a cellophane membrane and a Ti-mesh. One is the sink-type cell (S-cell, Fig. S5a†). In this cell, the sandwiched CF-biocathode is sunk in the electrolyte solution, through which O2 is supplied to the CF-biocathode. The other is the open-air type (OA-cell, Fig. S5b†). In this cell, the sandwiched CF-biocathode constitutes the bottom of the electrochemical cells and is exposed to air. Therefore, O2 can diffuse from air through the Ti-mesh into the CF-biocathode.
Electrochemical measurements for biofuel cells
We constructed a passive-type biofuel cell (Fig. 2) in which cellophane was sandwiched between CF-bioanode and CF-biocathode. 1.0 M PBS (pH 7.0) containing 0.4 M glucose was used as the fuel solution to characterize the biofuel cells. The current densities of CF-bioanode and CF-biocathode (open circle in Fig. S11†) were evaluated by using the electrochemical cell for the CF-bioanode (Fig. S2b†) and CF-biocathode (OA-cell, Fig. S5b†), respectively (Fig. S11†). Furthermore, we fabricated a passive-type multi-stacked biofuel cell unit (Fig. 3) composed of a pair of the single biofuel cells (Fig. 2) connected in parallel. The CF-sheets, cut into 30 × 55 mm rectangles, were used for the electrodes of the cell units. A pouched cellophane membrane was used to control the solution supply into the CF-biocathode for fast mass transfer of O2 from air and to hold the anode and the fuel solution. We also used spacers with many holes to penetrate the fuel and O2 and retainer plates to fix the biofuel cell.Other measurements for bioanodes and biocathodes
The pH values in the solution within the CF-bioanode and CF-biocathode after CA were evaluated by using a pH meter (TYPE PCE308S-SR, Toko Chemical Laboratory Ltd., Japan) with a flat surface. The amount of water within the CF-biocathode after CA was measured by using the Karl Fischer method and a moisture meter (CA-200, Mitsubishi Chemical Co., Japan).Results and discussion
We describe herein the fabrication of a passive-type biofuel cell unit (Fig. 4) with a multi-stacked structure, being able to generate enough power for electronic devices by introducing the main three technologies: (1) an enzyme/mediator immobilization method on the anode, (2) an electrolyte solution containing the buffer, and (3) an electrode structure of the cathode.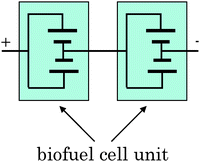 | ||
| Fig. 4 Circuit diagram of biofuel cell units composed of two multi-stacked biofuel cell units (Fig. 3) in series. | ||
We focused on NAD-dependent glucose dehydrogenase (GDH, EC 1.1.1.47) that catalyzes a two-electron-oxidation of glucose by NAD+ (Fig. 1). Since NADH has a large overpotential in direct electrochemical oxidation on electrode,18 we used diaphorase (DI, EC 1.6.99.-) as a bioelectrocatalyst for efficient NADH oxidation. 2-Methyl-1,4-naphthoquinone (VK3) was adopted as an electron-transfer mediator from DI (reduced) to the electrode, because VK3 is a promising mediator for the DI-catalyzed NADH oxidation system in view of its fast kinetics and small thermodynamic loss.11,17 In order to achieve enough catalytic current density, it is necessary to entrap GDH, NADH, DI, and VK3 (four components) on the electrode densely without reducing their biological and electrochemical activities.4 We adopted a polyion complex (PIC) method19,20 to immobilize the four components on a glassy carbon (GC) plate electrode using electrostatic interactions between polyacrylic acid as a polyanion and poly-L-lysine (PLL) as a polycation (GC-bioanode).
In the absence of glucose, the GC-bioanode in cyclic voltammetry (CV) yielded a pair of oxidation and reduction waves of immobilized VK3 with a midpoint potential of −0.17 V vs. Ag|AgCl in a 0.1 M phosphate buffer solution (PBS) at pH 7.0 (wave A in Fig. 5). In the presence of glucose, a catalytic wave of the glucose oxidation appeared (waves B–G in Fig. 5) and the current increased with the increase in the glucose concentration. In a glucose concentration more than 20 mM, the GC-bioanode showed a steady-state wave with the maximum current density, 0.7 mA cm−2 at 0.4 M glucose (inset in Fig. 5), where the reaction rate would be determined by the enzymatic reaction.11 The shape of the waves didn't change during multiple scans of voltage, indicating that the four components were stably and densely entrapped within the immobilized layer with the enzymatic activities retained.
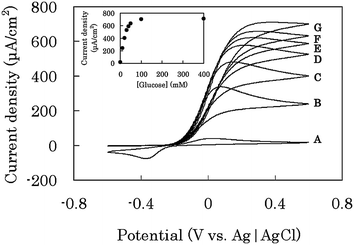 | ||
| Fig. 5 CV on a GC-bioanode in 0.1 M phosphate buffer solution (pH 7.0) containing (A) 0, (B) 10, (C) 20, (D) 30, (E) 40, (F) 50, and (G) 100 mM glucose. The data were taken at scan rate of 10 mV s−1 by using the electrochemical cell (Fig. S2a†). The inset shows the dependence of the current density at 0.6 V vs. Ag|AgCl upon glucose concentration. | ||
In order to increase the current density further, we employed a carbon-fiber (CF) sheet instead of the GC, because the CF has a higher surface area and a higher porosity, allowing the transport of glucose (CF-bioanode) not to be disturbed. The catalytic current density on the CF-bioanode was 4.1 mA cm−2 after 1 min chronoamperometry (CA) at 0.1 V under the same conditions with GC-bioanode (see the value at 0.1 M in Fig. 6). The value was only 5.9 times higher than that on the GC-bioanode, in spite of the fact that the evaluated surface area of the CF-electrode is about 20 times larger than that of the GC-electrode. We presumed that the low catalytic current density on the CF-bioanode was because of insufficient proton diffusion in the CF-bioanode. This may cause the increase of the proton concentration within the immobilized layer by the glucose oxidation reaction (glucose → gluconolactone + 2 H+ + 2 e−), which leads to suppression of the enzymatic activities of GDH and DI.
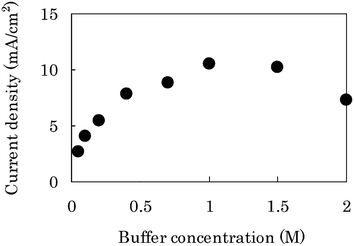 | ||
| Fig. 6 Plot of current density of the CF-bioanode against the buffer concentration by using the electrochemical cell (Fig. S2b†). The current was recorded at 1 min in CA at 0.1 V vs. Ag|AgCl in the presence of 0.4 M glucose under quiescent conditions. | ||
In order to solve this problem, we examined the effects of buffer concentration in the electrolyte solution at 0.4 M glucose. The current density after 1 min CA increased with the increase of the buffer concentration up to 1.0 M (Fig. 6). The current density reached the maximum value, 10.5 mA cm−2 in 1.0 M PBS, which was about 15 times higher than that on the GC-bioanode in 0.1 M PBS, and decreased gradually with the increase of the buffer concentration higher than 1.0 M. In the course of the measurements, pH change within the CF-bioanode was monitored: during 5 min, it dropped from 6.85 to 6.20 in 0.1 M PBS, which is more significant than those in 1.0 M (6.85 → 6.62) or in 2.0 M (6.85 → 6.79) (Fig. S6a†). The pH-drop causes decrease of the current density on the CF-bioanode (Fig. S7a†), due to the fact that reduction of the enzymatic activities depended on the pH. These results clearly show that the higher concentration buffer could suppress the pH-drop within the CF-bioanode.
On the other hand, the current density after 1 min CA decreased in the higher concentration of 1.0–2.0 M, presumably because enzymatic activities of GDH and DI in solution decreased with the increase of buffer concentration from 0.4 to 2.0 M (Fig. S8†). Thus, we revealed that there are two rate-determining steps; the insufficient proton diffusion in the lower buffer concentration (<1.0 M) and the enzymatic inactivation in the higher buffer concentration (>1.0 M). The immobilized layer on the CF-electrode may be stable up to 2.0 M, because the CV dependence on the buffer concentration is small (Fig. S4a, b†) although the data were taken at the GC-bioanode.
In order to make a high-power biofuel cell, we must also develop a cathode corresponding to the CF-bioanode. Bioelectrocatalytic systems involving multi-copper oxidases as promising enzymes for the four-electron reduction of O221 allows diffusion-controlled reduction of O2 at a current density as high as 10 mA cm−2 under convective conditions.15,22,23 In this work, bilirubin oxidase (BOD, EC 1.3.3.5), which is one of the multi-copper oxidases, was used as a suitable bioelectrocatalyst at neutral pH,24 and the Fe(CN)63−/4−redox couple was used as an electron-transfer mediator to enhance the catalytic current density25 (Fig. 1). BOD and K3[Fe(CN)6] were immobilized by using PLL on the CF-electrode with high surface area according to the literature25 (CF-biocathode).
The current density on the CF-biocathode decreased quickly during CA at 0 V in 1.0 M PBS (pH 7.0) and was close to zero after 1 min CA (curve A in Fig. 7) in a sink-type cell (S-cell, Fig. S5a†), where O2 is supplied through the bulk solution phase. The drastic decrease of the current density is due to insufficient O2-supply; the concentration and diffusion coefficient of O2 dissolved in aqueous solutions being 0.229 mM26 and 2.07 ± 0.66 × 10−5 cm2 s−1,27 respectively. In order to develop an efficient O2-supplying system, we designed a biocathode exposed to air; an open-air type cell (OA-cell, Fig. S5b†) with a cathode structure in a polymer electrolyte fuel cell or an air battery, because the concentration (9 mM) in air and the diffusion coefficient (order of 10−1 cm2 s−1) in gas28 are much higher than those in aqueous solution. In this cell, a cellophane membrane as a separator was located between the electrolyte solution of the anode and the cathode, to control the solution supply into the immobilized layer on the cathode. The current density of the OA-cell was 14.1 mA cm−2 after 1 min CA (curve B in Fig. 7), while that of the S-cell was almost zero. These results indicate that the OA-cell is effective in O2-supply. Actually, the amount of water within the CF-biocathode of the OA-cell was only 8.28 mg cm−2 after 1 min CA (Fig. S10†), indicating that the partially wet situation of the CF-biocathode is important for enhancing the mass transfer of O2 from air to the cathode and for effective enzyme reaction in the immobilized layer.29
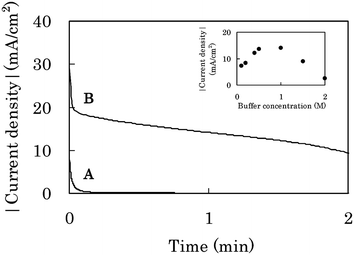 | ||
| Fig. 7 CA response on a CF-biocathode at 0 V vs. Ag|AgCl in 1.0 M phosphate buffer solution (pH 7.0) containing 0.4 M glucose under quiescent conditions. (A) and (B) were taken by using a sink-type cell (S-cell, Fig. S5a†) and an open-air type cell (OA-cell, Fig. S5b†), respectively. The inset shows the dependence of the current density after 1 min CA at 0 V upon the buffer concentration (pH 7.0) by using the OA-cell. | ||
The current density on the CF-biocathode also depended on the buffer concentration (inset in Fig. 7). This profile is very similar to that of the CF-bioanode (Fig. 6), where the optimum was found at 1.0 M PBS as well, although the optimum depends inherently on the kinds of enzymes, electrodes, immobilized layers and electrolytes. It can also be explained by the trade-off relationship between the insufficient proton diffusion and the enzymatic inactivation in the CF-biocathode (Fig. S6b, Fig. S7b and Fig. S9†).
By developing the main three technologies, both the bioanode and the biocathode have achieved a current density over 10 mA cm−2 under quiescent conditions. We constructed a passive-type glucose/O2 biofuel cell (Fig. 2) composed of these electrodes, and evaluated its performance in 0.4 M glucose/1.0 M PBS (pH 7.0). The power densities (triangles in Fig. 8) were estimated from the current densities (circles in Fig. 8) after 1 min CA at each voltage of the single biofuel cell (Fig. 2). The maximum power density was 1.45 ± 0.24 mW cm−2 at 0.3 V, the open-circuit voltage was 0.8 V, and the short-circuit current density was 11 mA cm−2. The anodic and cathodic current densities estimated at each potential of the cell were almost identical with those by using the electrochemical cell for the CF-bioanode (Fig. S2b†) and CF-biocathode (OA-cell, Fig. S5b†), respectively (Fig. S11†) This means that the potential drop due to the cell resistance is negligible even under mA-order current density.
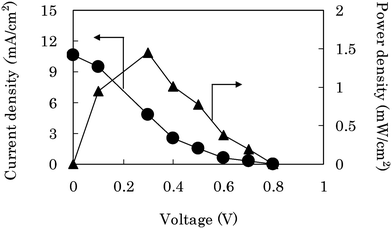 | ||
| Fig. 8 The performance of a passive-type biofuel cell (Fig. 2) composed of the CF-bioanode and the CF-biocathode in 0.4 M glucose/1.0 M phosphate buffer solution (pH 7.0). The current density (●) was obtained after 1 min CA at each voltage of the cell, and the power density (▲) was evaluated from the current density. | ||
Finally, we fabricated a passive-type biofuel cell unit with multi-stacked structure (Fig. 3, over 50 mW, 40 cm3) composed of two biofuel cells (Fig. 2) connected in parallel. These cell units with compact and simple structure can be connected to each other either in series or in parallel. In fact, we could readily construct passive-type biofuel cell units (Fig. 4, over 100 mW, 80 cm3, 39.7 g including 16.1 g of fuel solution) composed of two multi-stacked cell units (Fig. 3) in series and successfully operated a radio-controlled car (16.5 g) (Fig. S12a–c, video 1†) as well as the memory-type Walkman (Website†). The Walkman and the radio-controlled car were continuously operated over at least 2 h. We issued a press release about the development of a high-power biofuel cell in Aug. 2007. These demonstrations would be the first step towards applying biofuel cells to practical applications.
Conclusion
To the best of our knowledge, the power of our cell units (over 100 mW/80 cm3) is the highest in the biofuel cells reported to date, including those with convective systems. The bioanode proposed here is applicable for a wide variety of fuels just by replacing GDH with the corresponding dehydrogenases.7,8,17 For example, ethanol can be oxidized to acetate by alcohol dehydrogenase and acetaldehyde dehydrogenase, concomitantly converting two molecules of NAD+ into NADH.8 In the future, a number of enzymatic reactions can be introduced in the biofuel cell; such as a pentose phosphate cycle or a tricarboxylic acid cycle can completely oxidize sugars or organic acids to CO2 in a biomimetic fashion.30 If one molecule of glucose is completely converted to 6 molecules of CO2, the 24 electrons can be extracted as electric energy corresponding to 3600 A h kg−1.In order to make a practical biofuel cell with a power and durability enough to operate a robot or other devices, many efforts, such as improving immobilization, electrode structure and electrolyte, and introducing multi-enzyme system and stabilization technique for enzymes are underway.
Acknowledgements
We thank Satomi Murasawa of Fullcast Technology Corporation, Hiroki Mita, Ryuhei Matsumoto, Taiki Sugiyama, Atsushi Sato, Hideyuki Kumita, Masaya Kakuta, Yoshio Goto, Takashi Tomita, Yoshio Nishi of Sony Corporation, for their appropriate advice, their profitable discussion and their technical assistance.References
- A. Heller, Miniature biofuel cells, Phys. Chem. Chem. Phys., 2004, 6, 209–216 RSC.
- S. C. Barton, J. Gallaway and P. Atanassov, Enzymatic biofuel cells for implantable and microscale devices, Chem. Rev., 2004, 104, 4867–4886 CrossRef CAS.
- G. T. R. Palmore and G. M. Whitesides, Microbial and enzymatic biofuel cells, ACS Symp. Ser., 1994, 556, 271–290.
- K. Kano and T. Ikeda, Bioelectrocatalysis, Powerful means of connecting electrochemistry to biochemistry and biotechnology, Electrochemistry (Tokyo, Jpn.), 2003, 71(2), 86–99 CAS.
- E. Katz and I. Willner, A biofuel cell with electrochemically switchable and tunable power output, J. Am. Chem. Soc., 2003, 125, 6803–6813 CrossRef CAS.
- Y. Kamitaka, S. Tsujimura, N. Setoyama, T. Kajino and K. Kano, Fructose/dioxygen biofuel cell based on direct electron transfer-type bioelectrocatalysis, Phys. Chem. Chem. Phys., 2007, 9(15), 1793–1801 RSC.
- G. T. R. Palmore, H. Bertschy, S. H. Bergens and G. M. Whitesides, A methanol/dioxygen biofuel cell that uses NAD(+)-dependent dehydrogenases as catalysts: Application of an electro-enzymatic method to regenerate nicotinamide adenine dinucleotide at low overpotentials, J. Electroanal. Chem., 1998, 443, 155–161 CrossRef CAS.
- S. Topcagic and S. D. Minteer, Development of a membraneless ethanol/oxygen biofuel cell, Electrochim. Acta, 2007, 51, 2168–2172.
- S. Tsujimura, M. Fujita, H. Tatsumi, K. Kano and T. Ikeda, Bioelectrocatalysis-based dihydrogen/dioxygen fuel cell operating at physiological pH, Phys. Chem. Chem. Phys., 2001, 3, 1331–1335 RSC.
- N. Mano, F. Mao and A. Heller, Characteristics of a miniature compartment-less glucose–O2 biofuel cell and its operation in a living plant, J. Am. Chem. Soc., 2003, 125, 6588–6594 CrossRef CAS.
- M. Togo, A. Takamura, T. Asai, H. Kaji and M. Nishizawa, An enzyme-based microfluidic biofuel cell using vitamin K3-mediated glucose oxidation, Electrochim. Acta, 2007, 52, 4669–4674 CrossRef CAS.
- S. K. Chaudhuri and D. R. Lovley, Electricity generation by direct oxidation of glucose in mediatorless microbial fuel cells, Nat. Biotechnol., 2003, 21(10), 1229–1232 CrossRef.
- B. E. Logan, P. Aelterman, B. Hamelers, R. Rozendal, U. Schröder, J. Keller, S. Freguia, W. Verstraete and K. Rabaey, Microbial fuel cells: Methodology and technology, Environ. Sci. Technol., 2006, 9(3), 5181–5192 CrossRef.
- R. A. Bullen, T. C. Arnot, J. B. Lakeman and F. C. Walsh, Biofuel cells and their development, Biosens. Bioelectron., 2006, 21, 2015–2045 CAS.
- N. Mano, J. L. Fernandez, Y. Kim, W. Shin, A. J. Bard and A. Heller, Oxygen is electroreduced to water on a “wired” enzyme electrode at a lesser overpotential than on platinum, J. Am. Chem. Soc., 2003, 125, 15290–15291 CrossRef CAS.
- A. Sato, K. Kano and T. Ikeda, Diaphorase/naphthoquinone derivative-modified electrode as an anode for diffusion-controlled oxidation of NADH in electrochemical cells, Chem. Lett., 2003, 32, 880–881 CrossRef CAS.
- K. Takagi, K. Kano and T. Ikeda, Mediated bioelectrocatalysis based on NAD-related enzymes with reversible characteristics, J. Electroanal. Chem., 1998, 445, 209–217.
- L. Gorton and E. Domínguez, Electrocatalytic oxidation of NAD(P)H at mediator-modified electrodes, Rev. Mol. Biotechnol., 2002, 82, 371–392 CrossRef CAS.
- Y. Lvov, K. Ariga, I. Ichinose and T. Kunitake, Assembly of multicomponent protein films by means of electrostatic layer-by-layer adsorption, J. Am. Chem. Soc., 1995, 117, 6117–6123 CrossRef CAS.
- E. Kokufuta, Functional immobilized biocatalysts, Prog. Polym. Sci., 1992, 17, 647–697 CrossRef CAS.
- S. Shleev, J. Tkac, A. Christenson, T. Ruzgas, A. I. Yaropolov, J. W. Whittaker and L. Gorton, Direct electron transfer between copper-containing proteins and electrodes, Biosens. Bioelectron., 2005, 20, 2517–2554 CrossRef CAS.
- N. Mano, H.-H. Kim, Y. Zhang and A. Heller, An oxygen cathode operating in a physiological solution, J. Am. Chem. Soc., 2002, 124, 6480–6486 CrossRef CAS.
- S. Tsujimura, M. Kawaharada, T. Nakagawa, K. Kano and T. Ikeda, Mediated bioelectrocatalytic O2 reduction to water at highly positive electrode potentials near neutral pH, Electrochem. Commun., 2003, 5, 138–141 CrossRef CAS.
- S. Tsujimura, H. Tatsumi, J. Ogawa, S. Shimizu, K. Kano and T. Ikeda, Bioelectrocatalytic reduction of dioxygen to water at neutral pH using bilirubin oxidase as an enzyme and 2,2′-azinobis (3-ethylbenzothiazolin-6-sulfonate) as an electron transfer mediator, J. Electroanal. Chem., 2001, 496, 69–75 CrossRef CAS.
- T. Nakagawa, S. Tsujimura, K. Kano and T. Ikeda, Bilirubin oxidase and [Fe(CN)6]3−/4− modified electrode allowing diffusion-controlled reduction of O2 to water at pH 7.0, Chem. Lett., 2003, 32, 54–55 CrossRef CAS.
- Solubility Data Series, IUPAC, Pergamon Press7 Search PubMed.
- M. Tsushima, K. Tokuda and T. Ohsaka, Use of hydrodynamic chronocoulometry for simultaneous determination of diffusion coefficients and concentrations of dioxygen in various media, Anal. Chem., 1994, 66, 4551–4556 CrossRef CAS.
- C. T. Lynch, Handbook of Materials Science, CRC Press, 1974, p. 1 Search PubMed.
- S. C. Barton, Oxygen transport in composite mediated biocathodes, Electrochim. Acta, 2005, 50, 2145–2153 CrossRef.
- J. Woodward, M. Orr, K. Cordray and E. Greenbaum, Enzymatic production of biohydrogen, Nature, 2000, 405, 1014–1015 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Supplementary Fig. S1–S12, Website and Video. See DOI: 10.1039/b809841g |
| This journal is © The Royal Society of Chemistry 2009 |