A sweet out-of-the-box solution to the hydrogen economy: is the sugar-powered car science fiction?
Y.-H. Percival
Zhang
*abc
aBiological Systems Engineering Department, Virginia Polytechnic Institute and State University, 210-A Seitz Hall, Blacksburg, VA 24061, USA. E-mail: ypzhang@vt.edu; Fax: (+540) 231-3199; Tel: (+540) 231-7414
bInstitute for Critical Technology and Applied Sciences (ICTAS), Virginia Polytechnic Institute and State University, Blacksburg, VA 24061, USA
cDOE BioEnergy Science Center (BESC), Oak Ridge, TN 37831, USA
First published on 23rd January 2009
Abstract
The hydrogen economy presents a compelling future energy picture, especially for the transportation sector. The obstacles, such as low-cost hydrogen production, lack of high-density hydrogen storage approaches, costly infrastructure, and safety concerns are prohibiting its large-scale implementation. To address the above challenges, we propose a new solution – use of starch or cellulose (C6H10O5) from biomass as a hydrogen carrier. This new solution is based on the invention of complete conversion of glucans (starch and cellulose) and water to hydrogen and carbon dioxide as C6H10O5 (aq) + 7H2O (l) → 12H2 (g) + 6CO2 (g). The production of hydrogen from carbohydrates is a nearly carbon-neutral process based on the whole carbon cycle. The use of low-cost renewable carbohydrate as a high hydrogen density carrier (14.8 H2 mass %) may solve problems such as hydrogen production, storage and distribution, as well as address safety concerns. Increasing hydrogen generation rate (power density) and decreasing costs are two major tasks prior to this technology's wide implementation. Analysis based on past scientific knowledge and technical achievements suggests that sugar-powered vehicles could become real in the future with intensive R&D efforts. Here we are calling for international R&D collaborations to pursue the holy grail of the carbohydrate hydrogen economy.
 Y.-H. Percival Zhang Y.-H. Percival Zhang | Yi-Heng Percival Zhang was born in Wuhan, China. He received his BE and MS degrees from East China University of Science and Technology (Shanghai, China), and then obtained his Ph.D. of chemical engineering from Dartmouth College (USA) under supervision by biofuels pioneer Prof. Lee R. Lynd in 2002. He is an assistant professor at Virginia Polytechnic Institute and State University. His current research is focused on efficient cellulose solvent-based lignocelluloses fractionation followed by saccharification by engineered cellulases as well as sugar-to-biofuels (e.g., hydrogen, electricity) generation through an in vitro synthetic biology approach – synthetic enzymatic pathway engineering. |
Broader contextSynthetic biology is an emerging interdisciplinary area that combines science and engineering in order to design and build novel biological functions and systems. Cell-free synthetic biology through in vitroassembly of a number of enzymes and coenzymes has been designed to implement unnatural reactions as C6H10O5 (aq, starch or cellodextrins) + 7 H2O (l) → 12 H2 (g) + 6 CO2 (g). This new sugar-to-hydrogen technology promises to address several obstacles to the hydrogen economy – cheap hydrogen production , high hydrogen storage density (14.8 H2 mass%), and costly hydrogen infrastructure, and to eliminate safety concerns about mass utilization of hydrogen. Also, these reactions can produce more chemical energy output as hydrogen than chemical energy input stored in polysaccharides for the first time. |
1. Introduction
Human society has smoothly passed through two transportation energy revolutions from animal forces relying on living plant biomass to external combustion engines (steam engines) driven by solid coal to internal combustion engines (ICE) driven by liquid gasoline and diesel.1 Transportation ability often reflects civilization level. Without it, cities could not exist; families would have to live close to the land, gathering and growing their own food; materials, medicines, medical cares, manufacturing, and electricity generation all depend on transportation.2Currently, liquid fuels (gasoline, diesel, and jet fuel), along with internal combustion engines, are widely used to propel vehicles, trains, ships, and jet planes because of several advantages: (1) relatively low fuel prices (until more recently); (2) very high energy storage densities (MJ per kg of fuel and MJ per litre of fuel); (3) high power density (kW per kg of engine); (4) easy storage, distribution, transportation, and refilling for liquid fuels; (5) relatively low costs for ICE ($ per kW of output); and (6) safety for mass utilization. But the concerns pertaining to soaring prices of crude oil, depleting fossil fuels, net CO2 emissions, climate change, national energy security, global and local food security, (rural) economic development, energy utilization efficiency, and wealth transfer are motivating the development of sustainable alternative transportation fuels. Second generation biofuels such as cellulosic ethanol, butanol, algae biodiesel, hydrocarbons, and synthetic diesel, can be integrated well with current infrastructures for liquid fuels and ICE systems but the ICE systems have relatively low energy efficiencies, since the efficiencies of heat engines are restricted by the second law of thermodynamics.
In the long term, improving energy utilization efficiency through hydrogen-fuel cell/electricity systems will be vital for sustainable transportation. Distinct from first generation fuels (e.g., solid coal) and second generation fuels (e.g., liquid gasoline, diesel), third generation transportation fuels include hydrogen and electricity, both of which work as energy carriers that can be converted to kinetic work efficiently without the restriction of the second law of thermodynamics. Both hydrogen and electricity will be generated from various primary energy sources, such as biomass, solar energy, wind energy, geothermal energy, tidal energy and so on. The hydrogen-fuel cell-electricity system will play a predominant role because of (1) very high energy conversion efficiency through fuel cells, (2) minimal pollutants generated, (3) much higher energy storage densities than rechargeable batteries alone, and (4) diverse hydrogen-producing means from primary energy resources. But large-scale implementation of the hydrogen economy must break four technological hurdles – low cost hydrogen production from any primary energy resources, high hydrogen density storage means (>9 mass%), affordable fuel distribution infrastructure, and affordable fuel cells throughout the whole life cycle.3–5 In addition, hydrogen is a flammable, odorless, colorless gas. Any significant hydrogen explosion accident could prevent the public from accepting hydrogen as a transportation fuel.
Transportation fuels are and will be mainly produced by four primary resources – crude oil, natural gas, lignocellulosic biomass, and starchy crops like corn. Based on energy contents ($ per gigajoule, GJ), delivered lignocellulosic biomass at $60 per dry ton ($3.60 per GJ) is least costly among all primary energy sources – compared to natural gas ($7.58 per GJ, $8 per mbtu), crude oil ($15 per GJ, $80 per barrel), and corn kernels ($13 per GJ, $4.5 per bushel) (Fig. 1). Although coal energy content ($1.54 per GJ, $50 per ton) is lower than that of lignocellulosic biomass, the conversion of coal to liquid transportation fuels is economically and environmentally prohibitive, except in special times or areas (e.g., Germany during World War II and South Africa).
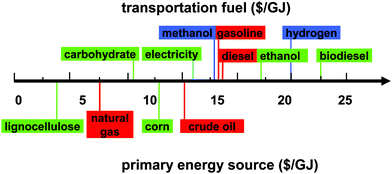 | ||
| Fig. 1 Cost comparison of primary energy resources and potential transportation fuels. The prices of energy resources and fuels vary in a relatively large range and the values only represent likely recent prices. | ||
Comparison of different current and potential transportation fuels is very complicated, involving a number of factors – fuel costs, resource availability, infrastructure availability, costs and lifetime of the engine/motor, environmental impacts, etc. Direct price comparison of transportation fuels, such as gasoline, diesel, ethanol, biodiesel, methanol, hydrogen, or even electricity, is relatively straightforward for end-users because their prices include costs associated with feedstock, processing, capital depreciation, distribution, profits, and taxes. Fig. 1 shows the energy contents of potential fuels in an increasing order from carbohydrate ($10.6 per GJ, $0.18 per kg), electricity ($16.7 per GJ, $0.04 per kWh), methanol ($17.8 per GJ, $0.35 per kg), gasoline ($17.6 per GJ, $2.5 per gallon), diesel ($19.5 per GJ, $2.7 per gallon), ethanol ($22.1 per GJ, $2 per gallon), hydrogen ($25.0 per GJ, $3 per kg), to biodiesel ($27.4 per GJ, $3.5 per gallon). Carbohydrates isolated from corn kernels, sugarcane or cellulosic materials will be the least costly. Further conversion of carbohydrates to other fuels, such as ethanol, hydrogen or even synthetic bio-oil, will lead to higher prices. Electricity, a universal energy currency, can be generated from a number of resources – coal, natural gas, wind energy, nuclear energy, hydroelectric energy, and so on. Regardless of its generation means, electricity prices vary in a relatively narrow range after numerous conversions and grid distribution.
In this perspective, we briefly review the challenges for the hydrogen economy, propose an out-of-the-box solution that could systematically solve several of these challenges, discuss its technical feasibility, and emphasize future research directions.
2. The hydrogen economy
The hydrogen economy will be a linked network of processes that produces hydrogen, stores hydrogen chemically or physically, and converts the stored hydrogen to electrical energy at the point of use.3,6–8Hydrogen is advantageous over electricity stored in rechargeable batteries for the transportation sector because stored hydrogen has a ∼20-fold to >100-fold higher energy storage density than electricity stored in rechargeable batteries in terms of GJ per kg.9,10 Battery-only electric vehicles have a much shorter driving distance per recharging than hydrogen fuel cell systems.Hydrogen can be produced from water and other hydrogen-containing compounds such as CH4 and carbohydrates by a number of chemical, biological, electrical, photochemical, and photobiological approaches. Most hydrogen is currently produced from natural gas by a combination of steam reforming and water shift reactions, accompanied with a net release of CO2 to the atmosphere. Because of soaring prices of fossil fuels, hydrogen production costs were more than $2.70 per kg of hydrogen in 2005;11 a situation that has clearly deteriorated since then.
Gaseous hydrogen storage is still the largest challenge. It can be stored (1) in high-pressure gas cylinders; (2) as liquid hydrogen in cryogenic tanks (at 21 K); and (3) in solid forms (e.g., adsorption on large specific surface area solid materials or hydrides (e.g., LiAlH4, NaAlH4, NaBH4) or by the reaction of light metals and water.4,12 As for approaches 1 and 2, considerable energy is lost in hydrogen compression (∼10–15%) or hydrogen liquefaction (∼33%). Both also have low hydrogen storage densities, for example, liquid hydrogen has a hydrogen density of only 70.8 kg/m3 (i.e., less than 7 mass H2%). Generally speaking, large scale high-pressure and cryogenic hydrogen storage systems are impractical for vehicular application due to safety concerns and volumetric constraints.13 Solid hydrogen storage technologies require high-gravimetric hydrogen density, adequate hydrogen-dissociation energetics, or stable and low-cost hydrogen carriers.12,13 Therefore, the US Department of Energy (DOE) set hydrogen storage goals at 6 mass% and 9 mass% for 2010 and 2015, respectively.5 Recently, possible hydrogen-storage materials meeting FreedomCar requirements (e.g., density, refilling rate, refilling time, and reuse cycle time), such as metal-organic frameworks with potential densities of 10 H2 mass%, have been proposed in the DOE 2008 annual merit review and peer evaluation.14
Hydrogen, a small and energetic molecule, can diffuse through container materials or react with materials. For example, hydrogen cannot be simply delivered by today's natural gas pipeline systems because of steel embrittlement, accompanied with increased maintenance costs, leakage rates, and material replacement costs. Hydrogen pipelines will be much more expensive than electric transmission lines and natural gas pipelines. Proponents of the hydrogen economy propose local hydrogen stations based on local sources.15,16 Unfortunately developing these stations in high demand urban areas will have many challenges, including NIMBY (not in my backyard) backlash. Finally, a huge investment in the infrastructure is required for storing and distributing hydrogen, costing at least one trillion of dollars in the USA alone.15,17
In order to solve the challenges associated with gaseous hydrogen storage and costly infrastructure, high-energy-density liquid fuels – such as methanol, ethanol, liquefied petroleum gas, gasoline, or biodiesel – have been proposed as hydrogen carriers. The vehicles must have an onboard chemical converter to reform them to hydrogen. Methanol, a liquid fuel, can be converted to hydrogen very easily viareforming or can be converted to electricity through direct methanol fuel cells (DMFC). The challenges faced by the DMFC technology include methanol crossover, high catalyst costs, low power density, poor efficiency, and short operation life.18–20Ethanol and hydrocarbons can be converted to hydrogen and CO2 plus some COviacatalytic steam reforming, partial oxidation, or auto-thermal reforming.21,22 Since a small amount of CO as a side-product of chemical catalysis can poison the catalysts of proton exchange membrane (PEM) fuel cells,22 extra purification steps are required to remove CO before entering PEM fuel cells. Carbon monoxide clean-up can be done in several ways – water gas shifting, selective CO removal, methanation, and Pd alloy membranes.21 These reformers have been shown to be highly complicated, difficult to operate, bulky, and expensive.23 In order to avoid CO poisoning, ammonia, an easily-liquefied carbon-free gas, has been proposed as a hydrogen carrier. Production of NH3 from pure hydrogen and the consequential conversion of ammonia to hydrogen is not energy- and cost-efficient. Obviously, any current high-temperature on-board reformers result in system complexity and some energy loss during such conversions, implying their infeasibility for vehicular applications.
Low-temperature PEM fuel cells are used primarily for transportation applications due to their fast startup time, low sensitivity to orientation, high energy conversion efficiency, low-operating temperature (below 100 °C), and favorable power-to-weight ratio (lightweight and compact). In contrast, high-temperature fuel cells are not amenable to transportation propulsion.24 Therefore, nearly all the major automakers have fuel cell projects based on PEM technology with an electric motor, but the challenge of gaseous hydrogen storage results in a shorter driving range compared to gasoline-powered vehicles (300–400 miles driving distance per tank). In contrast, the Nobel Prize winner George A. Olah advocates the methanol economy,25 but DMFC may be good only for low power applications, such as portable electronics.19
Fig. 2 presents different possible scenarios of the future hydrogen economy for the transportation sector, including hydrogen production, storage, distribution, fuel cell, and end users – vehicles. Hydrogen can be produced from diverse primary energy sources, such as solar energy, biomass, fossil fuels, tidal energy, geothermal energy, and so on. Once gaseous hydrogen is produced, its storage and distribution will lead to big challenges, as described above. The use of hydrogen carriers, such as methanol, hydrocarbons, or even ammonia, may be more promising in principle than direct use of gaseous hydrogen. But the system complexity of CO removal from the thermal reformers is a show stopper for the carbon-containing hydrogen carriers through on-board reforming. Therefore, the demonstration vehicle systems based on liquid hydrocarbon on-board reforming systems followed by PEM fuel cells have been abandoned. We propose a new solution – the on-board carbohydrate-to-hydrogen-PEM fuel cell system (Fig. 2).
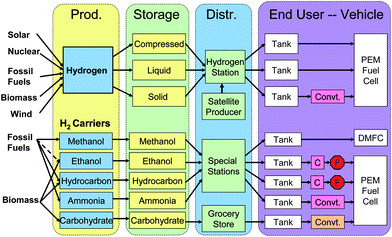 | ||
| Fig. 2 Comparison of the different scenarios of the hydrogen economy. | ||
3. An out-of-the-box solution for the hydrogen economy
We propose solid polymeric carbohydrates (C6H10O5, 14.8 H2 mass%) as a hydrogen carrier, based on the new in vitro synthetic biology approach.26 The use of low-cost, sustainable biomass as the primary energy source for producing transportation fuels (e.g., cellulosic ethanol and hydrogen) provides benefits to the environment, economy, and national security.1,6,27–38 Biomass is an enriched chemical energy source that can solve the scale-up and storage challenges associated with low-power density solar radiation.39 A number of biomass-to-hydrogen production approaches have been investigated previously:1. gasification,40,41 (fast or flash) pyrolysis,42–46 or aqueous phase reforming;47–51
2. anaerobic hydrogen fermentation8,31,52–57 and/or a bioelectrochemically assisted microbial fuel cell reactor that can convert acetate to hydrogen with the help of a little electricity;58,59
3. cell-free synthetic enzymatic pathways;26,60 and
4. combinatorial biological and chemical catalysis: polysaccharide hydrolysis31,38,61,62 and glucose–ethanol fermentation or consolidated bioprocessing31,63–65 followed by chemical catalysis – ethanol partial oxidation reforming.22,66
The carbohydrate-to-hydrogen conversion by the cell-free synthetic enzymatic pathways (a new in vitro synthetic biology approach) features (i) mild reaction conditions, (ii) no CO side-product, (iii) complete conversion, and (iv) potentially high reaction rates. This allows us to propose an out-of-the-box solution for the hydrogen economy: the use of sugars as a hydrogen carrier. Potential applications include stationary power providers, local hydrogen stations, refillable sugar batteries, sugar-powered automobiles, air-independent-propulsion submarines, or even electric aircraft.
3.1. Novel hydrogen production
The novel synthetic enzymatic pathways have been designed to produce 12 moles of hydrogen per mole of glucose equivalent of glucans (starch and cellulose) and water.26,60 The idea is to utilize the energy stored in polysaccharides to split water and stepwise release all energy of carbohydrates in the form of hydrogen under mild reaction conditions (≪ 100 °C and ∼1 atm) as below| C6H10O5 (aq) + 7H2O (l) → 12H2 (g) + 6CO2 (g) | (1) |
These synthetic catabolic pathways that do not exist in nature are comprised of 13 enzymes in one pot (Fig. 3). Most of the reactions in the pathway catalyzed by the enzymes are reversible. The removal of gaseous products from the aqueous phase favors the unidirectional overall reaction. In addition, enzymatic biochemical reactions are well-known for their 100% selectivity at modest reaction conditions. Thermodynamic analysis suggests that the overall reaction is a spontaneous process (i.e., ΔG° = −49.8 kJ mol−1) and is an endothermic reaction (i.e., ΔH° = 598 kJ mol−1).60 The negative value of Gibbs free energy at 25 °C suggests a nearly complete conversion. The Gibbs energy of this reaction decreased greatly with an increase in temperature, suggesting higher conversion at elevated temperatures. This reaction is driven by entropy gain rather than enthalpy loss. Another well-known entropy-driven reaction is acetate fermentation from glucose [C6H12O6 (aq) + 2 H2O (l) → 2 CH4O2 (aq) + 2 CO2 (g) + 4 H2 (g)]. In addition, the removal of both gaseous products from the aqueous reactants at mild reaction condition (< 100 °C and ∼1 atm) drives the reaction forward to completion.60 This entropy-driven chemical reaction can generate more output chemical energy in the form of hydrogen than input chemical energy in polysaccharides by adsorbing ambient-temperature thermal energy.26,60
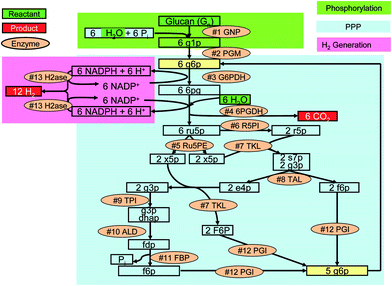 | ||
| Fig. 3 The synthetic metabolic pathway for complete conversion of glucan and water to hydrogen and carbon dioxide. PPP, pentose phosphate pathway taken from ref. 26. The enzymes are: #1 GNP, glucan phosphorylase; #2 PGM, phosphoglucomutase; #3 G6PDH, G-6-P dehydrogenase; #4 6PGDH, 6-phosphogluconate dehydrogenase; #5 R5PI, phosphoribose isomerase; #6 Ru5PE, ribulose 5-phosphate epimerase; #7 TKL, transketolase; #8 TAL, transaldolase; #9 TPI, triose phosphate isomerase; #10 ALD, aldolase; #11 FBP, fructose-1,6-bisphosphatase; #12 PGI, phosphoglucose isomerase; and #13 H2ase, hydrogenase. The metabolites and chemicals are: g1p, glucose-1-phosphate; g6p, glucose-6-phosphate; 6pg, 6-phosphogluconate; ru5p, ribulose-5-phosphate; x5p, xylulose-5-phosphate; r5p, ribose-5-phosphate; s7p, sedoheptulose-7-phosphate; g3p, glyceraldehyde-3-phosphate; e4p, erythrose-4-phosphate; dhap, dihydroxacetone phosphate; fdp, fructose-1,6-diphosphate; f6p, fructose-6-phosphate; and Pi, inorganic phosphate. | ||
The first proof-of-principle experiment has been conducted to validate whether or not hydrogen can be produced from starch and water.26,67 A number of enzymes, isolated from animal, plant, bacterial, and yeast sources, plus an archaeal hyperthermophilic hydrogenase, are put together in one pot. Although each of them has a different optimal pH, temperature, and cofactor, the compromised conditions used are 0.1 M HEPES buffer (pH 7.5) containing 5 mM thiamine pyrophosphate, 4 mM phosphate, 2 mM NADP+, 10 mM MgCl2, and 0.5 mM MnCl2 at 30 °C. Under these conditions, each enzyme remains active but is believed to be far from its optimal activity. The first reaction mediated by substrate phosphorylases plays an important role in producing glucose-1-phosphate by shortening polysaccahrides without the use of ATP.26,63,68,69 Utilization of substrate phosphorylase enzymes is far superior to any kinase reaction involving hexokinase and ATP because of (1) no costly ATP regeneration system; (2) no accumulation of phosphate, an inhibitor of several enzymes (e.g., fructose biphosphatase);70 (3) no Mg2+ precipitation,70 since Mg2+ is a key co-factor of several enzymes; and (4) a more homostatic pH.
Fig. 4 shows that hydrogen is produced as expected, a little later than CO2 evolution, consistent with the designed mechanism in Fig. 3. A lag phase of hydrogen production is attributed to the initial addition of NADP+ as a cofactor. When NADPH is used, there is no lag phase for hydrogen generation. This proof-of-principle experiment has been conducted by using off-the-shelf enzymes without any optimization so that the reaction rates are very low, far from the demands of practical applications.26 Recently, the hydrogen production rate has been increased by 8.2 fold starting from cellulosic materials as compared to the previous results by (i) increasing the rate-limiting hydrogenase concentration, (ii) increasing the substrate concentration, and (iii) elevating the reaction temperature slightly from 30 to 32 °C (Table 1). Under the current system parameters, the measured production rate of H2 is higher than those for photobiological systems and comparable to those reported for dark fermentations.54 Further enhancement in hydrogen production rates will be discussed in Section 4.
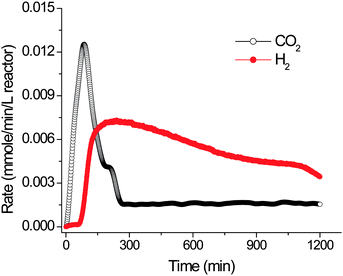
3.2. Special features
The complete conversion of sugars and water to hydrogen and carbon dioxide mediated by these synthetic enzymatic pathways26,60 provides a number of special features suitable for mobile PEM fuel cells.1. Highest energy efficiency. Enzymatic hydrogen production is the only one that can produce nearly 12 moles of hydrogen per mole of glucose equivalent. In addition to extracting all the chemical energy stored in the substrate sugars, the overall reaction is endothermic, i.e., some of low-temperature thermal energy is absorbed and converted to chemical energy in the form of hydrogen (22% combustion energy gain during this bioreforming).
2. High hydrogen storage density. Polysaccharides have a chemical formula C6H10O5 with a reaction of C6H10O5 (aq) + 7 H2O (l) → 12 H2 (g) + 6 CO2 (g). As a result, hydrogen storage density in polysaccharides is 24/162 = 14.8 H2 mass%, where water can be recycled from PEM fuel cells.
3. Mild reaction conditions (≪100 °C and ∼1 atm), which do not require bulky, costly pressure reactors. The reactor temperatures are at the same range of those of PEM fuel cells, good for coupling these endothermic and exothermic reactions.
4. Nearly no costs for product separation (gas/liquid). This reaction only produces two gaseous products – CO2 and hydrogen. Under mild reaction conditions, the reactants (sugar and water) plus the enzymes and the cofactor remain in the aqueous phase. Separation of the gaseous products and aqueous reaction is easy and nearly cost-free. Critically, the removal of the reaction products also drives the reactions forward and avoids product inhibition.
5. Clean products for PEM fuel cells along with easy power system configuration.
6. Simple and safe distribution and storage of solid sugars. Therefore, investment for upgrading infrastructure and distribution of solid sugars would be minimal.
3.3. Future applications
These enzymatic sugar-to-hydrogen reactions have several potential applications from local hydrogen generation stations to low-cost electricity generators, to high energy-density batteries, as well as sugar-powered vehicles, all of which require faster hydrogen production rates as this nascent technology is improved and optimized.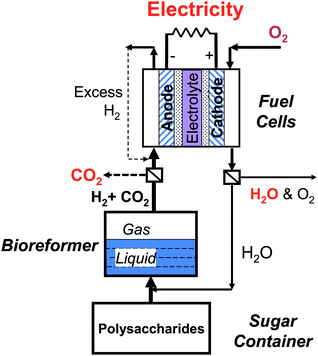 | ||
| Fig. 5 Conceptual sugar-to-electricity system. | ||
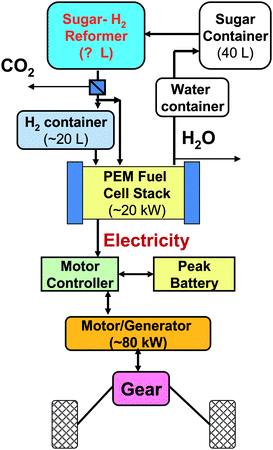 | ||
| Fig. 6 Conceptual hybrid power train system including on-board sugar-to-hydrogen converter, PEM fuel cell and rechargeable battery. | ||
Small-size hydrogen fuel cell vehicles need hydrogen production rates of ∼1–2 kg per hour. Producing sufficient hydrogen at rapid rates from a small bioreformer is the number one technological challenge. Producing one kg of hydrogen per hour will need a reaction volume of 130 m3 based on the current reaction rate of 3.92 mmole of hydrogen per hour per litre, implying that this application is technically impractical. But we expect to be able to increase the hydrogen production rate by several orders of magnitude through a combination of known technologies (see Section 4). To our knowledge, the highest biohydrogen production rate is 21.8 moles of hydrogen per litre per hour,71 ∼5600 times higher than the enzymatic hydrogen process.60 If we can increase the rate by 2000-fold, the volume of the bioreformer will be as small as 65 litres, which will be small enough to replace small-size internal combustion engines. If 4–10 kg of hydrogen is needed for driving more than 300 miles before refilling, that means that 27–67.6 kg of sugar will be stored in the vehicles, occupying a volume of 38.6–96.6 litres or 10.2–25.5 gallons.
The proposed power train systems would have a very high energy conversion efficiency (overall, 55%; carbohydrate–hydrogen, 122%; hydrogen–PEM fuel cell, 50%; electricity–motor, 90%), ∼3.0 times higher than that of ethanol-internal combustion engines (overall, 18.2%; carbohydrate–ethanol, 90%; internal combustion engine, 25%; transmission, 85%). This proposed energy efficiency would be the highest among all power-train systems, including internal combustion engines, standard hydrogen-fuel cell systems, gas turbines, etc. If the USA's biomass resource through bioethanol-internal combustion engines replaced 30% of transportation fuels in 2030,72 the same amount of biomass through hydrogen–PEM fuel cell systems would achieve at least 90% transportation fuel independence through this new technology without reliance on any other energy sources.
Seemingly competitive technology –aqueous phase reforming47–51 – is not suitable for on-board PEM fuel cell systems because it has poor hydrogen selectivity, low yield, and dirty products (e.g., CO), and requires high temperature (∼250 °C) and pressure (e.g., ∼50 atm) reactors. Therefore, on-board reformation though aqueous phase reforming appears not to be technically feasible. Similar situations occur with on-board hydrocarbon-to-hydrogen reforming.
As compared to current developing enzymatic biofuel cells,10,73–75 the hypothetic sugar–hydrogen–PEM fuel cell systems have several advantages: (1) much higher energy extracting efficiency (122% vs. 15–20%), (2) several orders of magnitude higher energy output density (W m−2), and (3) minimal product inhibition. Many attempts at enzymatic biofuel cells have been made recently to extract all the chemical energy in biofuels and convert it to electricity.76,77 All sugar batteries must overcome the challenges, such as enzyme costs and enzyme stability.74,78 For example, one kg of industrial immobilized thermostable glucose isomerase can convert at least 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500000 kg of glucose to fructose or have a turn-over number of ∼800000000.79,80 A startup company, Akermin, has claimed enzyme stabilization technology for three years by encasing enzymes in a proprietary, protective polymer structure. Another example is the more than one year shelf-life of glucose dehydrogenase at room temperature used in the blood sugar strips for diabetes patients. Obviously, the collaborations for enzyme and cofactor stabilization among groups of enzymatic biofuel cells, biosensors, and the hypothesized sugar-to-hydrogen–PEM fuel cell systems are expected.
500000 kg of glucose to fructose or have a turn-over number of ∼800000000.79,80 A startup company, Akermin, has claimed enzyme stabilization technology for three years by encasing enzymes in a proprietary, protective polymer structure. Another example is the more than one year shelf-life of glucose dehydrogenase at room temperature used in the blood sugar strips for diabetes patients. Obviously, the collaborations for enzyme and cofactor stabilization among groups of enzymatic biofuel cells, biosensors, and the hypothesized sugar-to-hydrogen–PEM fuel cell systems are expected.
4. Research and design perspectives
Before the above-mentioned applications are implemented, two major technical challenges must be overcome – (i) slow hydrogen production rate and (ii) high production cost.Increasing the hydrogen production rate is the number one technological challenge because it is a requirement for all future applications. The proof-of-principle biohydrogen production experiment by the synthetic enzymatic pathway conducted by using off-the-shelf enzymes with some optimization has a reaction rate of 3.92 mmole of hydrogen per litre of reaction volume per hour.60 The first significant improvement in reaction rates can be made by optimizing the enzyme ratio. We have estimated a potential improvement of at least ∼20-fold by optimization of the rate-limiting step enzyme ratios and increasing substrate levels.81 Second, another significant improvement will be implemented by increasing the reaction temperature. Currently, we are lacking thermostable enzymes. The rule of thumb suggests that most enzymatic reaction rates usually are doubled with every 10 °C increase (i.e., Q10 effect). Therefore, an increase in the reaction temperature from 30 °C to 80 °C could result in another ∼32 fold improvement. For example, the hyperthermophilic P. furious hydrogenase exhibits < 1% of its potential activity in the proof-of-principle experiment (32 °C). Increasing reaction temperature will decrease hydrogenase use and increase the overall reaction rate. Third, a 100-fold increase in enzyme concentration could lead another potential rate enhancement by 20–100 fold. Fourth, when the overall enzyme concentration is high, macromolecular crowding effects could lead to metabolite or substrate channeling between the cascade enzymes, which could contribute to another reaction rate enhancement by ∼2–100 fold, which is observed sometimes, especially in macromolecular crowding conditions.82–84 Finally, there will be a great enhancement potential in the turnover numbers for each enzyme by several orders of magnitude, because their catalytic efficiencies are still much lower than those catalytically perfect enzymes with a kcat/Km of 108–109 per M per s.85,86 Based on the above analysis, an increase in hydrogen production rate by at least 3 orders of magnitude from the current levels will be reachable after intensive R&D efforts within several years. Comparatively, the power density of microbial fuel cells has been improved by greater than 104–106 fold during the past 10 years.58,87
To our knowledge, the highest biological hydrogen production rate is 11.8 moles of hydrogen per litre of reactor volume per hour, which is mainly implemented by using two combinatorial technologies: high enzyme loading and high substrate concentration.71 This rate is high enough for some high power applications, for example, hydrogen–PEM fuel cell devices. Given the same reaction rate, a high-power vehicle equipped with a 100 kW (134 hp) PEM fuel cell stack would need an on-board bioreformer having a reasonable volume of 210 litres, plus a peak battery with a several hundred kW electric motor.
High hydrogen production costs are associated with three key components – costly and unstable enzymes, the coenzyme (NADP+), and the substrates. Decreasing the enzyme costs can be carried out by two main approaches – decreasing enzyme production costs and extending enzyme lifetime. The former can be mainly implemented by (a) producing recombinant enzymes rather than purifying them from natural biological entities,88 (b) over-expressing the target enzymes,88,89 (c) implementing high-cell density fermentation by using low-cost nutrients,38 and (d) decreasing enzyme purification costs.90–92 The latter (i.e., stabilization of the enzymes) can be implemented by (a) immobilization on traditional materials or nano-materials,93–99 (b) thermostable enzyme replacement,100–103 (c) enzyme formulation,104–106 and (d) enzyme engineering by directed evolution or rational design.107–113 Recently, a hyperthermostable 6-phosphogluconate dehydrogenase (#4 enzyme) from the hyperthermophilic bacterium Thermotoga maritima has been over-expressed in E. coli with a yield of more than 200 mg per litre of culture. It is found to retain >90% of its activity at 80 °C for more than 48 hours (manuscript under preparation). Stabilization of one enzyme or multiple enzymes on solid supporters is a widely-known technology.74,114 With the rapid development in nano-materials with much larger surface areas (i.e., more enzymes can be immobilized), examples of ultra-stable immobilized enzymes have been reported to be active for one to several months.93,96,98,115,116 It is expected that these combinatorial technologies will stabilize the enzymes for several months or even longer at ambient temperatures and at the evaluated temperature for more than 200 hours in the near future.
NAD(P) is not a stable under certain circumstances117,118 but its stability can be enhanced greatly by chemical modifications or immobilization.114,119 Asymmetric synthesis mediated by enzymes involving NAD(P)H regeneration is becoming more and more competitive in the pharmaceutical industry.120,121 The reported total turnover number for cofactors is as high as 600000122 or even more than 1 million,123 suggesting the economical feasibility of recycling NAD(P)H for hydrogen production.
Starch is food and animal feed, and its supply is becoming more restricted again. Cellulosic material is the most abundant renewable resource; the yearly energy production is ∼6 fold of all human energy consumption.124,125 If a small fraction of yearly cellulosic material (e.g., 10%) is used for transportation, transportation fuel independence will be reached. Cellulose has the same chemical formula as starch except with different glucosidic bond linkage between anhydroglucose units.61 Producing hydrogen from cellulosic materials must overcome two obstacles: (1) increasing cellulose reactivity for fast reaction rates and (2) discovery or development of cellulose phoshorylases that can phosphorolyze β-1,4-glucosidic bonds. With regard to obstacle 1, the crystalline cellulose structure can be completely broken by using cellulose solvents, such as concentrated phosphoric acid,126–128 ionic liquids129–131 and so on. The presence of lignin and hemicellulose in natural lignocellulose negatively influences cellulose hydrolysis rates and digestibility. The best lignocellulose pretreatment will be implemented if (1) hemicellulose and lignin can be removed efficiently, (2) crystalline cellulose can be converted to amorphous cellulose, (3) low processing costs are attained, and (4) low capital investment is used. Recently, a new cellulose solvent- and organic solvent-based lignocellulose fractionation (COSLIF) technology that combines a cellulose solvent (concentrated phosphoric acid) and a organic solvent featuring modest reaction conditions (e.g., 50 °C and atmospheric pressure) aims at lignin, hemicellulose, and cellulose at the same time.128,132 Very high cellulose digestibilities (∼97%) by cellulase are obtained for a number of feedstocks (e.g., corn stover, switchgrass and hybrid poplar) within a short hydrolysis time of 24 hours. With regard to obstacle 2, cellobiose and cellodextrin phoshosphorylases63,69,133–135 may be the starting enzymes for creating unnatural or undiscovered cellulose phosphorylase.
Costs of hydrogen production from carbohydates (e.g., $0.18 per kg of carbohydrate) would be as low as ∼$2 per kg of H2, assuming that feedstock costs account for 60% of overall costs and enzymes and co-enzymes account for 40%. In general, approximately 40–75% of commodity prices, such as gasoline from crude oil, hydrogen from natural gas, and ethanol from corn kernels, come from feedstock costs.136 If the enzymes were produced as cheaply as industrial enzymes (e.g., cellulase, amylase, protease), and their stability was enhanced to the same level of immobilized glucose isomerase,80 the estimated hydrogen production costs through this enzymatic biocatalysis would be far lower than $2 per kg of hydrogen.
An alternative way to decrease the costs of enzymes and coenzyme for hydrogen production is to put the synthetic enzymatic pathway containing 13 over-expressed enzymes into a minimal bacterium137 or create a new super hydrogen production microorganism by total synthesis of the whole genomic sequence.138 But the implementation of the hypothesized new bacteria will take a long time, the hydrogen yields must be a little lower than 12 H2 per glucose unit due to cellular biomass synthesis, and the hydrogen production rates could be very slow for some applications due to membrane blockage.67,139
To implement sugar-powered cars, a number of process engineering challenges have to be overcome, for example, warm-up of the bioreformer, shut-down of the bioreformer, temperature controlling for the coupled bioreformer and fuel cells, mixing and gas release control for the bioreformer, and re-generation of used enzymes and co-enzymes in the bioreformer, to name a few. But such technical challenges can be solved if the great potential is widely realized.
5. Conclusion
Hydrogen production by synthetic enzymatic pathways is the most efficient way to convert the energy stored in renewable sugars to hydrogen energy.26,60 In addition, an endothermic reaction at ambient temperature means absorption of some low-temperature heat energy and conversion to a high-quality chemical energy carrier – hydrogen.26,60,67Hydrogen production from the enriched chemical energy source – sugars produced from photosynthesis – suggests minimal challenges for scale-up and storage of feedstocks. We now need to address both increasing the hydrogen production rates and decreasing the hydrogen production costs. With technological improvements, this carbohydrate-to-hydrogen technology will address the challenges associated with hydrogen production, storage, safety, distribution, and infrastructure in the hydrogen economy.26We envision that we will drive sugar-powered vehicles having a driving distance of >300 miles per refill. Solid sugar (∼27–68 kg of sugars or 4–10 kg of hydrogen per refilling) will be added at local outlets such as grocery stores and the like. The on-board bioreformer with a volume of several tens or hundreds of litres containing a number of stabilized enzyme cocktails will convert sugar syrup to hydrogen, which will be converted to electricity quickly with very high energy efficiency and high power density via the PEM fuel cell. As a result, driving tomorrow with renewable sugars will no longer be viewed as science fiction! These systems will be the most energy efficient and greenest power-train with high power density and high energy storage density. This ambitious project of the sugar-powered vehicle will become a hen that will lay golden eggs for various sub-directions – enzyme engineering, enzyme immobilization, synthetic biology, fuel cells, battery, powertrain system integration, and so on.
Acknowledgements
This work was supported by the Air Force Office of Scientific Research (FA9550-08-1-0145), the Department of Defense Contract (W911SR-08-P-0021), USDA, DOE BioEnergy Science Center (BESC), and DuPont Young Professor Award. The author also appreciated the useful discussion and critical reading from Dr Mielenz Jonathan at the Oak Ridge National Laboratory and Joe Rollin at Virginia Tech.References
- Y.-H. P. Zhang, J. Ind. Microbiol. Biotechnol., 2008, 35, 367–375 CrossRef CAS.
- Y.-H. Zhang, Public Service Rev.: Sci. Technol., 2008, 1, 126 Search PubMed.
- B. C. H. Steele and A. Heinzel, Nature, 2001, 414, 345–352 CrossRef CAS.
- L. Schlapbach and A. Zuttel, Nature, 2001, 414, 353–358 CrossRef.
- DOE, Basic Research Needs for the Hydrogen Economy, 2004, http://www.sc.doe.gov/bes/hydrogen.pdf Search PubMed.
- M. I. Hoffert, K. Caldeira, G. Benford, D. R. Criswell, C. Green, H. Herzog, A. K. Jain, H. S. Kheshgi, K. S. Lackner, J. S. Lewis, H. D. Lightfoot, W. Manheimer, J. C. Mankins, M. E. Mauel, L. J. Perkins, M. E. Schlesinger, T. A. Volk and T. M. Wigley, Science, 2002, 298, 981–987 CrossRef CAS.
- A. Sartbaeva, V. L. Kuznetsov, S. A. Wells and P. P. Edwards, Energy Environ. Sci., 2008, 1, 79–85 RSC.
- W. Lubitz, E. J. Reijerse and J. Messinger, Energy Environ. Sci., 2008, 1, 15–31 RSC.
- J. M. Tarascon and M. Armand, Nature, 2001, 414, 359–367 CrossRef CAS.
- M. Armand and J. M. Tarascon, Nature, 2008, 451, 652–657 CrossRef CAS.
- DOE, DOE announces new hydrogen cost goal, July 14, 2005 Search PubMed.
- H. Cheng, L. Chen, A. C. Cooper, X. Sha and G. P. Pez, Energy Environ. Sci., 2008, 1, 338–354 RSC.
- S. i. Orimo, Y. Nakamori, J. R. Eliseo, A. Zuttel and C. M. Jensen, Chem. Rev., 2007, 107, 4111–4132 CrossRef CAS.
- DOE, DOE Hydrogen Program, 2008, http://www.hydrogen.energy.gov/annual_review08_proceedings.html Search PubMed.
- P. M. Grant, C. Starr and T. J. Overbye, Sci. Am., 2006, 295, 76–83 CrossRef CAS.
- R. Agrawal, M. Offutt and M. P. Ramage, AIChE J., 2005, 51, 1582–1589 CrossRef CAS.
- R. B. Moore and V. Raman, Int. J. Hydrogen Energy, 1998, 23, 617–620 CrossRef CAS.
- A. Heinzel and V. M. Barragan, J. Power Sources, 1999, 84, 70–74 CrossRef CAS.
- T. F. Fuller, 2005.
- D. R. Palo, R. A. Dagle and J. D. Holladay, Chem. Rev., 2007, 107, 3992–4021 CrossRef CAS.
- L. Carreette, K. A. Friendrich and U. Stimming, Fuel Cells, 2001, 1, 5–39 CrossRef CAS.
- G. A. Deluga, J. R. Salge, L. D. Schmidt and X. E. Verykios, Science, 2004, 303, 993–997 CrossRef CAS.
- T. Schultz, S. S. Zhou and K. Sundmacher, Chem. Eng. Technol., 2001, 24, 1223–1233 CrossRef CAS.
- R. Borup, J. Meyers, B. Pivovar, Y. S. Kim, R. Mukundan, N. Garland, D. Myers, M. Wilson, F. Garzon, D. Wood, P. Zelenay, K. More, K. Stroh, T. Zawodzinski, J. Boncella, J. E. McGrath, M. Inaba, K. Miyatake, M. Hori, K. Ota, Z. Ogumi, S. Miyata, A. Nishikata, Z. Siroma, Y. Uchimoto, K. Yasuda, K. i. Kimijima and N. Iwashita, Chem. Rev., 2007, 107, 3904–3951 CrossRef CAS.
- G. A. Olah, Angew. Chem., Int. Ed., 2005, 44, 2636–2639 CrossRef CAS.
- Y.-H. P. Zhang, B. R. Evans, J. R. Mielenz, R. C. Hopkins and M. W. W. Adams, PLoS One, 2007, 2, e456 CrossRef.
- L. T. Angenent, K. Karim, M. H. Al-Dahhan, B. A. Wrenn and R. Domiguez-Espinosa, Trends Biotechnol., 2004, 22, 477–485 CrossRef CAS.
- A. L. Demain, M. Newcomb and J. H. D. Wu, Microbiol. Mol. Biol. Rev., 2005, 69, 124–154 CrossRef.
- L. R. Lynd, Annu. Rev. Energy Env., 1996, 21, 403–465 Search PubMed.
- L. R. Lynd, J. H. Cushman, R. J. Nichols and C. E. Wyman, Science, 1991, 251, 1318–1323 CrossRef CAS.
- L. R. Lynd, P. J. Weimer, W. H. van Zyl and I. S. Pretorius, Microbiol. Mol. Biol. Rev., 2002, 66, 506–577 CrossRef CAS.
- B. Kamm and M. Kamm, Appl. Microbiol. Biotechnol., 2004, 64, 137–145 CrossRef CAS.
- K. Caldeira, A. K. Jain and M. I. Hoffert, Science, 2003, 299, 2052–2054 CrossRef CAS.
- M. I. Hoffert, K. Caldeira, A. K. Jain, E. F. Haites, L. D. D. Harvey, S. D. Potter, M. E. Schlesinger, S. H. Schneider, R. G. Watts, T. M. Wigley and D. J. Wuebbles, Nature, 1998, 395, 881–884 CrossRef CAS.
- T. E. Wirth, C. B. Gray and J. D. Podesta, Foreign Affairs, 2003, 82, 125–144 Search PubMed.
- A. J. Ragauskas, C. K. Williams, B. H. Davison, G. Britovsek, J. Cairney, C. A. Eckert, W. J. Frederick, Jr., J. P. Hallett, D. J. Leak, C. L. Liotta, J. R. Mielenz, R. Murphy, R. Templer and T. Tschaplinski, Science, 2006, 311, 484–489 CrossRef.
- G. W. Bush, State of the Union Address by the President, White House, Washington D. C., 2006 Search PubMed.
- Y.-H. P. Zhang, M. Himmel and J. R. Mielenz, Biotechnol. Adv., 2006, 24, 452–481 CrossRef.
- Y.-H. Zhang, Public Service Rev.: Sci. Technol., 2008, 1, 150 Search PubMed.
- M. J. Antal, S. G. Allen, D. Schulman, X. Xu and R. J. Divilio, Ind. Eng. Chem. Res., 2000, 39, 4040–4053 CrossRef.
- Y. Matsumura, T. Minowa, B. Potic, S. R. A. Kersten, W. Prins, W. P. M. van Swaaij, B. van de Beld, D. C. Elliott, G. G. Neuenschwander, A. Kruse and M. Jerry Antal Jr, Biomass Bioenergy, 2005, 29, 269–292 CrossRef CAS.
- R. M. Navarro, M. A. Pena and J. L. G. Fierro, Chem. Rev., 2007, 107, 3952–3991 CrossRef CAS.
- S. Czernik and A. V. Bridgwater, Energy Fuels, 2004, 18, 590–598 CrossRef.
- T. Carlson, T. Vispute and G. W. Huber, ChemSusChem, 2008, 1, 397–400 CrossRef CAS.
- P. Dauenhauer, B. Dreyer, N. Degenstein and L. Schmidt, Angew. Chem., Int. Ed., 2007, 46, 5864–5867 CrossRef CAS.
- J. R. Salge, B. J. Dreyer, P. J. Dauenhauer and L. D. Schmidt, Science, 2006, 314, 801–804 CrossRef CAS.
- G. W. Huber, J. W. Shabaker and J. A. Dumesic, Science, 2003, 300, 2075–2077 CrossRef CAS.
- W. B. Kim, T. Voitl, G. J. Rodriguez-Rivera and J. A. Dumesic, Science, 2004, 305, 1280–1283 CrossRef CAS.
- J. Chheda, G. Huber and J. Dumesic, Angew. Chem., Int. Ed., 2007, 46, 7164–7183 CrossRef CAS.
- R. D. Cortright, R. R. Davda and J. A. Dumesic, Nature, 2002, 418, 964–967 CrossRef CAS.
- R. R. Davda and J. A. Dumesic, Angew. Chem., Int. Ed., 2003, 42, 4068–4071 CrossRef CAS.
- M. W. W. Adams and E. I. Stiefel, Science, 1998, 282, 1842–1843 CrossRef CAS.
- P. C. Hallenbeck and J. R. Benemann, Int. J. Hydrogen Energy, 2002, 27, 1185–1193 CrossRef CAS.
- D. Das and T. N. Veziroglu, Int. J. Hydrogen Energy, 2001, 26, 13–28 CrossRef CAS.
- M. Momirlan and T. N. Veziroglu, Ren. Sus. Energy Rev., 2002, 6, 141–179 Search PubMed.
- T. Maeda, V. Sanchez-Torres and T. K. Wood, Microbial Biotechnol., 2008, 1, 30–39 Search PubMed.
- R. Kleerebezem and M. C. M. van Loosdrecht, Curr. Opin. Biotechnol., 2007, 18, 207–212 CrossRef CAS.
- B. E. Logan and J. M. Regan, Environ. Sci. Technol., 2006, 40, 5172–5180 CAS.
- S. Cheng and B. E. Logan, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 18871–18873 CrossRef CAS.
- X. Ye, Y. Wang, R. C. Hopkins, M. W. W. Adams, B. R. Evans, J. R. Mielenz and Y.-H. P. Zhang, ChemSusChem, 2009 DOI:10.1002/cssc.2009200017.
- Y.-H. P. Zhang and L. R. Lynd, Biotechnol. Bioeng., 2004, 88, 797–824 CrossRef CAS.
- Y.-H. P. Zhang and L. R. Lynd, Biotechnol. Bioeng., 2006, 94, 888–898 CrossRef CAS.
- Y.-H. P. Zhang and L. R. Lynd, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 7321–7325 CrossRef CAS.
- Y. Lu, Y.-H. P. Zhang and L. R. Lynd, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 16165–16169 CrossRef CAS.
- Y.-H. P. Zhang and L. R. Lynd, J. Bacteriol., 2005, 187, 99–106 CrossRef CAS.
- A. Haryanto, S. Fernando, N. Murali and S. Adhikari, Energy Fuels, 2005, 19, 2098–2106 CrossRef CAS.
- Y.-H. Zhang, Y. Wang and X. Ye, in Biotechnology: Research, Technology and Applications ed. F. Columbus, Nova Science Publishers, Hauppauge, NY, USA, 2008, pp. 1–16 Search PubMed.
- Y.-H. P. Zhang and L. R. Lynd, in Biomass Recalcitrance: Deconstructing the Plant Cell Wall for Bioenergy, ed. M. E. Himmel, Blackwell Publishing, 2008, pp. 480–494 Search PubMed.
- Y.-H. P. Zhang and L. R. Lynd, Appl. Environ. Microbiol., 2004, 70, 1563–1569 CrossRef CAS.
- J. L. Donahue, J. L. Bownas, W. G. Niehaus and T. J. Larson, J. Bacteriol., 2000, 182, 5624–5627 CrossRef CAS.
- A. Yoshida, T. Nishimura, H. Kawaguchi, M. Inui and H. Yukawa, Appl. Environ. Microbiol., 2005, 71, 6762–6768 CrossRef CAS.
- M. E. Himmel, S.-Y. Ding, D. K. Johnson, W. S. Adney, M. R. Nimlos, J. W. Brady and T. D. Foust, Science, 2007, 315, 804–807 CrossRef CAS.
- S. C. Barton, J. Gallaway and P. Atanassov, Chem. Rev., 2004, 104, 4867–4886 CrossRef CAS.
- J. Kim, H. Jia and P. Wang, Biotechnol. Adv., 2006, 24, 296–308 CrossRef CAS.
- S. D. Minteer, B. Y. Liaw and M. J. Cooney, Curr. Opin. Biotechnol., 2007, 18, 228–234 CrossRef CAS.
- R. Arechederra and S. D. Minteer, Electrochim. Acta, 2008, 53, 6698–6703 CrossRef CAS.
- D. Sokic-Lazic and S. D. Minteer, Biosens. Bioelectron., 2008, 24, 939–944 CrossRef CAS.
- M. Moehlenbrock and S. Minteer, Chem. Soc. Rev., 2008, 37, 1188–1196 RSC.
- W. Tischer and V. Kasche, Trends Biotechnol., 1999, 17, 326–335 CrossRef CAS.
- S. Bhosale, M. Rao and V. Deshpande, Microbiol. Rev., 1996, 60, 280–300 CAS.
- X. Ye and Y. H. P. Zhang, AIChE's 2007 Annual Meeting Salt Lake City, Utah, 2007.
- D. K. Srivastava and S. A. Bernhard, Science, 1986, 234, 1081–1086 CAS.
- D. K. Srivastava and S. A. Bernhard, Annu. Rev. Biophys. Biophys. Chem., 1987, 16, 175–204 CrossRef CAS.
- T. Maier, M. Leibundgut and N. Ban, Science, 2008, 321, 1315–1322 CrossRef CAS.
- R. Wolfenden and M. J. Snider, Acc. Chem. Res., 2001, 34, 938–945 CrossRef CAS.
- S. G. Burton, D. A. Cowan and J. M. Woodley, Nat. Biotechnol., 2002, 20, 37–45 CrossRef CAS.
- A. Rinaldi, B. Mecheri, V. Garavaglia, S. Licoccia, N. P. Di and E. Traversa, Energy Environ. Sci., 2008, 1, 417–429 RSC.
- J. L. Hartley, Curr. Opin. Biotechnol., 2006, 17, 359–366 CrossRef CAS.
- G. Barnard, G. Henderson, S. Srinivasan and T. Gerngross, Protein Expression Purif., 2004, 38, 264–271 CrossRef CAS.
- J. Hong, Y. Wang, X. Ye and Y.-H. P. Zhang, J. Chromatogr., A, 2008, 1194, 150–154 CrossRef CAS.
- J. Hong, X. Ye, Y. Wang and Y.-H. P. Zhang, Anal. Chim. Acta, 2008, 621, 193–199 CrossRef CAS.
- D. W. Wood, W. Wu, G. Belfort, V. Derbyshire and M. Belfort, Nat. Biotechnol., 1999, 17, 889–992 CrossRef CAS.
- U. T. Bornscheuer, Angew. Chem., Int. Ed., 2003, 42, 3336–3337 CrossRef CAS.
- T. B. Uwe, ChemInform, 2003, 34.
- C. P. Govardhan, Curr. Opin. Biotechnol., 1999, 10, 331–335 CrossRef CAS.
- J. Lee, E. Hwang, B. Kim, S.-M. Lee, B.-I. Sang, Y. Choi, J. Kim and M. Gu, Appl. Microbiol. Biotechnol., 2007, 75, 1301–1307 CrossRef CAS.
- B. C. Kim, S. Nair, J. Kim, J. H. Kwak, J. W. Grate, S. H. Kim and M. B. Gu, Nanotechnology, 2005, 16, S382–S388 CrossRef.
- K. M. Polizzi, A. S. Bommarius, J. M. Broering and J. F. Chaparro-Riggers, Curr. Opin. Chem. Biol., 2007, 11, 220–225 CrossRef CAS.
- B. El-Zahab, H. Jia and P. Wang, Biotechnol. Bioeng., 2004, 87, 178–183 CrossRef CAS.
- C. Vieille, J. Gregory and G. J. Zeikus, Microbiol. Mol. Biol. Rev., 2001, 65, 1–43 CrossRef CAS.
- L. D. Unsworth, J. van der Oost and S. Koutsopoulos, FEBS J., 2007, 274, 4044–4056 CrossRef CAS.
- M. J. Danson, D. W. Hough, R. J. M. Russell, G. L. Taylor and L. Pearl, Protein Eng., 1996, 9, 629–630 CrossRef CAS.
- A. Razvi and J. M. Scholtz, Protein Sci., 2006, 15, 1569–1578 CrossRef CAS.
- Y. Wang and Y.-H. Zhang, 2009, submitted.
- T. Hancock and J. Hsu, Biotechnol. Bioeng., 1996, 51, 410–421 CrossRef CAS.
- D. Fedunová and M. Antalík, Biotechnol. Bioeng., 2006, 93, 485–493 CrossRef CAS.
- A. Korkegian, M. E. Black, D. Baker and B. L. Stoddard, Science, 2005, 308, 857–860 CrossRef CAS.
- U. T. Bornscheuer and M. Pohl, Curr. Opin. Chem. Biol., 2001, 5, 137–143 CrossRef CAS.
- H. Chautard, E. Blas-Galindo, T. Menguy, L. Grand'Moursel, F. Cava, J. Berenguer and M. Delcourt, Nat. Methods, 2007, 4, 919–921 CrossRef CAS.
- C. Roodveldt, A. Aharoni and D. S. Tawfik, Curr. Opin. Struct. Biol., 2005, 15, 50–56 CrossRef CAS.
- O. J. Miller, K. Bernath, J. J. Agresti, G. Amitai, B. T. Kelly, E. Mastrobattista, V. Taly, S. Magdassi, D. S. Tawfik and A. D. Griffiths, Nat. Methods, 2006, 3, 561–570 CrossRef CAS.
- K. D. Wittrup, Curr. Opin. Biotechnol., 2001, 12, 395–399 CrossRef CAS.
- D. N. Rubingh, Curr. Opin. Biotechnol., 1997, 8, 417–422 CrossRef CAS.
- W. Liu and P. Wang, Biotechnol. Adv., 2007, 25, 369–384 CrossRef CAS.
- J. Lee, J. Kim, J. Kim, H. Jia, M. Kim, J. Kwak, S. Jin, A. Dohnalkova, H. Park, H. Chang, P. Wang, J. Grate and H.T., Small, 2005, 1, 744–753 CrossRef CAS.
- N. St. Clair, Y.-F. Wang and A. L. Margolin, Angew. Chem., Int. Ed., 2000, 39, 380–383 CrossRef.
- C.-H. Wong and G. M. Whitesides, J. Am. Chem. Soc., 1981, 103, 4890–4899 CrossRef CAS.
- S. L. Johnson and P. T. Tuazon, Biochemistry, 1977, 16, 1175–1183 CrossRef CAS.
- A. Ohno, S. Oda, Y. Ishikawa and N. Yamazaki, J. Org. Chem., 2000, 65, 6381–6387 CrossRef CAS.
- J. C. Moore, D. J. Pollard, B. Kosjek and P. N. Devine, Acc. Chem. Res., 2007, 40, 1412–1419 CrossRef CAS.
- S. M. A. D. Wildeman, T. Sonke, H. E. Schoemaker and O. May, Acc. Chem. Res., 2007, 40, 1260–1266 CrossRef.
- U. Kragl, D. Vasicracki and C. Wandrey, Indian J. Chem. B: Org. Chem. Med. Chem., 1993, 1, 103–117 Search PubMed.
- R. Kazandjian and A. Klibanov, J. Am. Chem. Soc., 1985, 107, 5448–5450 CrossRef.
- W. A. Hermann, Energy, 2006, 31, 1685–1702 CrossRef CAS.
- W. H. Wise, Energy resources: Occurrence, production, conversion and use, Springer-Verlag, New York, 2000 Search PubMed.
- Y.-H. P. Zhang, J.-B. Cui, L. R. Lynd and L. R. Kuang, Biomacromolecules, 2006, 7, 644–648 CrossRef CAS.
- J. Hong, X. Ye and Y. H. P. Zhang, Langmuir, 2007, 23, 12535–12540 CrossRef CAS.
- G. M. Moxley, Z. Zhu and Y.-H. P. Zhang, J. Agric. Food Chem., 2008, 56, 7885–7890 CrossRef CAS.
- A. P. Dadi, S. Varanasi and C. A. Schall, Biotechnol. Bioeng., 2006, 95, 904–910 CrossRef CAS.
- R. P. Swatloski, S. K. Spear, J. D. Holbrey and R. D. Rogers, J. Am. Chem. Soc., 2002, 124, 4974–4975 CrossRef CAS.
- S. Zhu, Y. Wu, Q. Chen, Z. Yu, C. Wang, S. Jin, Y. Ding and G. Wu, Green Chem., 2006, 8, 325–327 RSC.
- Y.-H. P. Zhang, S.-Y. Ding, J. R. Mielenz, R. Elander, M. Laser, M. Himmel, J. D. McMillan and L. R. Lynd, Biotechnol. Bioeng., 2007, 97, 214–223 CrossRef CAS.
- J. K. Alexander, J. Biol. Chem., 1968, 2899–904, 2899.
- J. K. Alexander, Methods Enzymol., 1972, 130, 944–948 Search PubMed.
- Y.-H. P. Zhang and L. R. Lynd, Appl. Microbiol. Biotechnol., 2006, 70, 123–129 CrossRef CAS.
- L. R. Lynd, C. E. Wyman and T. U. Gerngross, Biotechnol. Prog., 1999, 15, 777–793 CrossRef CAS.
- J. I. Glass, N. Assad-Garcia, N. Alperovich, S. Yooseph, M. R. Lewis, M. Maruf, C. A. Hutchison, III, H. O. Smith and J. C. Venter, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 425–430 CrossRef CAS.
- D. G. Gibson, G. A. Benders, C. Andrews-Pfannkoch, E. A. Denisova, H. Baden-Tillson, J. Zaveri, T. B. Stockwell, A. Brownley, D. W. Thomas, M. A. Algire, C. Merryman, L. Young, V. N. Noskov, J. I. Glass, J. C. Venter, C. A. Hutchison, III and H. O. Smith, Science, 2008, 319, 1215–1220 CrossRef CAS.
- E. J. Allain, J. Chem. Technol. Biotechnol., 2007, 82, 117–120 CrossRef CAS.
- J. Woodward, M. Orr, K. Cordray and E. Greenbaum, Nature, 2000, 405, 1014–1015 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2009 |