Development of alternative photocatalysts to TiO2: Challenges and opportunities
María D.
Hernández-Alonso
a,
Fernando
Fresno
b,
Silvia
Suárez
a and
Juan M.
Coronado
*c
aEnvironmental Applications of Solar Energy, CIEMAT-PSA, Avenida Complutense 22, Building #42, 28040, Madrid, Spain. Fax: +34 91 346 6037; Tel: +34 91 346 6177
bSolar Concentrating Systems, CIEMAT-PSA, Avenida Complutense 22, Building #42, 28040, Madrid, Spain
cIMDEA-Energía, URJC, Laboratorios III, C/Tulipán s/n.E-28933 Móstoles, Madrid, Spain. E-mail: juanmanuel.coronado@imdea.org; Fax: +34 91 488 85 64; Tel: +34 91 614 76 11
First published on 24th August 2009
Abstract
Since the early development of this technology in the 1970s, TiO2 constitutes the archetypical photocatalyst due to its relatively high efficiency, low cost and availability. However, during the last decade a considerable number of new photocatalytic materials, either semiconductor or not, have been proposed as potential substitutes of TiO2, particularly in the case of solar applications, for which this standard photocatalyst is not very suitable because of its wide band gap. Semiconductors based on cations with d0 configuration such Ta5+ or Nb5+, as well as oxides or nitrides of d10 elements such as Bi3+, In3+ or Ga3+ are among the most successful novel photocatalysts, but non-semiconductor solids like cation-interchanged zeolites also produce interesting results. In addition, some classical semiconductors like ZnO or CdS, initially discarded as a consequence of their poor stability under irradiation, have been reconsidered as feasible photocatalysts for particular applications. This growing body of data requires new analysis of the challenges and opportunities facing photocatalysis in order to assess which of the photoactive materials are best for each particular application. In this review, we summarize, with an historical perspective, the main achievements obtained with photocatalyst alternatives to TiO2 in the three main niches for this technology: water splitting for hydrogen production, decontamination and disinfection processes, and organic synthesis.
 María D. Hernández-Alonso María D. Hernández-Alonso | María D. Hernández-Alonso received her doctoral degree in 2006 from the Universidad Autónoma of Madrid, conducting her PhD research at the Institute of Catalysis and Petrochemistry of the National Council of Scientific Research (CSIC). This was followed by two-years of postdoctoral work at the Catalysis Engineering Department of Delft University of Technology. She is now working in the Environmental Applications of Solar Radiation group at CIEMAT. Her research interests relate to pollutant abatement by photocatalytic processes, with special focus on obtaining improved TiO2-based photocatalysts. |
 Fernando Fresno Fernando Fresno | Fernando Fresno received his PhD in Chemistry in 2006 from the Autónoma University of Madrid, working in the field of heterogeneous photocatalysis at the Institute of Catalysis and Petrochemistry of the National Council of Scientific Research (CISC). He then moved to the Center for Research on Energy, Environment and Technology (CIEMAT), where he is currently working as a postdoctoral researcher on solar hydrogen production by thermochemical cycles. |
 Silvia Suárez Silvia Suárez | Silvia Suárez obtained a PhD in Chemistry from the University of Alcalá in 2002, working at the Institute of Catalysis and Petrochemistry, CSIC. She moved to the Environmental Application of Solar Radiation Unit PSA-CIEMAT in 2006. Awarded a Ramón y Cajal contract as a researcher at CIEMAT in 2008, her research interests are in the areas of environmental heterogeneous chemistry, photocatalysis, development of novel supported catalysts and chemical technology. |
 Juan M. Coronado Juan M. Coronado | Juan M. Coronado conducted his doctoral studies at the Catalysis and Petrochemistry Institute (ICP-CSIC) and he received his PhD in Chemistry from the Complutense University of Madrid in 1995. He was recently appointed as a Senior Researcher at IMDEA-Energia, a new centre created by the Madrid regional government for energy studies. Previously, he worked as a scientist at CIEMAT and CSIC. His research interests currently focus on solar photocatalysis, and the production of renewable fuels. |
Broader contextPhotocatalysis potentially can provide solutions for many of the environmental challenges facing the modern world because it provides a simple way to use light to induce chemical transformations. Pollution control, either in aqueous solutions or air, is very likely the most studied application of photocatalysis, although commercial uses relate mainly to self-cleaning surfaces. Besides this, photocatalysis can be also applied to the production of fuels like hydrogen or as a green route to obtain fine chemicals. Currently, TiO2 is by far the most widely used photocatalyst because it comprises the best balance of properties among the known or assayed semiconductors. However, it still presents some disadvantages such as limited activity and reduced sensitivity to sunlight. Therefore, in the last few years significant effort has been devoted to the search for new materials that may overcome the limitations of TiO2. This review gives an overview of photocatalysts, different to TiO2, that have been tested for the most relevant photocatalytic applications: water splitting, detoxification and disinfection, and organic synthesis. |
1. Introduction
Early investigations in photocatalysis were devoted to the study of semiconductors based on metal oxides, like ZnO1–3 and NiO,4 or sulfides like CdS.5 However, the publication in 1971 of the work by Formenti et al.,6 followed one year later by A. Fujishima and K. Honda's7 renowned article prompted a true revolution based on the extensive use of TiO2 as a photocatalyst. Ever since this decade interest in this semiconductor, first in academia and then in industry, has grown exponentially. During this time, photocatalysis with TiO2 was applied with varied success to a number of processes, including hydrogen production, effluents detoxification and disinfection, and organic synthesis.8–13 The relatively high quantum yield and elevated stability of TiO2 are the key reasons for the preponderance of this semiconductor, which has become a virtual synonym for photocatalyst. Nowadays, the market for photochemical applications of TiO2 is thriving, especially in Asian countries such as Japan, South Korea and China.14–16 Photocatalytic coatings deposited on external building elements, like windows, along with air purifiers are the main products of this emerging business, but dozens of other commercial uses have been proposed and some experimental devices are already on the market.14–16Despite these achievements and remarkable advantages, heterogeneous photocatalysis with TiO2 has to cope with significant limitations. In general, photocatalytic reactions rates are moderate, and consequently this technology is not appropriate for high throughput processes, as for example in the decontamination of heavily polluted industrial effluents.9,10,17 Increment of photon flux increases the reaction rate, but saturation is usually achieved at relatively low irradiance, and consequently energetic efficiency of the process drops.17,18 However, the most important drawback of photocatalysis is derived from the mismatch between the TiO2 band gap energy and the sunlight spectra, which overlap only in the UVA (400–320 nm) and UVB (320–290 nm) ranges. As a consequence, this technology can only take advantage of less than 6% of the solar energy impinging on the Earth's surface, and its potential as a sustainable technology cannot be entirely fulfilled.8–13,18 This fact has profoundly influenced research in photocatalysis, so that modification of TiO2 to achieve efficient photoactivation in the visible spectrum is an active field of research.8–13,19,20
During the last few years an increasingly great number of new photocatalysts have been synthesised and tested as possible alternatives to TiO2. These materials are not derived from TiO2 by any of the usual modifications such as doping, coupling with an additional phase, or morphological changes, instead they are completely different compounds with distinct composition and structure. In this context, the feasibility of using some well-known photocatalysts like ZnO21,22 or CdS,23,24 have been reconsidered in light of recent advances in nanotechnology. More interestingly, a great variety of entirely novel photoactive semiconductors have been developed in the last few years as possible substitutes for TiO2.25–27Among these, mixed oxides of transition metal like Nb, V or Ta, or with main group elements such as Ga, In, Sb or Bi have been extensively investigated as alternative photocatalysts.25–27 Besides, sulfides and nitrides of different metals have been frequently selected to obtain materials with photoactivity in the visible range.25 In addition, some high surface area solids, such as cation interchanged zeolites, have been also evaluated as photocatalysts, despite the fact that they do not present semiconductor properties.28 The growing number of publications devoted to these different kinds of photocatalyst in the recent years is displayed in Fig. 1. This clearly shows an increase in interest by the scientific community in the three types of photocatalysts considered. Especially significant is the increase in the number of contributions dedicated to ZnO, which historically has been a close competitor of TiO2.1–3 Even so, photocatalysis is still dominated by TiO2, and in 2008 all these advanced materials combined represented less than 25% of the articles related to semiconductors' photocatalysis.
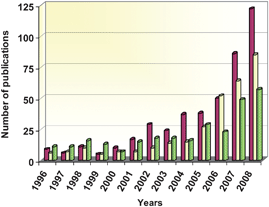 | ||
Fig. 1 Annual evolution of the number of publications devoted to photocatalysis with alternative materials to TiO2. ZnO ( ), sulfides ( ), sulfides ( ) and mixed oxides containing Nb, V, Ti or Ta ( ) and mixed oxides containing Nb, V, Ti or Ta (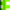 ). Data source: ISI web of knowledge. ). Data source: ISI web of knowledge. | ||
In this review, we summarize the main achievements obtained with photocatalyst alternatives to TiO2 in order to assess the feasibility of substituting this paradigmatic semiconductor by other materials with enhanced properties. As a survey of the bibliography reveals, there are clear differences in emphasis devoted to the development of these innovative photocatalysts depending on each specific application. In this regard, it is worth noting that most of the novel photocatalytic materials have been applied to water splitting.25–27 This highlights the relevance of this technology on the search for environmentally acceptable fuels. Accordingly, in this review we will discuss separately the three main potential niches of photocatalysis: hydrogen production, detoxification of effluents, and photosynthesis. Nevertheless, in order to evaluate the possible advantages of the alternative photoactive materials, it is important to fully understand the basis of photocatalysis with TiO2, because it constitutes the obvious benchmark. Therefore, we first briefly discuss the grounds for the preponderance of TiO2. Here it is essential to establish which material is considered as a chemical modification to TiO2 and which is taken as an entirely new photocatalyst, because in some cases the boundary between these two situations may not be sharply delimited. A good example of this situation is the progressive transition from anatase TiO2 to cubic TiN under thermal treatment in NH3.29 So, to remove possible ambiguities we have considered photocatalysts different to TiO2i.e. those solids that have neither anatase nor rutile structures. According to this criterion, we focus on describing the photoactivity of compounds such as SrTiO3 (perovskite) or Y2Ti2O7 (cubic pyrochlore), while materials like TiO2−xNx will be considered only for comparison. Furthermore, we deal exclusively with solid photocatalysts, and consequently do not discuss homogeneous processes like photo-Fenton30 or processes based on the use of free metalloporphyrins,31 or polyoxometallates.32,33 Those readers interested in these homogeneous treatments are referred to excellent reviews on these topics.30,33,34
2. Fundamental aspects of photocatalysis
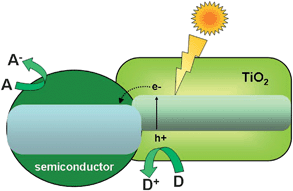
The initial step of photocatalysis is the absorption of photons of wavelength adequate to match energy levels of the photoactive material. In the case of semiconductors, illumination induces electron promotion from the valence to the conduction band if the energy of the photons exceeds the band width.8–13 This process leaves an unoccupied state, or electron hole in the valence band, which can be conveniently described as a particle in its own right. Most of these electron–hole pairs recombine, releasing the absorbed energy as light or, more frequently, as heat. However, a small percentage of these carriers migrate to the surface where they can be captured by adsorbed molecules to start the catalytic cycle.8–13 A generic scheme of these processes can be seen in Fig. 2. Conduction band electrons can reduce electron acceptors, like oxygen molecules or H+. On the other hand, valence band holes are oxidants that are able to attack donor species such as organic molecules or OH−groups. In addition, the incorporation of a co-catalyst is crucial to fully exploit the potential of these solids in reactions like water splitting.25–27 The main role of this component is to facilitate the reduction of protons and prevent the back reaction of O2 and H2. Noble metals like Pt, Pd or Rh or metal oxides like NiO, RuO2 are frequently used for this function.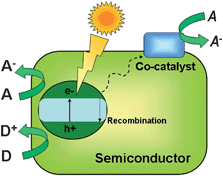 | ||
| Fig. 2 Pictorial diagram showing the main events of photocatalysis over semiconductors. | ||
Photoactivity is determined by the interplay between surface and electronic characteristics, and is often estimated by quantum yield. This parameter is defined as the ratio between the rate of photoinduced events and the flux of absorbed photon.8–13 However, an accurate estimation of absorbed photons in real systems is often impossible to obtain due to extensive light scattering. Consequently, for practical reasons many studies assume that all the radiation is absorbed, and an alternative performance index, the photonic efficiency, is defined as the quotient between the rate of the photocatalytic events and the photon flux.8–13
Photocatalysts have the ability to convert light power into chemical energy through a series of electronic processes and surface reactions. This fact challenges the conventional definition of catalysis because it implies the promotion of non-spontaneous reaction (ΔG > 0), and for that reason some authors prefer the term photosynthesis for this kind of process.35,36 This feature endows photocatalysis with the potential to be utilised for the accumulation of solar power. In fact this is the goal of water splitting which aims to store sunlight energy in the form of H2.25–27 In contrast oxidation reactions, such as those of detoxification treatments are downhill processes (ΔG < 0) and do not accumulate energy, although they must overcome the activation barrier for the rupture of strong molecular bonds like C–C. In any case, this energy flux requires materials with specific characteristics of energy levels, specifically sufficient lifetime of the excited state. On the other hand, in contrast with other catalytic processes, in a typical experiment to remove aqueous pollutants the molar ratio between the photocatalysts and the molecule degraded is larger than 10. This fact implies that photocatalysts should be reused many times without a significant lack of efficiency in order to be considered a catalyst rather than a reagent.36
Most photocatalysts are semiconductors, due to the relatively high stability and mobility of charge carriers on these solids, which facilitates their transport to the surface where they can interact with adsorbed molecules.8–13 However, photocatalysis is also possible using isolated photoactive centres dispersed in a non-absorbing solid matrix, such as metal-loaded zeolites. These materials have been denominated single-site photocatalysts by Anpo and collaborators28 and this term will be used in this review to distinguish these materials from semiconductors. In contrast to TiO2, the electronic levels of these photocatalysts are discrete, and therefore all processes occur in the photoactive centre without any transportation of the charge carriers. These active sites are constituted by isolated or highly dispersed transition metal cations (see Fig. 3), and photoactivation implies charge transference transitions within d levels.28,37 A scheme of a possible photocatalytic oxidation cycle on these kind of materials is shown in Fig. 3 for CO photo-oxidation. Nevertheless, depending on the nature of the photoactive site and the process, alternative mechanisms can be described. An important feature of these materials is that the photoactive centres must be located on the surface in order to interact with reagent molecules.37 Therefore, highly specific areas and elevated dispersions of cationic sites are expected to have a direct impact on the performance of this kind of photocatalyst.
 | ||
| Fig. 3 Examples of single site photocatalytic centres (A), and pictorial diagram (B) showing the mechanism of CO photo-oxidation over single-site photocatalysts with Mo centres. (Based on ref. 28). | ||
3. Why use TiO2? Pros and cons of this benchmark photocatalyst
3.1. Influence of structural and electronic characteristics on TiO2 photoactivity
Four TiO2 polymorphs were prepared using conventional synthesis conditions: anatase (tetragonal), rutile (tetragonal), brookite (orthorhombic) and TiO2 (B) (monoclinic).8,11,39 However, these last two phases have been scarcely used as photocatalysts, and consequently we will focus exclusively on anatase and rutile. All TiO2 forms can be described as arrangement of slightly elongated TiO6 octahedra connected in different ways by vertices and edges. 8,11,20,39Although rutile is generally considered the most stable polymorph, differences in the Gibbs free energy of formation between rutile and anatase are small (lower than 15 KJ mol−1).38,39 Consequently, anatase can be easily obtained by synthesis at low temperature treatments (below 400 °C), but rutile frequently starts to appear at moderate temperatures (400–600 °C) and it becomes the predominant phase after annealing at higher temperatures.8,39Due to the presence of a small amount of oxygen vacancies, which are compensated by the presence of Ti3+ centres, TiO2 is an n-type semiconductor. The valence band of this material is mainly formed by the overlapping of the oxygen 2p orbitals, whereas the lower part of the conduction band is mainly constituted by the 3d orbitals, with t2g symmetry, of Ti4+ cations. The band gaps are 3.2 and 3.0 eV for anatase and rutile, respectively.8–13,39Interband transitions of TiO2 are indirect39 (i.e. implying both electronic levels and lattice phonons) but factors like the crystalline size or the presence of dopants can modify the type of transition and somewhat conflicting reports are found in the literature.40,41 This characteristic directly affects the photonic efficiency because indirect semiconductors present a reduced photon absorption and consequently require a higher mass of photocatalyst to obtain the same effect. In any case, it is worth emphasizing that as much as a 90% of the electron–hole pairs recombine in less than 10 ns and consequently photogenerated carriers available for surface reactions are quite limited.39 Values of quantum yield vary broadly with the process considered, for TiO2 reactions in solution they are typically lower than 1%,42 but they can exceed 25% for some gas phase reactions.43 These values depend, among other factors, on electronic transferences in the interface and surface characteristics, but considering exclusively the photoactivation process, TiO2 shows a limited performance. Thus, in contrast with silicon, which presents an internal quantum efficiency (IQE) close to 100% under illumination at 600 nm,44 with TiO2 the absorbed photon-to-current efficiency (APCE, parameter equivalent to quantum yield) is about 30% at 360 nm.45
The chemical potentials of photogenerated electrons and holes depend on the position of the energy levels in the semiconductor. More specifically, the redox potential of a donor species adsorbed on the surface of the photocatalyst needs to be more negative (higher in energy) than the valence band position of the semiconductor in order to replenish the electron vacancies. Similarly, acceptor molecules must have a redox potential more positive (lower in energy) than the conduction band.8,13In view of this, one of the key advantages of TiO2 among other semiconductors is that its electronic structure is such that it allows both the reduction of protons (ENHE(H+/H2) = 0.0 eV) and the oxidation of water (ENHE(O2/H2O) = 1.2 eV), which are key processes for water splitting.8,10,11,39 This can be appreciated in Fig. 4. Therefore, in contrast to other semiconductors, which are efficient for either water reduction like Si, or for water oxidation like SnO2, TiO2 is suitable for activating both reactions simultaneously. Surface OH−groups can act as donor species, reacting with a valence band holes to yield hydroxyl radicals, OH˙. These species have a very high oxidation potential (ENHE(OH˙/H2O) = 2.27 eV) and are considered the key intermediate in the photo-oxidation reactions with TiO2.8–13
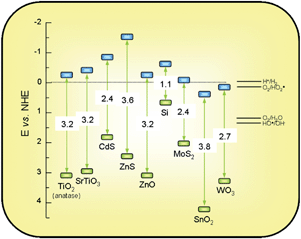 | ||
| Fig. 4 Band gaps (eV) and redox potentials, using the normal hydrogen electrode (NHE) as a reference, for several semiconductors (Based on the data in ref. 8,10–12). | ||
Although some discrepancies can be found in the literature, anatase is usually considered the most photoactive phase of TiO2.8–13 In this regard, a recent study by Choi et al. on the photocatalytic degradation of an ample selection of pollutants in aqueous solution indicated that pure rutile is more efficient than anatase exclusively for some specific substrates, such the dye Orange 7.46Fig. 5 shows the photoactivity of different TiO2 samples in the degradation of four pollutants in aqueous solution. Several reasons are proposed to explain differences in photoactivity between anatase and rutile structures, including variations in electronic (e.g. Fermi level position,electron mobility…) or surface (e.g.hydroxyl concentration…).8–12 In addition, rutile generally presents lower surface area than anatase, due to the larger crystalline size imposed by thermodynamic constraints38, and this fact may also contribute to the lower photoactivity of this phase. In any case, numerous studies have also shown that elevated rates for organic pollutants degradation are achieved with anatase–rutile mixtures, such as the benchmark photocatalyst Degussa P25.47,48Fig. 5 also provides some examples of the superior performance of biphasic TiO2. This behaviour has been attributed to the formation of n–p junctions due to the contact of the crystals of both phases, which improves efficiency of charge separation.47,48
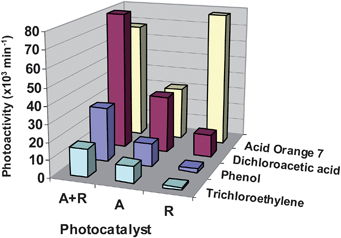 | ||
| Fig. 5 Photoactivity expressed as the initial rate for the decomposition of three different pollutants in aqueous solution using different TiO2 commercial photocatalysts with the structures: anatase (A) supplied by Millenium, rutile (R), or a mixture of rutile and anatase (R + A; Degussa P25). Data taken from ref. 46. | ||
Corrosion of semiconductors can be induced by irradiation, if the photogenerated charge carriers attack the solid constituents. This phenomenon is known as photo-corrosion, but it does not affect to TiO2 because water oxidation, is thermodynamically more favoured than the formation of molecular oxygen from oxide anions. This stability, which is maintained over a large pH range, is a crucial feature which explains the widespread use of TiO2 in solution.8–13
3.2. Influence of surface properties on TiO2 photoactivity
Water molecules dissociate on the surface of TiO2 to yield hydroxyls groups, which saturate the coordination sphere of Ti4+, and protons, which bind to the bridging oxygen.20,49 This process reduces the excess energy caused by the abrupt discontinuity of the solid structure at the edge of the crystal and it occurs even with air moisture. Consequently, adsorption of reagent molecules is determined, to a great extent, by the density and specific characteristics of hydroxyl groups. Many organic substances like aromatic compounds are attached to the TiO2 surface by hydrogen bonding,50 but stronger interactions with formation of new bonds can be observed for other molecules such as alcohols vapours or acidic gases.51 In aqueous solution, it is necessary to consider the amphoteric character of the >Ti–OH species, because the surface charge depends on the pH. Thus, at low pH values the TiO2 surface is positively charged, while in alkaline conditions it is negatively charged. The pH value at which the concentration of negative and positive centres is the same constitutes the so-called of zero charge point (ZPC), which varies slightly with the crystalline phase or the preparation method. As an example, a ZPC value of 6.4 is reported for Degussa P25.52 In the case of photocatalytic reactions with ionic species, this parameter is crucial, because it defines the pH interval at which adsorption is facilitated or hindered by electrostatic interactions.52As in any heterogeneous catalytic process, surface area is also a relevant parameter for photocatalysis, because adsorption capacity is related to its magnitude. Nevertheless, the extensive literature on this subject indicates that photoactivity is relatively insensitive to the increment of specific surface, and consequently its effect on reaction rates is modest.46 The reasons for this behaviour are not entirely clear and most likely are diverse, but a possible explanation is that the rate limiting step of the photocatalysis corresponds to electronic processes rather than surface reactions. In this regard, it is worth emphasizing that any increment of surface area is often associated with a reduction of crystallinity which may increase the density of recombination centres.
3.3. Safety and availability of TiO2
Titanium dioxide is a material with a great number of technological applications (such as pigments, sensors, photovoltaic cells, catalysts…) and its annual production for whitening products as diverse as paper, plastics or paints was more than 5.28 millions tons in 2008, representing an expansion of about 2% with respect to the previous year.53 In spite of this high consumption, a shortage of TiO2 is not foreseen in the near future. Titanium is the ninth most abundant element and constitutes about 0.63% in weight of the Earth's crust. Minerals rutile and ilmenite, FeTiO3, are the main ores of this element and they are found in large deposits in Norway, Australia, China, Canada and many other countries.8,53The toxicity of TiO2 is low and it has been approved as a food colorant (E-171 in EU legislation).8 In fact many everyday products such as toothpaste, pill coatings and chewing gum contain TiO2.8 Nevertheless, new concerns have arisen recently as a consequence of the extensive research on TiO2 particles of nanometric size. Nanomaterials present risks associated with their small size which greatly facilitates intake by inhalation. In the case of TiO2, oral administration of a high dose of nanoparticles (5g Kg−1) does not cause acute toxicity, although evidence of hepatic damage was found.54 More severe effects have been reported in the case of inhalation of high concentrations of TiO2nanoparticles, which caused inflammation in rats' lungs.55 Nevertheless, although most of the studies show that the toxicity associated with TiO2nanoparticles seems to be relatively mild, further studies are necessary to definitively set the limits of safety for TiO2 nanomaterials.56
3.4. Strategies for improving TiO2 photoactivity
Considerable effort has been devoted to improve TiO2 photocatalytic efficiency. Although dozens of different approaches have been adopted with this aim, all of them consist of either morphological modifications,39 such as increasing surface area and porosity, or the incorporation of additional components, such as metals or a second semiconductor phase. However, contradictory reports abound in the literature devoted to TiO2 improvement, very likely because changing composition or morphology unavoidably modifies other parameters which also affect efficiency. Besides, as experiments are not carried out under standardized conditions (e.g. with constant irradiance and homogeneous light distribution), the assessment of the real progress achieved with modified TiO2 is often difficult. In addition, comparisons between pristine TiO2 and enhanced photocatalyts are frequently biased, because samples selected as reference materials present a relatively low photoactivity. Nevertheless, from a literature survey, it seems that TiO2 can be tailored in different fashions to obtain moderate increments of photocatalytic rate, as discussed below. Table 1 summarizes the performance of some selected examples of modifications to TiO2.| Modification of TiO2 | Process | Relative reaction ratea | Ref. | |
|---|---|---|---|---|
| Morphological | Chemical | |||
| a With respect to TiO2. b With respect to Pt/TiO2 P25. | ||||
| Nanoparticles (6 nm) | Trichloroethylene degradation in gas phase | 4.2 | 35 | |
| Nanoparticulated film (7 nm) | Isopropanol degradation in gas phase | 1.6 | 57 | |
| Foam | Acetaldehyde degradation in gas phase | 3.2 | 59 | |
| Mesoporous | Acetone in the gas phase | 1.6 | 60 | |
| Ti0.93Fe0.07O2 | Phenol aqueous solution | 2.8 | 62 | |
| Ti0.92Zr0.08O2 | 4-Chlorophenol aqueous solution | 1.5 | 63 | |
| 10% SnO2/TiO2 | Methyl orange in aqueous solution | 1.3 | 65 | |
| TiO2−x Nx | Acetaldehyde degradation in gas phase | 3.0 (463 nm) | 66 | |
| 1.15 (351 nm) | 66 | |||
| Mesoporous | Pt/TiO2 | Water splitting using CH3OH as sacrificial agent | 1.16b | 61 |
Physical modifications of TiO2, in the form of nanoparticles,40,57nanotubes,58 foams59 mesoporous60,61 phases, or other morphologies have shown different degrees of photoactivity improvement with respect to unmodified TiO2. Monodispersed nanoparticles usually present an optimal diameter for which the benefits of small crystalline size (high surface area, reduced bulk recombination) overcome the detrimental effects (surface recombination, low crystallinity).40
Metal-doping of TiO2 has been extensively explored as a way to improve photoactivity under visible light.8,11 Nevertheless, foreign cations frequently acts as recombination centres and therefore significant improvements are only possible at low concentration of dopants, and using careful synthesis methods to limit lattice distortion.62,63 On the other hand, coupling of TiO2 with another semiconductor is another widely used approach to increase the photonic efficiency, because, if the band structure of both materials is adequate, charge carriers become physically separated upon generation and therefore the recombination rate greatly decreases.8,10,11 This fact implies the formation of n–p junctions analogous to those established in photovoltaic cells. Fig. 6 displays a diagram of this process in the particular case of a semiconductor with wider band gap than TiO2, although the reverse situation is also possible. As this scheme suggests, the two phases must be in close contact in order to make possible the electronic transference. Consequently, synthesis procedures must ensure the interaction of the two phases. Some of the semiconductors more frequently coupled with TiO2 are SnO2, WO3, and CdS, although many other oxides and sulfides have been tested.64 Experimentally, it is usually found that higher photoactivity is achieved when the loading of the second semiconductor is lower than 15%.65
 | ||
| Fig. 6 Mechanism for charge separation on TiO2 coupled with a different semiconductor. | ||
Among the many chemical modifications adopted for shifting the TiO2 band gap to lower energy, currently the most promising route seems to be the partial substitution of oxygen with N and other elements like C and S, as proposed by Asahi and co-workers in 2001.66 In particular, most of these studies have focused on TiO2−xNx materials, which show remarkable photoactivity under visible illumination. The origin of this photoresponse at higher wavelengths is the mixing of the 2p nitrogen level with the oxygen 2p orbitals to form the valence band, which results in a lower band gap.20 This material has been applied to several processes including water splitting and pollutant removal.67,68 Thus, 1% of nitrogen doping results in ten-fold increment of the rate by visible light excitation with respect to pristine TiO2. However, it is worth noting that photoactivity under visible light is often significantly lower than under UV.66 In this regard, it has been reported that degradation of trichloroethylene in an air stream using N-doped TiO2 under solar irradiation is mainly due to UVA activation.69
4. Alternative photocatalyst for water splitting
Dissociation of the water molecule to yield hydrogen and oxygen occurs according to the equation:| H2O(l) → ½O2(g) + H2(g) ΔG = +237 kJ mol−1 | (1) |
This apparently simple process has gathered a great deal of interest from an energetic point of view because it holds the promise of obtaining a clean fuel, H2, from a ubiquitous and cheap resource, H2O.70 The difficulties of applying this process to energy storage arise from the endothermic character of the reaction, which would require a temperature of 2500 K to obtain ca. 5% dissociation at atmospheric pressure.71 This fact makes a direct attempt to split water impractical. In contrast, photochemical decomposition of water is a feasible alternative because photons with a wavelength shorter than 1100 nm have the energy (1.3 eV) to split a water molecule. However, a purely photochemical reaction has to overcome a considerable energy barrier and it is only relevant for irradiation with wavelengths lower than 190 nm.72 The use of a photocatalyst reduces appreciably this activation energy and makes the process feasible with photons within solar spectrum. In this respect, the classical work by A. Fujishima and K. Honda proved that photoelectrochemical decomposition of water is possible over TiO2electrodes provided a chemical bias, caused by pH differences between the two halves of the cell.7 Subsequent investigations showed that an external potential was not required to split the water molecule and that the photoactivity could be boosted if a co-catalyst (e.g.Pt) was incorporated.73 Considering that a small percentage of the sunlight power reaching the earth surface (3.7 × 1018MJ year−1) can fulfil the current energy consumption of mankind (4.1 × 1014MJ year−1), the enormous importance of developing efficient systems to harvest solar energy is easily understood.70,71 Consequently, the interest in photocatalytic water splitting has not decreased since the 1970's and it has recently increased due to the growing concern about global warming, caused by worldwide power generation which relies largely on fossil fuels. In fact, although water splitting over TiO2 based photocatalysts still represents more than 50% of the articles published in this field, about 140 different semiconductors have been evaluated with the aim of producing H2 more efficiently by solar photocatalytic processes.25–27
One of the most important limitations of photocatalysis for water dissociation is that the process with pure water is rather inefficient. This is related to the fact that reduction of water is a complex multistep reaction which involves four electrons. Using sacrificial molecules as electron donors can improve remarkably H2 production, as holes are scavenged by these molecules and recombination is greatly reduced. Furthermore, as O2 is not produced, the back reaction to produce water is suppressed, increasing H2 yield and avoiding a gas separation stage. Simple alcohols like CH3OH or NaS/Na2SO3 mixtures are added to the aqueous solution with this purpose. However, these additives generate waste products, which must be eliminated and consequently the sustainability of the process decreases. Therefore, the utilization of sacrificial molecules is only environmentally sensible when they come from biomass (e.g.bioethanol) or from residues than must be disposed (e.g. residual sulfur compounds from the oil industry).74 On the other hand, an electron acceptor like AgNO3 is sometimes utilized to enhance the photocatalytic production of O2, but the interest in this process is usually academic rather than practical.26,28
Considering that sunlight photons with wavelengths lower than 1100 nm can be used for the photocatalytic dissociation of water molecules, more than 800 W m−2 (equivalent to 2.8 mmol s−1m−2)75 of the available solar energy could be potentially stored as H2. Obviously, this figure is a thermodynamically unachievable limit, which assumes complete power conversion, but it emphasizes the huge potential of solar hydrogen. Furthermore, Fig. 7 illustrates the importance of engineering the energy levels of the photocatalyst to overlap as much as possible with the sunlight spectrum. Thus, shifting the band gap edge from 360 to 500 nm can suppose a ten-fold increment of the accumulated energy keeping the quantum yield constant. In fact, poor absorption in the visible range is the main drawback not only of TiO2 but also of other very efficient semiconductors like NaTaO3.76 Therefore, achieving high efficiency under visible light illumination remains as a major challenge for the production of hydrogen by photocatalysis. In this respect, Maeda and Domen25 have proposed that, before commercial development, photocatalysts must reach a quantum yield of 30% at 600 nm. The current reported record for pure water splitting is by (Ga0.88Zn0.12)(N0.88O0.12) combined with Rh2−xCrxO3 as co-catalyst, with a quantum yield of 5.9% at 420–440 nm (see Fig. 7).77 Therefore, there is still a long way to go before solar photocatalysis for H2 production becomes a reality.
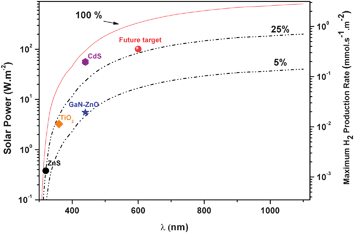 | ||
| Fig. 7 Incident solar power as a function of the photons wavelength as obtained from the integration of the 1.5 AM reference spectrum74. Dashed lines represent the solar power accumulated at each wavelength considering photon efficiencies of 5% and 25%. The different symbol marks some of the current or future achievements of photocatalysis for H2 production using TiO2, ZnS, CdS or (Ga0.88Zn0.12)(N0.88O0.12) as photocatalyst. It should be noted that these values are significantly overestimated because total photon absorption is considered and consequently H2 production rates would be appreciably lower in practice. | ||
Basically all photocatalysts developed for H2 production are oxides, sulfides or nitrides of metal cations with either d0 (Ti4+, V5+, Nb5+…) or d10 (Zn2+, Cd2+, Ga3+…) configuration. This fact has been attributed to the fact that partly filled d orbitals of other cations can act as recombination centres.25–27 These metals contribute to formation of the conduction band with their d or sp orbitals, while representative elements contribute to valence band with their corresponding p orbitals. Finally alkaline, alkaline earth or lanthanide cations in some mixed oxides semiconductors do not participate appreciably in the band structure but maintain the electroneutrality of the crystal lattices as in the case of perovskites.27 However, the distortion of the lattice induced by the size of alkaline cations is detrimental to photoactivity because it hinders the migration of electron–hole pairs through the solid, and modifies the band gap.27 Finally it is important to highlight that co-catalysts, either noble metals (Pt, Pd, Au…) or oxides (NiO, RuO2…) greatly enhance photoactivity, and their incorporation must be thoroughly controlled to maximize the dispersion and facilitate electronic transferences.26,27
In this section we have outlined the main achievements obtained with photocatalyst alternatives to TiO2 for water splitting in three different situations of increasing complexity: using sacrificial agents, in pure water and employing visible light as excitation source. Since recent extensive reviews have accounted for most of the photoactive materials used for this purpose,25–27 and many of the photocatalysts evaluated present limited performance, we focused exclusively on the most promising semiconductors. For further details readers are referred to these papers .25–27
4.1. Water splitting with sacrificial agents
Some niobates of the Dion–Jacobsen series and general formula ABn−1NbnO3n+1 (A = K, Rb, Cs; B = Ca, Sr, Na, Pb; n = 3–4) have shown a remarkable photoactivity for H2 production using CH3OH as the sacrificial molecule.26,27,78 These materials comprise perovskite layers of NbO6 corner-sharing octahedra with B cations placed in the centre of the cube delimited by 8 of these niobium polyhedra. Cations A are sandwiched between these slabs to compensate the charge and are rather mobile. In fact, interchange of the alkaline cations of the A positions by protons has been used to boost H2 generation under UV irradiation.79 Thus, HCa2Nb3O10 modified with Pt produces as much as 19 mmol h−1 g−1, which is more than 40 times larger than the rate obtained with Pt/TiO2.26,80 Enhancement of photoactivity upon protonation of these layered perovskites, has been related to the widening of the interlayer gap, which favours the interaction with methanol,. Further enlargement of the space between slabs of Pt/HCa2Nb3O10 can be achieved using SiO2 pillars, and accordingly a remarkable increment in the H2 production rate can be obtained.81 On the other hand, the photoactiviy KCa2Nb3O10 can be significantly improved if the surface area is increased by alternating acid and alkaline treatments, which results in a restacking of perovskite sheets.82The utilization of sacrificial agents is warranted when using sulfides as photocatalyst, because in pure water they experience extensive photocorrosion, which in the case of ZnS takes place according to the reaction:26,27,74
| ZnS + 2h+ → 2Zn2+ + S | (2) |
In contrast, this semiconductor (band gap 3.66 eV) is rather stable if a Na2S/Na2SO3 mixture is incorporated to the solution. Under these conditions a quantum yield as high as 90% has been obtained with ZnS using Pt as co-cotalyst under UV illumination (λ > 300 nm).83 Furthermore, stability was maintained for at least 34 h.83 Bearing in mind the abundance of sulfur residues generated by the petroleum industry,74 the use of sulfides with absorption in the visible range has attracted considerable interest due to its low cost. An example of these photocatalysts is Rh/AgGaS2 which can achieve a quantum yield of 25% at 440 nm.84 Solid solutions of sulfides like those of the system ZnS–CuInS2–AgInS2, are very attractive for solar applications because the band gap can be modulated in the whole visible range by simply changing the composition.85 Nevertheless, very likely the most investigated sulfide photocatalyst is CdS, which has wurtzite structure and presents a band gap of 2.4 eV and a suitable potential of the photoelectrons for proton reduction (see Fig. 4). This semiconductor, either on its own23 or in combination with other sulfides, such as Ag2S and ZnS,86 or oxides like CdO,73 presents a remarkable photoactivity when using noble metal such as Pt or Ru co-catalysts. Thus, a 37% of quantum yield at 440 nm is obtained with high surface are photocatalysts of the Ag-Zn-Cd-S system.86 However, the current record of photoactivity in the visible range using sacrificial molecules corresponds to the nanostructured CdS (in the form of nanosheets or hollow nanorods) with Pt nanocrsytals as co-catalyst, which achieves a quantum yield of 60% at 440 nm.23 On the other hand, CdS can be used to form pillars in layered semiconductors like K2Ti4O9, and the formed heterojunction show a significant photoproduction of H2 under visible light excitation.87
4.2. Photocatalytic splitting of pure water under UV light
Although simple perovskite titanates such as SrTiO3 show a reduced activity for the cleavage of the water molecule,88 those with layered structure show a remarkable efficiency for splitting pure water. Among them, La2Ti2O7, which is formed by slabs Fig. 8 with a perovskite-like array separated by La3+ layers,26 shows a photonic efficiency of 12% for water dissociation in H2 and O2.89 A significant increment of photoactivity of this semiconductor can be achieved by doping with BaO, so as to reach a 50% photonic efficiency using a NaOH solution. These results were obtained using NiO as co-catalyst, because poorer H2 production are attained using Pt.90 In contrast, the maximum quantum yield obtained with TiO2 was 29%, utilising Rh as co-catalyst.80 Another layered perovskite titanate with interesting photocatalytic properties for water splitting is K2La2Ti3O10, which presents a good rate for the stoichiometric production of H2 and O2 using NiOx as co-catalysts.91,92 Utilization of gold as co-catalyst can be more efficient than NiOx under visible light illumination (λ = 419 nm), if during the incorporation of the metal the crystallinity of the semiconductor is preserved.93 More recently, high photoactivity for water dissociation were obtained with NiOx/BaLa4Ti4O15 which has a quantum yield of 15%.94 Similarly, some layered niobates, such as K4Nb6O1795 or Ba5Nb4O15,94 which also have a layered structure, show a significant photoactivity for water cleavage. | ||
| Fig. 8 SEM images of (a) Au and (b) NiOx loaded on BaLa4Ti4O15 photocatalysts by a photo-deposition method. (Reproduced from ref. 94). | ||
Mesostructured crystalline Ta2O5 (orthorhombic), prepared using silicone scaffolding, is very efficient for water splitting when loaded with 3% of NiOx96 Tantalates in general are rather efficient photocatalyst for water dissociation. Among them, one of the most remarkable is Ba5Ta4O15, which presents a (111)-layered perovskite structure, which reaches its maximum yield when it is impurified with a small amount of Ba0.5TaO3 and using NiOx as co-catalyst.97 However, the best results of pure water splitting are attained with perovskites of general formula MTaO3, where M represents an alkaline earth.98 Incorporation of K+ in the A position of the perovskite results in a rather poor photocatalyst, but, in contrast, a very high activity is observed for NaTaO3.26,27,98 Further improvement can be obtained if this semiconductor is doped with La76 due to the morphological changes of the semiconductor. Thus, lanthanum leads to a lower particle size and results in stepped NaTaO3 surfaces, which facilitates a physical separation of reduction and oxidation processes.99
Recently, it has been reported that Ga2O3 doped with Zn present a noticeable efficiency for water splitting using NiOx as co-catalysts.100 Non-oxidic semiconductors like β-Ge3N4 (phenacite structure) have also been applied. The photocatalytic cleavage of water is obtained with a quantum yield of 9% when using RuO2 as co-catalyst.101 Finally, among non-semiconductors photocatalyst, a significant rate for water dissociation can be achieved with the mesoporous material MCM-41 loaded with 0.3% of Ce, although oxygen generated is lower than stoichiometric amounts.102
4.3. Photocatalytic splitting of pure water under visible light
Efficient hydrogen production from water using sunlight and without sacrificial additives is one of the most challenging goals of photocatalysis and just a few materials have reported significant activity (see Table 2). Under broad range light source (310–800 nm) it has been reported that the semiconductor TiS2 (20% amorphous phase) generates H2 without co-catalyst with an energy efficiency of 4%, but O2 is retained on the solid.103 Similarly, it has been reported that CuAlO2, which present a delafossite structure (hexagonal) can reach a yield of roughly 10% for the photothermal conversion of solar energy into hydrogen, although formation of oxygen is not stated.104 This fact suggests that the process may not be entirely photocatalytic. Therefore, rather than the use of sunlight it would be desirable to measure the action spectrum of these two semiconductors to separate thermal from photochemical conversion.36| Photocatalyst | Band gap/eV | Co-catalysts | Conditions | Activity | Ref. | ||
|---|---|---|---|---|---|---|---|
| Sacrificial agent | Lamp | H2 production rate/µmol h−1 g−1 | QY (%) | ||||
| TiO2 | 3.2 | Rh | none | 500 W Xe | 1497 | 29 | 80 |
| HCa2Nb3O10 | 3.3–3.5 | Pt | CH3OH | 450 W Hg | 19000 | 79 | |
| ZnS | 3.66 | Pt | S2−/SO32− | 200 W Hg | 32500 | 90 | 83 |
| AgGaS2 | 2.6 | Rh | S2−/SO32− | 300 W Xe | — | 25.0 (440 nm) | 84 |
| CdS (nanostructured) | 2.25 | Pt | S2−/SO32− | 300 W Xe | 73330 | 60.3 (420 nm) | 23 |
| Ba:La2Ti2O7 | 3.8 | NiOx | none, NaOH soln | 450 W Hg | 5000 | 50 | 90 |
| BaLa4Ti4O15 | 3.85 | NiOx | none | 450 W Hg | 4600 | 15 | 94 |
| La:NaTaO3 | 4.0 | NiOx | none | 400 W Hg | 19800 | 56 | 99 |
| TiSi2 | 3.4–1.5 | none | none | halogen | 1000 | 4 | 103 |
| CuAlO2 | 3.01/1.87 | none | none | Sunlight 500 W m−2 | — | 10 | 104 |
| (Ga0.88Zn0.12)(N0.88O0.12) | 2.4–2.8 | Rh2−xCrxO3 | none | 300 W Xe | 3000 | 5.9 (420–440 nm) | 76 |
Purely photonic water splitting can be obtained with a narrow band semiconductor such as InTaO4, which has monoclinic wolframite structure.105 Optimal results are obtained with this semiconductor when indium is partly substituted by Ni, and NiOx is used as co-catalyst. However, as mentioned above, the higher photoactivity currently obtained under visible light (λ > 400 nm) corresponds to (Ga1−xZnx)(N1−xOx).25,77 Both constituent semiconductors, GaN and ZnO, present wurtzite structures and as the lattice parameters of both materials are comparable, a solid solution can be easily formed by heating the corresponding oxides in NH3. Surprisingly, the band gap of (Ga1−xZnx)(N1−xOx) is narrower (2.4–2.8 eV) than for the parent semiconductors and it shifts to longer wavelengths with increasing ZnO concentration.25,77 This fact provides the photo response in the visible range and it has been related to the repulsion between Zn 3d and N 2p levels in the valence band. Incorporation of a co-catalyst is crucial because the photoactivity of neat (Ga1−xZnx)(N1−xOx) is poor, but it is boosted remarkably with the addition of RuO2. However, the highest yield is achieved using Rh/Cr2O3 core/shell nanoparticles dispersed on the semiconductors surface.25,77 Therefore, optimization of the co-catalyst characteristics and interactions with the photoactive phase are key aspects for the development of photocatalysts for water splitting. On the other hand, the solid solution (Zn1+xGex)(N2−xOx) also shows a significant photoactivity for hydrogen production in the visible range using RuO2 as the co-catalyst.106
4.4. Future challenges of water splitting photocatalysts
Significant progress has been attained in recent years in the development of novel photocatalysts for water splitting. Nevertheless, performance, especially under visible light, has to improve considerably before the development of practical solar powered systems. In this respect, it is important to emphasize that many semiconductors are prepared at high temperatures and presumably they present low surface area, although, in general, this parameter is not stated. Surprisingly, attempts to synthesize these photocatalysts as nanostructures are still relatively scarce.85,96 Predictably, significant research effort will be devoted to enhance the photoactivity by means of morphological modifications of the new semiconductors. Furthermore, as progress achieved in this area has been attained by only a few groups, it is desirable to perform more ample investigations in order to fix the gained insight and to guarantee reproducibility of the characteristics of these novel photocatalysts. In particular, thorough investigations of the interaction between the semiconductor and the co-catalysts, which are frequently complex heterogeneous materials, are warranted.On the other hand, it is worth noting that if future photocatalysts are to be based on tantalum oxides or gallium nitrides, the scarcity of these elements could be a relevant issue. The price of tantalum oxide is strongly dependent on demand from the electronic industry, but it is about 60 times higher than TiO2.53 Furthermore, high purity gallium, which is mainly consumed in optoelectronic devices, has reached a value of $500 per kilogram.53 Similarly, the cost of noble metals used as co-catalysts may cause an economical drawback for the implementation of this technology.
5. Alternative photocatalysts for water and air detoxification
In this section we summarize the main achievements obtained with materials alternative to TiO2 in the three main fields of environmental photocatalysis: water treatment; gas decontamination and disinfection.5.1. Alternative photocatalysts for water detoxification
Recently, increasing research activity has been dedicated to investigating photocatalytic materials that differ from TiO2 for detoxification of aqueous effluents, most aimed to achieve more efficient usage of solar light. A considerable variety of material alternatives to TiO2, most of them oxides but also some sulfides, have been tested for aqueous pollutants degradation. Regarding pollutants, organic dyes are the most employed model compounds for photocatalytic activity tests. However, it should be noted that, when using these kind of molecules, dye sensitization of the semiconductor instead of direct excitation of the photocatalyst cannot be ruled out as a possible reaction mechanism.36 The mechanism when using these pollutants is not generally assessed experimentally, and further demonstration with other types of target compounds, especially when reactions are carried out under visible light, will sometimes be needed.The fundamentals of photocatalytic water detoxification have been reviewed.8–13 Unsurprisingly, the thermodynamic requirements of the photocatalyst for pollutant degradation are different to those for water splitting. For the mineralization of organic compounds, the commonest case, the reduction potential of the electrons in the conduction band must be negative enough to reduce adsorbed molecular oxygen to superoxide. As for the valence band holes, their reduction potential must be positive enough to react either directly with organic matter, or with OH−groups to produce strongly oxidizing OH˙ radicals.8–13
| Photocatalyst | Characterization | Photocatalytic activity | Ref. | ||||
|---|---|---|---|---|---|---|---|
| Structure | Band gap/eV | Surface area/m2g−1 | Pollutant | Conditionsa | Rate | ||
| a All the experiments were carried out in batch reactors. b Estimated by the authors from C/C0 − t data in the original reference | |||||||
| ZnO (Merck) | Wurtzite | — | — | Triclopyr (herbicide) | UV light (4 × 15 W fluorescent lamp) | r 0 (ZnO) = 1.54 × 10−5 mol L−1 min−1 | 109 |
| catalyst: 2 g L−1 | r 0 (TiO2) = 6.86 × 10−6 mol L−1 min−1 | ||||||
| pollutant: 10 mg L−1 | |||||||
| pH ≈ 4 | |||||||
| CeO2 | Fluorite | 2.95 | 42 | Acid orange 7 | Visible light (1000 W halogen lamp) | 1st order | 119 |
| cat.: 1 g L−1 | k′ = 0.011 min−1 | ||||||
| poll: 70 mg L−1 | k′ = krate Kads | ||||||
| pH = 2.96 | |||||||
| Pt–WO3 | Triclinic WO3 (2% Pt) | — | 16.4 | Phenol | UV light (125 W Hg lamp, 13.5 mW cm−2) | 1st order kobs (min−1) | 123 |
| cat: 2.4 g L−1 | Pt (2%)–WO3: 0.035: P25 TiO2: 0.025 | ||||||
| poll.: 50 mg L−1 | |||||||
| pH = 5.7 | |||||||
| BiVO4 | Scheelite (monoclinic) | 2.38 | 0.2 | 4-n-Nonylphenol | Visible light (500 W Xe lamp, 9 mW cm−2) | pseudo-first order kinetics | 128 |
| cat.: 8 g L−1 | O2 satur. soln: k = 0.027 min−1 | ||||||
| poll.: 2 × 10−4 M | |||||||
| pH = 13 | |||||||
| Bi2WO6 | Russellite (orthorhombic) | 2.69 | 0.64 | CHCl3 | Visible light (Xe lamp) | 1.2 × 10−7 mol L−1 min−1 (from CO2 formation) | 138 |
| cat: 1 g L−1 | |||||||
| poll: 0.12 M | |||||||
| pH? | |||||||
| Bi2MoO6 | Koechlinite (orthorhombic) | 2.59 | 4.55 | Rhodamine B | Visible light (Xe lamp) | 1st order k = 0.022 min−1 | 149 |
| cat: 1 g L−1 | |||||||
| poll: 5 mg L−1 | |||||||
| pH? | |||||||
| CaIn2O4 | Orthorhombic | — | 1.27 | Methylene blue | Visible light (Xe lamp) | initial MB degradation rate 1.88 × 10−6 mol L−1 min−1 | 151 |
| cat: 3 g L−1 | |||||||
| poll: 15.3 mg L−1 | |||||||
| pH? | |||||||
| BiOCl | Tetragonal | 3.46 | — | Methyl orange | UV light (300 W Hg lamp) | k (BiOCl) = 0.28 min−1 | 159 |
| cat: 2 g L−1 | k (P25) = 0.20 min −1b | ||||||
| poll: 10 mg L−1 | |||||||
| pH? | |||||||
| BiOBr | Tetragonal | 2.54 | 24.45 | Methyl orange | Visible light (Xe lamp) | k = 0.012 min−1b | 161 |
| cat: 1 g L−1 | |||||||
| poll: 10 mg L−1 | |||||||
| pH? | |||||||
ZnO has been frequently considered as an alternative to TiO2 for photocatalytic applications, since it shows similar activity in certain conditions.10 However, it suffers from anodic photocorrosion and, differently to TiO2, this reaction is not inhibited by water oxidation. In addition, it is soluble in strong acids and alkalis, which limits the pH range in which it can be used, as well as in the presence of Zn2+ chelating agents. These properties make ZnO scarcely practical for water treatment but, in spite of this, a considerable number of works have dealt with the degradation of aqueous pollutants using ZnO. Poulios et al. compared the photocatalytic activities of ZnO (Merck) and TiO2 (Degussa P25) for the degradation of aqueous solutions of the herbicide triclopyr (3,5,6-trichloro-2-pyridiyloxyacetic acid)109 with a higher initial degradation rate (r0) value for ZnO than for TiO2. However, the cited photocorrosion was observed in the case of ZnO, especially at low pH values. In addition, the mineralization extent was remarkably lower with ZnO which also occurred in degradation of the insecticide methyl parathion.110
Several approaches have been employed to tune the photocatalytic properties of ZnO and overcome its drawbacks. Colón et al. prepared nanosized ZnO samples by precipitation of Zn2+ with triethylamine followed either by a thermal treatment for crystallization or by hydrothermal treatment followed by calcination.111 In general, higher photocatalytic reaction rates were obtained with ZnO samples than with TiO2 Degussa P25, in spite of the low surface area of the ZnO samples. The authors attributed this elevated efficiency to the structural characteristics of the ZnO photocatalysts, which preferentially expose the unstable (100) face. The coupling of ZnO with other semiconductor has been also attempted in order to improve its photocatalytic properties. Thus, ZnO/SnO2 and ZnO/ZnO2 coupled oxides have been evaluated for photocatalytic degradation of aqueous methyl orange (MO) solutions,112,113 which resulted in faster degradation kinetics with respect to ZnO itself. The deposition of Fe2O3, WO3 and CdS onto ZnO substrates also led to higher activities compared to bare ZnO.114 The increase in activity in these coupled systems is ascribed to charge separation at the interface, as it has been described for other semiconductor couples.47,62
Similar to TiO2, another limitation of ZnO is its wide band gap (3.2 eV), which restricts light absorption to the UV region.10 A possible strategy to extend ZnO absorption to visible light may be the modification of its valence band position by anionic doping, as has been achieved for TiO263 Lin et al. used this approach to obtain visible light active ZnO powders by means of thermal plasma synthesis.115 As a result of N-doping, an absorption edge was observed in the visible region depending on the preparation conditions of the samples, but the influence of this on the activity under visible light (tested with MB) seems unclear. As far solar photocatalytic experiments are concerned, ZnO, Fe2O3 and TiO2 were evaluated under different irradiance conditions.116 Experiments carried out with a sodium lamp, as well as those conducted under non-concentrated solar radiation, resulted in phenol degradation rates in the order TiO2 > ZnO > Fe2O3. In contrast, when concentrated solar radiation (40–50 suns) was used, the reactivity order was ZnO ≈ TiO2 > Fe2O3. Phenol photocatalytic degradation under sunlight irradiation was also performed more recently by Pardeshi et al.117 In this work, a decrease in degradation efficiency was observed after successive runs due to the loss of ZnO caused by photocorrosion.
In summary, many works have dealt with aqueous pollutants degradation over ZnO describing reaction kinetics sometimes faster than those obtained with TiO2. Some chemical modifications succeeded in increasing the intrinsic photoactivity of ZnO or extending its response to visible light. However, these modifications do not overcome the practical limitations of ZnO mentioned above, which make ZnO hardly practical for water detoxification.
CeO2 has also received attention as a photocatalyst because of its interesting properties: stability under illumination and strong absorption of both UV and visible light. However, this material has been generally found to be less active than TiO2 under UV irradiation.107,118 However, in contrast with titania, this oxide can be activated by visible (violet) light. Ji et al. prepared mesoporous CeO2 photocatalyst using MCM-48 as a template in a replication process.119 The mesoporous materials showed a high surface area and a blue shift in their light absorption with respect to bulk CeO2, as observed in their UV–vis spectra. In the photocatalytic degradation of Acid Orange 7 (AO7) under visible light, both bulk and mesoporous CeO2 led to faster degradation of the dye than TiO2 P25 from Degussa, but the mesoporous sample presented the best performance. No degradation of AO7 was observed in the absence of catalyst. More recently, the same group investigated the photocatalytic degradation of AO7 under visible light (λ > 420 nm) over CeO2nanoparticles.120 Higher activity is found in CeO2 with respect to TiO2 (P25), which is attributed to a higher adsorption capacity of the lanthanide oxide, with no comments on the influence of the different light absorption onset of both catalysts. The degradation of the dye Acidic Black over CeO2 under solar irradiation has also been studied.121
WO3 and SnO2 themselves have been scarcely used as photocatalysts for water detoxification, but they have been more widely employed as an additive to improve the photocatalytic behaviour of TiO2.62 Lettmann et al. compared the photocatalytic activities under visible light of series of doped TiO2, WO3 and SnO2 obtained by a combinatorial high-throughput synthesis technique.122 22 out of 71 doped SnO2 samples presented photocatalytic activity for the degradation of 4-chlorophenol under visible light irradiation, while bare SnO2 was totally inactive, as expected from its wide band gap energy (3.88 eV). Transition metals (e.g.Cr, Mn, Ru, Ir), rare earths (Ce, Tb, Ho) and main group metals (Bi, Ca) were identified as positive cases of SnO2 doping. Undoped WO3 was found to be inactive towards the same reaction, and only the Ir- and Cr-doped WO3 photocatalysts degraded the pollutant. As illustrated by this example, and in spite of its interesting electronic properties, bare WO3 presents a generally low photocatalytic activity for organic pollutants degradation that has been related to a high electron–hole recombination rate and/or to the difficulty to photoreduce O2 due to overpotential effects.123 Both doping122 and Pt loading123,124 have been tested to overcome these limitations with positive results. Yet, degradation rates of aqueous phenol obtained with Pt-loaded WO3 were slightly lower than those with TiO2, and mineralization was low.123 On the other hand, WO3 has been used for simultaneous photocatalytic reduction of Cr(VI) and oxidation of the MB dye in aqueous solutions.125
Some metal sulfides have shown high photocatalytic activities, but due to the already mentioned anodic photocorrosion, these materials are not relevant for aqueous pollutant degradation. In fact, the possible leaching of metal cations (e.g.Cd2+) from these semiconductors can be detrimental for the environment. Nevertheless, several works have been devoted to the study of these photocatalysts, among which the application of ZnS to the reduction of nitrates and nitrites,126 or the oxidation of hexafluorobenzene127 can be cited.
Chemical modification of BiVO4 has been studied as a way to improve its photocatalytic properties. Kohtani et al. have applied Ag-loaded BiVO4 to the photocatalytic degradation of various aqueous pollutants.133,134 In these materials, depending on the preparation method, Ag or Ag/AgO/Ag2O loading may be obtained. Ag-loaded vanadate showed a higher activity for 4-n-octyl- and 4-n-nonylphenol degradation than BiVO4, as well as a higher mineralization degree. Apparently, the increased activity is related to strong adsorption of the organic molecules on the silver oxide species covering the surface of Ag nanoparticles. Similar enhancement of the photoactivity of BiVO4 has been also observed for the degradation of different polycyclic aromatic hydrocarbons.134 In the same line, Ge has recently employed Pd- and Pt-loaded BiVO4 photocatalyst for the abatement of MO with better results than with the unloaded vanadate. 135,136 This improvement was ascribed to the enhancement of charge separation due to metal loading. CuO-loaded BiVO4 has also given rise to a faster degradation of MB than bare BiVO4.137
The photocatalytic activity of Bi2WO6 has been studied by several research groups. Tang and co-workers reported that Bi2WO6, which presents orthorhombic Aurivillius-type structure and has a band gap of 2.69 eV, could degrade aqueous CHCl3 under visible light (λ > 420 nm).138 Total mineralization of trichloromethane was achieved after illumination for 200 h. Zhu's group has studied the degradation of dyes over Bi2WO6 nanoplates prepared by hydrothermal synthesis.139 In comparison to Degussa P25 TiO2, much faster kinetics were obtained with Bi2WO6 nanoplates.140 Moreover, no deactivation of the tungstate catalyst was observed after 5 photocatalytic runs, although a drastic loss of activity occurred at low pH values due to the decomposition of Bi2WO6 into H2WO4 and Bi2O3. Spin-trapping electron paramagnetic resonance (EPR) experiments suggest that oxidation of organic matter occurs directly by the valence band holes at Bi2WO6. Modifications of these Bi2WO6 nanosized photocatalysts by both C60 deposition141 and F-doping142 have been successfully assayed to increase their activity. Electron migration from the conduction band of Bi2WO6 to the conjugated π system of C60 molecules improves charge separation and results in higher rates for dyes degradation under visible light.141 In fluorinated Bi2WO6, concurrence of bulk-doping and surface modification by F− produces a beneficial synergetic effect.142 Control of the morphologies of Bi2WO6 photocatalysts for water treatment have recently received considerable attention. Flower-shaped superstructures of this tungstate were obtained by Zhang et al. by a simple hydrothermal process without surfactants or templates.143 Apart from their visible light absorption, high surface area and transport paths created by the porous system of the superstructures were invoked to account for the high activity of flower-like Bi2WO6 particles (Fig. 9). Similarly, Wuet al. prepared “nest-like” hierarchical structures based on the crystal growth modifying effect of the polymer polyvinylpyrrolidone (PVP). They showed remarkable activity for visible-light photocatalytic degradation of RhB in aqueous solution, considerably higher than that of single Bi2WO6 nanoplates and TiO2 (P25).144 Amano and co-workers prepared similar Bi2WO6 “flake-ball” hierarchical structures by means of a hydrothermal method without structure-directing agents.145,146 Interestingly, the flake-ball particles could induce the decomposition of acetic acid under visible light (λ > 400 nm) irradiation, whereas TiO2 ST-01 showed negligible activity for this reaction under the same conditions.
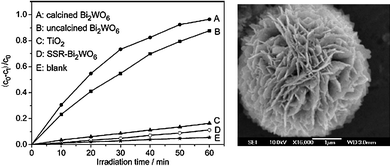 | ||
| Fig. 9 Left panel: Photocatalytic degradation of aqueous rhodamine B over flower-shaped Bi2WO6 (A); uncalcined (B); TiO2 Degussa P25 (C); and solid-state synthesized Bi2WO6 (D). Right: SEM image of uncalcined hydrothermally treated Bi2WO6 superstructure. Figure adapted from ref. 143. | ||
Other tungstates have also been investigated for aqueous pollutants degradation. Zhu's group compared the activities of ZnWO4 and PbWO4 with that of Bi2WO6 for RhB degradation under simulated solar light and visible light irradiation.147 Under both illumination conditions, ZnWO4 and PbWO4 exhibited lower activity than Bi2WO6 for this reaction. MO and RhB were used to evaluate the photocatalytic activity of CdWO4 for aqueous pollutant degradation.148 Its activity was similar to that of ZnWO4 for both reactions and was also similar to that of TiO2 under UV irradiation, but activity slightly declined after 5 photocatalytic runs.
Isostructural to Bi2WO6, Bi2MoO6 has been also studied for wastewater treatment. Martínez de la Cruz et al. investigated the photocatalytic degradation of RhB over nanosized Bi2MoO6.149 Different to other cases, the band gap energy values (2.33–2.59 eV) observed in nanosized samples were lower than those of solid-state synthesized Bi2MoO6 (2.64 eV), and increased with increasing crystal size. The best photoactivity observed for the degradation of RhB molecules corresponded to a Bi2MoO6 sample with 38 nm crystal size. Belver and co-workers also used RhB abatement to compare the photocatalytic activity of Bi2WO6 and Bi2MoO6 prepared by the Pechini technique.150 Bi2WO6 showed higher activity for this reaction than Bi2MoO6 under both UV–vis and visible light irradiation. In summary, these tungstates and molibdates appear interesting alternatives to TiO2 for visible-light photocatalytic detoxification, although further assessment of their activities with light insensitive target compounds seems necessary. Nevertheless, the results obtained with CHCl3 and acetic acid are very promising.
Alkali earth metal indates have been also studied for aqueous-phase photocatalytic reactions in several works. Tang et al. prepared MIn2O4 (M: Ca, Sr, Ba) photocatalysts by solid-state reaction which exhibited higher photocatalytic activity than TiO2 P25 for the degradation of MB dye under visible light, with CaIn2O4 showing the highest activity.151 DFT calculations showed that the smaller the radius of M2+ in MIn2O4 the higher the oxidizing power of the conduction band, which agrees with the order of photocatalytic activities. CaIn2O4/In2O3 core–shell composites were found to be more active than CaIn2O4 due to charge separation at the interface of the semiconductors.152 Some other mixed oxides that have been studied for photocatalytic degradation of aqueous pollutants are titanates,153,154 stannates,155 ferrites156 and titanoniobates.157
5.2. Alternative photocatalysts for air treatment
The destruction of pollutants in gas phase is another target for photocatalytic research. The photocatalytic treatment of air mainly centres on the mineralization of volatile organic compounds (hydrocarbons, chlorinated compounds, alcohols, molecules containing nitrogen and sulfur or siloxanes) and inorganic compounds (NO, NO2, N2O or O3) present at ppm or ppb concentrations. 8–13,162 Although air treatment has received relatively less attention than other applications, this is a rapidly changing scenario and the use of photocatalysts as an alternative to titania for gas phase decontamination is gaining momentum. Consequently, in this section, we describe alternative photocatalysts proposed for air applications, highlighting those studies where enough data to compare with TiO2 has been reported (Table 4).| Photocatalyst | Characterization | Photocatalytic activity | |||||
|---|---|---|---|---|---|---|---|
| Structure | Band gap/eV | Surface area/m2g−1 | Pollutant | Conditions | Photocatalytic activity (when possible compared to TiO2) | Ref. | |
| N–ZnO | Wurtzite | 2.5 | 59 | Acetaldehyde decomposition | UV light: 200W Hg–Xe Vis: 150 W Xe lamp | r 0 = 2.3 10−7 mol min−1 | 171 |
| Close circulation system (250 cm3) | r 0 = 1.5 10−8 mol min−1 | ||||||
| Catalysts: 0.05 g | |||||||
| Pollutant: 730 ppm | |||||||
| Pd/ZnO | Wurtzite | — | — | n-C7H16 | 400 W Hg lamp | C/Co(3 h) = 0.8 | 164 |
| Catalyst: 0.1 g | |||||||
| Reactor: 300 mL | |||||||
| ZrO2 | Tetragonal | — | 289 | Methanol oxidation | 8W UV lamp | X(methanol) = 31.6% | 165 |
| Hexane oxidation | Catalyst: 0.3 g | X(P25) = 40% | |||||
| Methanol: 1100 ppm. Hexane: 1700 ppm | X(hexane) = 31.6% | ||||||
| GHSV = 4000 h−1 | X(P25) = 17% | ||||||
| Ga2O3 | Monoclinic (α) | 4.56 | 58 | Benzene, toluene, ethylbenzene oxidation | 4-W UV lamp | β-Ga2O3 | 166 |
| Rhombohedral (β) | 4.7 | 80 | Benzene: 450 ppm. Toluene: 450 ppm. Ethylbenzene: 350 ppm | r 0(C7H8) = 0.52 µmol h−1m−2 | |||
| Cubic (γ) | 4.67 | 135 | Total flow rate = 20 ml min−1 | r 0(TiO2) = 0.15 µmol h−1m−2 | |||
| NiO/SrBi2O4 | Monoclinic | — | — | Acetaldehyde oxidation | Vis light: 350 W Xe lamp | C/C0(60 min) = 0.4 (1 wt % Ni) | 169 |
| Catalyst: 0.5 g | C/C0(P25) = 0 | ||||||
| Reactor: 550 ml | |||||||
| CH3CHO = 15 ppm | |||||||
| NaTaO3 | Cubic | 3.96 | — | Formaldehyde oxidation | UV light: 8 W lamp | C(30 min) = 100 ppm | 180 |
| Reactor volume: 400 ml | |||||||
| Catalyst: 0.06 g. | |||||||
| Pollutant: 1400 ppm | |||||||
| N–SrTiO3 | — | 3.18 | 14 | NO destruction | Vis light: 450W high pressure mercury arc. | X(NO) = 48% | 175 |
| 2.64 | Reactor: 375 ml | X(P25) = 34% | |||||
| Pollutant: 1 ppm | |||||||
| Flow rate: 200 ml min−1 | |||||||
| SrTiO3 (N,La) | Perovskite | 1.4 | 7–8 | 2-Propanol to acetone | Vis light: 250W xe lamp. UV 10W black light | N,LaSrTiO3 > SrTiO3 | 176 |
| Catalyst: 0.3 g | |||||||
| Reactor: 800 ml | |||||||
| Pollutant: 500 ppm | |||||||
| LaTiO2N | — | 2.41 | 26 | Acetone decomposition | Vis light: 150W Xe lamp | X(600 min) = 35% | 177 |
| Pollutant: 5 µl | |||||||
| Ag+/NbO2F | Cubic | 3.0 | 3 | 2-Propanol oxidation | UV-vis: Xe lamp | Vis: QE = 0.27% | 182 |
| Catalyst: 0.3 g | |||||||
| Reactor: 500 ml | |||||||
| Pollutant: 300 ppm | |||||||
| Ag+, Cu+/ZSM-5 | — | — | — | NO decomposition | UV irradiation, Hg lamp | Ag+/SZM5(600 min) | 187 |
| Yield N2 = 60 µmol g−1 | |||||||
| Cu+/ZSM-5 | |||||||
| Yield N2 = 5 µmol g−1 | |||||||
| Ag+, Cu+/ZSM-5 | — | — | — | N2O decomposition | UV irradiation, Hg lamp | Ag+/ZM5(250 min) | 189,190 |
| Yield N2 = 35 µmol g−1 | |||||||
| Cu+/ZSM-5 | |||||||
| Yield N2 = 17 µmol g−1 | |||||||
| M–Al–MCM-41 (Co, Cr, Mn, Cu) | — | — | 910–1190 | Acetaldehyde decomposition | UV-vis irradiation 1000W high pressure Hg lamp | UV Light(60 min) | 191 |
| Pollutant: 100 µl | −ln(Co/C) (P25) = 4 | ||||||
| −ln(Co/C) CrMCM41 = 3.5 | |||||||
Zirconia has also been used as a photocatalyst for gas phase applications although, due to its wide bad gap (5.1 eV), it has received less attention. In this way, amorphous ZrO2 photocatalysts synthesized at low temperature present better performance than TiO2 P25 for hexane photooxidation.165 Similarly, it has been found that β-Ga2O3 is a highly photoactive oxide for the mineralization of gaseous aromatic compounds (benzene, toluene, ethylbenzene) to CO2 and H2O, under dry air streams without the catalyst deactivation observed for TiO2.166 This polymorph of Ga2O3, which presents a monoclinic structure shows better performance than TiO2, although its large band gap (4.7 eV) constitutes an important limitation. On the contrary, photocatalytic oxidation of toluene vapours over CeO2 is less efficient than over TiO2, although mineralization of toluene is almost complete.118Bismuth compounds, such as SrBi2O4, are good candidates for visible light photocatalysis because the hybridation of the Bi 6s with O 2p orbitals push up the valence band position which reduces the band gap.167,168 This hybridation turns the valence band to largely dispersing, and this favours the mobility of holes. In this way, NiO/SrBi2O4 is used for the oxidation of acetaldehyde.169 The addition of 1.0% of NiO significantly improves the oxidation rate of acetaldehyde to CO2. The ESR spin-trap technique indicates that O2˙− and OH˙ radicals are produced on the surface of the NiO/SrBi2O4 and constitute the active species in the photocatalytic reaction.
SrTiO3 is another interesting photocatalyst, but it can be only activated in the UV region because of its high band gap.174 Therefore, extending the high reactivity of this material to the visible range has become a relevant research target. With this aim SrTiO3 has been doped with N, F or La and its photocatalytic activity has been evaluated mainly for NO destruction and 2-propanol oxidation. Nitrogen doped SrTiO3 has been prepared by a mechanochemical method by Wang et al. using different N-doping molecules and it has been tested for NO removal under visible light.175 The photocatalytic performace of this material was 1.4 times higher than commercial TiO2 P25. Further studies with higher NO concentrations are required to analyse potential applications for this material. For SrTiO3−xFx the substitution of O2− by F− is compensated by the formation of Ti3+ centres to maintain the electroneutrality, contributing to the higher light absorption in the visible range.175 Similarly Sr1−xLaxTiO3−xNx codoped with La and N were evaluated for photocatalytic oxidation of 2-propanol to acetone under UV and visible irradiation.176 The decrease of the number of oxygen vacancies which act as electron–hole recombination centres explains the higher performance of this material compared to SrTiO3−2xNx.
Nanoparticles of LaTiO2N with perovskite structure have been tested for decomposition of acetone under visible light.177 Spherical particles were obtained by NH3 treatment at temperature of 800°C and 1000°C with size of 10 nm and 40–50 nm, respectively (see Fig. 10). In order to explain the photocatalytic performance properties surface area and band gap size were discussed by the authors, although no definite conclusions were drawn. On the other hand, K2Ta2O6−xNx with a pyrochlore structure178 and the perovskite NaTaO3−xNx179,180 were used for formaldehyde degradation under visible light (λ > 400 nm).
 | ||
| Fig. 10 Transmission electron micrographs of the oxynitride nanoparticles ammonolyzed at (a) 800 °C and (b) 1000 °C. In (c) a high resolution TEM image of the sample treated in NH3 at 800 °C is shown. In (d) a typical electron energy loss spectrum clearly shows the presence of nitrogen in the crystallites. (Reproduced from ref. 177). | ||
Oxyfluorides with general formula MO2F are electrochromic materials with perovskite structure, ABO3, where the A site is vacant. NbO2F is more stable that TiOF2.181 This material can adsorb only UV light, but the incorporation of Ag+ to the vacant A site has been proposed to develop visible light activity.182 The shift of the photo response is due to the formation of a hybrid orbital between Ag 4d and O 2p in the valence band, which displaces the top of the band to a more negative potential.183 Consequently, Ag+/NbO2F can decompose gaseous 2-propanol to CO2 with acetone as an intermediate product under 400–530 nm light irradiation. β-SiC nanowires coated with amorphous SiO2 exhibited excellent photocatalytic activity in the photodegradation of acetaldehyde with UV irradiation. This material displays higher photocatalytic activity than bare SiC through the stronger adsorption of gaseous acetaldehyde and higher probability of trapping the excited electron in the conduction band.184
In order to shift the photoactivity to the visible region, alkali ion modifications of the highly dispersed V-oxides species was studied (λ > 390 nm).185,186 The incorporation of Na+, K+, and Rb+, led to photocatalytic activity for the partial oxidation of hydrocarbons under visible light irradiation. This was due to the formation of V-oxides species where two V–O bonds interact with an alkali metal ion.
Ag+/ZSM-5 presents high photoactivity in the decomposition of NO although N2O and NO2 were also detected. The electron transfer from excited Ag+ ion into the π antibonding molecular orbital of the NO molecule plays a significant role in the photocatalytic decomposition of NO.187 The higher performance of this material compared to Cu+/ZSM-5188 was assigned to the elevated chemical stability of the Ag+ ion and its efficient interaction with the NO molecule. Moreover, the performance of these materials in the photocatalytic decomposition of N2O into N2 and O2 has been studied and compared to the Ag+/Y-zeolite structure. 189,190In situ spectroscopic investigation of the Ag+/zeolite catalysts indicated that two-coordinate isolated Ag+ ions exist within the ZSM-5 zeolite cavities, while the aggregated Ag species were formed within the Y-zeolite. The analysis of the efficiency as a function of the wavelength of the UV light source indicated that photoexcitation of the Ag+–N2O adducts (λ ∼ 225 nm) is the key step of the photocatalytic decomposition of N2O.
Mesoporous materials, discovered by Mobil scientists in 1992, opened up new possibilities for modulating the selectivity in catalytic reactions. MCM-41 is one of the most commonly used mesoporous materials due to its large surface area and its arrangement of hexagonally ordered pores with diameters ranging from 2 to 10 nm. Owing to the lack of active sites in pure siliceous mesoporous solids, much effort has been devoted to increasing the activity by chemical modification. Mesoporous silicas incorporating different metals have been investigated as photocatalysts, including V and Mo,28Mn,191Cr,28,191,192Cu,191Co,191Ag,193 UO2+.194 Cr–Al–MCM-41 has been used in the photocatalytic degradation of acetaldehyde191 and trichloroethylene in the gas phase using UV and visible light.192 The Cr6+/Cr3+ redox couple plays a key role in the photocatalytic performance of this material, although it is lower than that of TiO2 P25 under UV illumination. Cr–HSM containing mesoporous silica shows photocatalytic reactivity in the decomposition of NO into N2 and O2 and the partial oxidation of propane under UV and visible light.28,195 AgBr/Al–MCM-41 has been also tested in the photodegradation of acetaldehyde obtaining similar results for both UV and visible light.193 Anpo et al. analysed the photocatalytic activity of Ti–, V–, and Mo–MCM-41 in the NO decomposition reaction.28Ti–MCM-41 showed the best results but in the presence of propene V– and Mo–MCM-41 displayed remarkable increases in photoactivity.28 The charge transfer excited states of V- and Mo-oxide species exhibited high reactivity for hydrogen abstraction from propane, accounting for the high efficiency.
5.3. Disinfection with alternative photocatalysts to TiO2
Disinfection implies the elimination of pathogenic microorganisms from water, air or surfaces. These organisms include bacteria, viruses, fungi, protozoa and algae. Chlorination, usually applied as NaClO, is the most widespread and cost-effective method for water disinfection, but it is not adequate to inactivate some spores and viruses.8 Furthermore, chlorinated chemicals may cause corrosion and are not appropriate for certain applications like air or culture media disinfection. Consequently, there is considerable interest to develop alternative technologies for microorganisms’ deactivation, especially if they do not involve the consumption of chemicals. Within this scenario in the last decade photocatalysis has emerged as a promising technology.196 The first heterogeneous photocatalytic disinfection was reported by Matsunaga et al. in 1988,197 although previously studies by the same group had probed the efficiency of Pt/TiO2 for this purpose, using using a photoeletrochemical process.198 Currently, research in this field is still dominated by TiO2.196 Only a few studies in the literature relate to disinfection by photocatalysts different from TiO2. The most relevant results are discussed in this section.ZnO presents similar photoactivity to TiO2 for the inactivation of a variety of microorganisms, although some fungi, like Aspergillium niger, show considerable resistance to this treatment.199NiO/SrBi2O4 powders prepared by co-precipitation methods were evaluated in the photocatalytic degradation of Escherichia coli under visible light irradiation and compared to TiO2 P25 and NiO/P25 (λ > 420 nm).169 The results showed that the monoclinic structure SrBi2O4 demonstrated visible light activity, which was greatly enhanced when NiO was loaded onto the semiconductor by impregnation. This was attributed to NiO promotion of electron–hole separation. The determination of intracellular K+ leakage originating from the inactivation of E. coli was verified as a consequence of the damage to the outer membrane of the cell. In contrast, no changes in K+ were observed for TiO2 P25 photocatalysts.
Clay materials have also been used as heterogeneous photocatalys for disinfection. Wyoming smectite clay minerals were used for this purpose with different types of metallic cations covering the surface: Ag+, Zn2+,Ti4+. 200 When the smectite catalysts were illuminated with a sodium lamp, they reduced the number of viable bacteria in surface water and achieved a disinfecting effect. On the whole, smectite–Ti catalysts had the best disinfection efficiency, followed by coupled smectite–Ag/Zn catalysts, smectite–Ag catalysts and smectite–Zn catalysts. UV-vis absorbing ruthenium(II) tris–chelate complex (2 g m−2) immobilised onto porous silicone, was studied as a generator of singlet molecular oxygen.201 This excited state (1Δg) of molecular oxygen, abbreviated as 1O2, is cytotoxic and can be exploited for disinfection. This type of photocatalyst used a CPC collector where the catalysts were configured in different geometries. E. coli (Gram negative) or Enterococcus faecalis (Gram positive) were subject to photocatalytic treatment for 5 h. Using a fin-type reactor and 104 to 102 CFU mL−1 initial concentrations, bacterial survival dropped to ca. 1% and 0.1% for E. coli and E. faecalis, respectively.
Dihydroxo and dimethoxo(tetraphenylporphyrinato) phosphorus(V) complexes, ([P(OR)2tpp]+) were immobilized on silica-gel powder and silica-gel beads affording visible-light driven photocatalysis.202 The bactericidal effect of these complexes on Escherichia coli was investigated. In the case of the hydroxo-complex, the amount of E. coli decreased linearly versus irradiation time and was more effective for sterilization than when methylated. However deactivation took place, most probably due to bacterial adsorption on the catalyst, as is suggested by kinetic analysis.
6. Alternative photocatalysts for organic synthesis
The use of photocatalysis for synthetic purposes, which allows reactions to be performed under mild conditions, has proved a promising alternative to conventional industrial methods, both for economical and environmental reasons; being especially attractive if solar light can be used as the irradiation source. Photocatalysis has been successfully applied to reduction processes (studies of CO2 photo-reduction are described in Section 7 because of their relevance to global warming and energy storage), isomerization reactions and the formation of C–C and C–N bonds.203–205 However, research on the use of this technology for the production of chemicals has been mainly focused on the oxidation of hydrocarbons by molecular oxygen,34 with most studies dealing with oxofunctionalization of aliphatic and aromatic alkanes and alkenes. To the best of our knowledge, photocatalysis is the only catalytic process able to oxidize light alkanes under ambient conditions to the corresponding carbonylic compounds. Photo-assisted oxidation of alcohols to obtain the corresponding ketones or aldehydes is also a matter of great interest, because of its potential industrial applications, which would prevent the use of strong oxidants and the generation of heavy metal wastes.206However, the mineralization effectiveness of TiO2 plays a negative role in synthesis reactions, due to the low selectivity to partial oxidation products often exhibited. Nevertheless, many other photocatalysts have been used in selective reactions to produce organics, although sometimes only to obtain very low quantum yields. Usually, for a specific catalytic reaction, an increase in selectivity is observed as the conversion decreases.207 It is worth pointing out the crucial role that H2O plays in this respect. In its presence total oxidation is promoted due to the formation of strong oxidizing OH˙ radicals, while in the absence of water different active species are generated, e.g. atomic oxygen species, O˙, that favours mild selective oxidation processes.208 The selection of semiconductor for a specific reaction is essential, because it will control not only the conversion but also the selectivity of the process, partly due to the different adsorptive properties of the photocatalyst surface. The location of the valence and conduction bands (see Fig. 4) also plays a key role in selectivity. For instance, Harada et al. showed that pyruvic acid was produced from lactic acid with a Pt/CdS photocatalyst, while with a Pt/TiO2catalyst decarboxylation took place and acetaldehyde was formed.209 The study of selective oxidation by other α-hydroxycarboxylic acids proved that, depending on the semiconductor used, evident differences in the product distribution could be obtained.210
This section summarizes the achievements of photocatalysis as a synthetic technology to produce chemicals with high selectivity using photocatalytic systems which do not contain TiO2. As the most studied class of reactions, photo-oxofunctionalization is preferentially taken into account. However, it is worth mentioning that any comparison, in quantitative terms, of the efficiency of the photocatalysts used in different studies is a difficult task, since reaction conditions differ from one to another and reference photocatalysts are not always used.
6.1 Oxides, sulfides and other semiconductors
Semiconductors such as ZnO, ZnS, CdS, SrTiO3, MoS2, GaP are frequently used as photocatalysts. However, until now, the number of studies for synthetic applications is much lower than those dealing with complete mineralization.In Table 5, some examples of the application of oxides, sulfides and other semiconductors as alternative photocatalysts for synthetic purposes are detailed. The synthesis of ammonia from water and nitrogen was favoured when the conduction band energy of the semiconductor became more negative.212 Higher ammonia yields were obtained when GaP and CdS were used, in comparison to those of TiO2 or ZnO (see Table 4). This behavior is explained by the greater ability of the photoexcited electron to reduce nitrogen in a more negative conduction band. Larger yields were reached when Pt black was also incorporated into the catalyst. Ammonia has been also obtained on SrTiO3- and BaTiO3-based catalysts,213although none managed to improve the yields of ammonia reported in the previously mentioned work.212
| Reactants | Product | Conditions | Catalyst | Yield | Ref. |
|---|---|---|---|---|---|
| μmol 0.3 g−1 cat (5 h) | |||||
| N2 + H2O | Ammonia (NH3) | 100 W Hg lampT = 311 °C | TiO2 | 1.6 | 212 |
| SrTiO3 | 1.9 | ||||
| ZnO | 2.1 | ||||
| CdS | 3.2 | ||||
| GaP | 4.6 | ||||
| GaP–Pt | 7.5 | ||||
| μmol 0.5 g−1 cat (3 h) | |||||
| N2 + H2O | Ammonia (NH3) | 450 W Hg lamp T = 323 °C | SrTiO3 | 0.41 | 213 |
| BaTiO3 | 0.87 | ||||
| RuO2–NiO–SrTiO3 | 2.51 | ||||
| RuO2–NiO–BaTiO3 | 2.61 | ||||
| μmol 0.3 g−1 cat (5 h) | |||||
| Glycolic acid + H2O | Glyoxylic acid (HCOCOOH) | 500 W Xe lamp | Pt/CdS | 36 | 210 |
| (H2C(OH)COOH) | T not measured | Pt/TiO2 | 494 | ||
| μmol 0.25 g−1 cat h−1 | |||||
| Methane (CH4) + O2 | Formaldehyde (HCHO) | 200 W Hg lamp | ZnO | 3.1 | 214 |
| T = 493 K | MoO3/ZnO | 4.8 | |||
| Ethane (CH3CH3) + O2 | Acetaldehyde (CH3CHO) | ZnO | 84 | 215 | |
| 200 W Hg lamp | TiO2 | 5.1 | |||
| Propane (CH3CH2CH3) + O2 | Propanal (CH3CH2CHO) | T = 493 K | ZnO | 57 | |
| TiO2 | 0.3 | ||||
| Ethene (CH2CH2) + O2 | Formaldehyde (HCHO) | 200 W Hg lamp | ZnO | Trace | 216 |
| T = 493 K | MoO3/ZnO | 7 | |||
| TiO2 | Trace | ||||
| μmol 0.1g −1 cat (24 h) | |||||
| 1-Pentanol + O2(CH3(CH2)3CH2OH) | Pentanal (CH3(CH2)3CHO) | 500 W Hg lamp | Nb2O5 | 2.43 (97% select.) | 211 |
| T = 323 K | TiO2 | 2.85 (83% select.) | |||
Selective photo-oxidation of light alkanes into oxygen-containing derivatives has been a subject of great interest. MoO3-loaded ZnO photocatalysts have been successfully used in the selective conversion of methane into formaldehyde214 at 493 K, improving the yield obtained with bare ZnO and hindering almost completely CO2 formation. On the other hand, ZnO was found to be one of the best catalysts for the photo-oxidation of ethane and propane with selectivities over 70% at 493 K, while TiO2 predominantly yielded carbon oxides.215 Furthermore, ZnO-based photocatalysts were also tested for the photo-oxidation of light alkenes, propene and ethane, and the results were similar to those previously obtained for methane, so that MoO3 or V2O5 loaded on ZnO greatly improved the selectivity for oxygenated organics, while with titania, mainly carbon dioxide was produced.216
Silicon nanostructures have showed promising results as photocatalysts. Kang et al. reported the photoactivity of Si quantum dots (SiQDs), proving that they can be use either for reduction, decomposition or selective oxidation reactions, due to their tunable size and band gap.217 Finally, CdS has also been utilized for the photocatalyzed synthesis of thio–organic compounds,218 the selective cyclization of amino acids219 and the formation of bromo-derivatives from phenol.220
6.2 Supported systems
Non-semiconducting metal oxides, like alumina and silica, have been frequently used for supporting photocatalysts. However, in the 1970s, Hubbard et al. used these materials as model to study the likelihood of the photocatalyzed production of biologically important compounds (e.g.formic acid, ammonium cyanate) under UV irradiation, as a part of the investigation of the pre-biotic synthesis on primitive Earth or on the surface of Mars.221–223Regarding the use of supported metal oxides as photocatalysts for organic synthesis, most of the studies that can be found in literature deal with the selective conversion of light alkanes to other valuable products of industrial interest. However, it is important to note that most of the works that have been cited have been produced by a very limited number of research groups and, consequently, the reproducibility is yet to be confirmed. A summary of some of the examples found in the bibliography are displayed in Table 6.
| Reactants | Product | Conditions | Catalyst | Yield | Ref. | |
|---|---|---|---|---|---|---|
| a Different preparation methods. b Irradiation time = 1 h. | ||||||
| μmol h−1 | ||||||
| Methane (CH4) | Formaldehyde (HCHO) | 200 W Hg lamp | SiO2 (0.050 g) | Trace | 224 | |
| T = 463 K | ||||||
| 200 W Hg lamp | MoO3(5 wt%)/SiO2 (0.025 g) | 5.8 | ||||
| T = 493 K | ||||||
| Ethane (CH3CH3) | Acetaldehyde (CH3CHO) | 200 Hg lamp | MoO3(2.5 wt%)/SiO2 (0.025 g) | 60 (CH3CHO) | 225 | |
| Formaldehyde (HCHO) | T = 493 K | 22 (HCHO) | ||||
| μmol 0.025 g−1 cat h−1 | ||||||
| Methane (CH4) | Formaldehyde (HCHO) | 200 W Hg lamp | V2O5/SiO2a | 62 | 227 | |
| Ethane (CH3CH3) | Acetaldehyde (CH3CHO) | T = 493 K | V2O5/SiO2a | 62 | ||
| Conversion (%) | Selectivity (%) | |||||
| Propane (CH3CH2CH3) | Acetone (CH3COCH3) | 250 W Hg lamp | V2O5/SiO2 | λ 1 = 63.4 | λ 1 = 16 | 186 |
| λ 1 > 310 nm | λ 2 = 5.0 | λ 2 = 15 | ||||
| λ 2 > 390 nm | Na–V2O5/SiO2 | λ 1 = 83.8 | λ 1 = 33 | |||
| λ 2 = 60.3 | λ 2 = 52 | |||||
| K–V2O5/SiO2 | λ 1 = 85.2 | λ 1 = 37 | ||||
| λ 2 = 67.0 | λ 2 = 51 | |||||
| Rb–V2O5/SiO2 | λ 1 = 88.5 | λ 1 = 33 | ||||
| λ 2 = 67.1 | λ 2 = 60 | |||||
| Propene (CH3CHCH2) | Propene oxide (CH3CHCH2O) | 250 W Hg lamp | SiO2 | — | 28.9 | 233 |
| mcat = 0.1 g | Mg/SiO2 | — | 50.8 | |||
| 250 W Hg lamp | Nb2O5/SiO2 | — | 33 | 232 | ||
| mcat = 0.5 g | ||||||
| 200 W Xe lamp | ZnO | 1.25 | 1.9 | 235 | ||
| mcat = 0.2 g | ZnO/SiO2 | 8.61 | 33.4 | |||
| 200 W Xe lamp | SiO2 | 1.5 | 22.3 | 236 | ||
| mcat = 0.2 g | TiO2b | 14.1 | 0.0 | |||
| LiOx/SiO2 | 0.8 | 37.9 | ||||
| MgOx/SiO2 | 4.7 | 22.8 | ||||
| SrOx/SiO2 | 1.2 | 37.0 | ||||
| TiOx/SiO2 | 24.4 | 19.2 | ||||
| ZnOx/SiO2 | 8.6 | 33.4 | ||||
| PbOx/SiO2 | 8.2 | 26.9 | ||||
| BiOx/SiO2 | 18.7 | 20.2 | ||||
| CrOx/SiO2 | 34.4 | 4.5 | ||||
| 200 W Xe lamp | SiO2 | 0.76 | 18.7 | 239 | ||
| mcat = 0.2 g | ZnO | 1.3 | 1.9 | |||
| ZnO/SiO2 (0.01 mol%) | 0.47 | 48.7 | ||||
| ZnO/SiO2 (0.1 mol%) | 1.9 | 45.3 | ||||
| ZnO/SiO2 (5.0 mol%) | 6.7 | 16.7 | ||||
| 300 W Xe lamp | SiO2 | 0.01 | 72.3 | 240 | ||
| mcat = 0.3 g | V2O5/SiO2 (0.5 wt%) | 0.38 | 13.7 | |||
| Rb–V2O5/SiO2 (Rb/V = 1) | 1.40 | 31.3 | ||||
| Cyclohexene (C6H10) | Cyclohexene oxide (C6H10O) | λ > 280 nm | TiO2 | 27 | 21 | 250 |
| Octene (CH3(CH2)5CHCH2) | Octene oxide (CH3(CH2)5CHCH2O) | Addition of MeCN | T–S (0.9 mol%) | 10 | 71 | |
| mcat = 0.01 g | T–OS (10 mol%) | 11 | 76 | 251 | ||
| TiO2 | 26 | 25 | 250 | |||
| T–S (0.9 mol%) | 11 | >99 | ||||
Watanabe and co-workers extensively studied the selective photo-oxidation of light alkanes into the corresponding aldehydes, at elevated reaction temperatures (>440 K), using MoO3 supported on silica photocatalysts.224–226 Selective photo-oxidation of methane and ethane into their corresponding aldehydes was also conducted over V2O5/SiO2227 and acetone was synthesized from propane186 and 2-methylpropane228over alkali-ion-modified V2O5/SiO2catalysts.
Selective oxidation of cyclohexane to cyclohexanone is another commercially important reaction, since cyclohexanone is used to obtain caprolactam for Nylon-6 production.229 V2O5/Al2O3, with isolated VO4, was found to be active for this reaction with high selectivity to partial oxidation products. Under visible light activation, highly dispersed Cr oxide species on silica were proved to promote efficiently the selective oxidation of cyclohexane to the ketone.230
Selective photo-oxidation of alkenes is also worth mentioning. For instance, Cr–SiO2 materials, containing highly dispersed chromate species, have proved to catalyze the photo-oxidation of different olefins under visible light irradiation with high selectivity to partially oxidized products, while TiO2 promoted complete decomposition.231 Direct epoxidation, and most specifically, obtaining propylene oxide from the selective photo-oxidation of propene, is of great importance since this compound is used in the production of, among other chemical compounds, polyurethane plastics. Pichat et al. reported that propylene oxide could be obtained from partial photocatalytic oxidation of propene over semiconductors such as TiO2, ZnO or SnO2, although complete mineralization was the predominant reaction.207 Yoshida et al.232–239 and Tanaka et al.240–242 have extensively investigated the selectivity to the epoxide of different metal oxides supported on silica (V2O5, Nb2O5, MgO, ZnO, CrOx). The presence of highly dispersed (isolated) tetrahedral metal oxide species seemed to promote the partial oxidation of propene to propylene oxide under UV irradiation.233–236,239 Yoshida and co-workers screened 50 types of silica-supported metal oxides for the photoepoxidation of propene,237 showing that the highest selectivities to the epoxide, together with the highest propene conversions, were achieved with the Ti, Zn, Pb and Bi systems. This study also revealed the possible influence of the acid–base properties on the propylene oxide yield.238CrOx/SiO2catalysts proved to be effective also under visible light irradiation.237 In addition to metal oxides, other materials supported on silica have been also tried for the selective photo-epoxidation of propene. For instance, Kanai et al. reported the high selectivity to propene oxide of several hydroxyapatite–silica composites.243
The selective photo-oxidation of alcohols into target compounds has also been studied. Tanaka et al. examined the use of silica-supported Nb2O5, acting simultaneously as acid catalyst and photocatalyst, that led to the formation of diethylacetal from ethanol with selectivities higher than 90%,244 and they reported the performance of Ta2O5/SiO2catalysts in the same reaction with analogous results. They concluded that ethanol is photo-oxidized to ethanal on the active sites and subsequently, on the same site, diethylacetal formation takes places by an acid-catalyzed process.245
Photocatalysis has also been applied to other synthetic reactions. For instance, Anpo and co-workers conducted the isomerisation of 2-butene over Zr–Si binary oxides catalysts246 and Soggiu et al. studied the photo-oxidation of organic sulfides to sulfoxides under visible irradiation using a sensitizer (DCA) covalently grafted on silica that led to a higher efficiency in comparison to the homogeneous reaction.247
A particular case among supported materials is constituted by the so-called single-site photocatalysts, already mentioned in Section 2. These materials can show high efficiency in selective photocatalytic reactions for synthetic purposes.248,249 Shiraishi et al. claimed to have achieved the highest selectivities to the corresponding epoxide in the photocatalytic epoxidation of olefins, using photocatalysts consisting of Ti-containing mesoporous silica (T-S) and Ti-containing mesoporous organosilica (T-OS)250,251 in the presence of MeCN. The results contrast with the low selectivities obtained on bulk TiO2, in spite of its higher olefin conversion. With the aim of obtaining materials that could operate under visible light irradiation, Cr and (Cr–Ti)-containing mesoporous silica were synthesized.230,249,252 Different mesoporous silicas, like FSM-16,253SBA-15,254MCM-41,255,256 or TUD-1257–259 have being used as matrix for obtaining different single-site photocatalysts. For instance, the effectiveness of Ti–TUD-1 was tested under UV-light activation,257 while V– and Cr–TUD-1 were proven to promote selective photo-oxidation reactions under visible irradiation.258,259
It is worth pointing out that some of the experiments on semiconductors and supported materials have been realized at relatively high temperatures, as shown in Table 5 and Table 6, implying an additional energy supply which could partly override one of the advantages of using a photocatalytic process. Furthermore, the requirement of heating suggests that not all the stages of the process are photoactivated but further research is necessary to clarify this point.
6.3 Zeolites
Among all the porous materials, zeolites deserve a special attention. Zeolites can act as microreactors with shape selectivity260 or hosts for the actual photoactive guest (a photosensitizer or a semiconductor). On the other hand, the incorporation of heteroatoms in the zeolite framework can turn the whole structure into a photocatalyst.261 After Frei and co-workers demonstrated in 1994 that small hydrocarbons could be selectively oxidized by visible light in the cation-exchange zeolite Y the possibilities of using different zeolite-based photocatalysts were also explored with, as stated by the authors, unprecedented selectivities.262–264 Frei et al. attributed this high selectivity to the stabilization of the hydrocarbon–O2 charge-transfer complex by electrostatic interaction with the zeolite framework that significantly lowered the energy required to activate both O2 and the hydrocarbon molecule. However, desorption of the strongly physisorbed carbonyl compounds inside the zeolite pores due to their polar character is still a challenge.264,265Myli et al. reported the production of benzaldehyde from toluene and acrolein from propene in zeolites X, Y and ZSM-5, emphasizing the shape-selective properties of the materials and successful removal of the products from the zeolite after their formation.266 The factors influencing product formation in the photo-oxidation of alkenes in zeolites have been studied by Xiang et al., who pointed out the importance of considering the light wavelength, thermochemistry and the type of zeolite used in the reaction.267 The performance of a series of transition metal-exchanged BEA zeolites on the selective photo-oxidation of benzene was studied by Shimizu et al.268 While phenol and CO2 were the main products detected on TiO2, selective formation of phenols or dihydroxybenzenes could be achieved depending on the metal immobilized on the BEA zeolite.
6.4 Polyoxometallates
Polytungstates have been used, not only in homogeneous reactions,269 but also as supported materials in heterogeneous reactions. The use of the latter makes it easier to reutilize the catalysts. For example, Fornal et al. examined the effect of catalyst loading on the photocatalytic activity of sodium deca-tungstates supported on ion-exchange resins in the oxidation of cyclohexane, finding that this factor determined the selectivity of the reaction. Lower loading of deca-tungstate favoured cyclohexanone production whereas higher loading promoted the formation of cyclohexyl hydroperoxide.270 Deca-tungstates supported on silica were efficiently used for the photo-oxidation of diols to ketones and aldehydes,271 with selectivity above 90%. The production of carbonylic compounds from the oxidation of primary and secondary benzylic alcohols, selectively and in high yields, was also achieved using (nBu4N)4W10O32,272 H3PW12O40,273 or H4SiW12O40 encapsulated into silica matrixes.274 Yields in the range 50–97% were obtained. Comparison with the unsupported materials showed that the composite photocatalysts were significantly more active.274 Deca-tungstates supported on ZrO2275 or Al2O3276 have also been utilized to catalyze these reactions, achieving high conversions and selectivities.6.5 Other photocatalysts
Metal Organic Frameworks (MOFs) are crystalline porous materials consisting of metal ions linked together by organic linkers that constitute a novel class of compounds, which deserves some attention. It has been experimentally demonstrated that the band gap of isoreticular MOFs can be tuned simply by changing the organic linker.277 This fact, together with their intrinsic size-selectivity properties and their world record surface areas, e.g. a surface area of 4500 m2 g−1 was reported for MOF-177 by Yaghi and co-workers,278 lead us to consider these compounds as a potential new class of photocatalysts. Although to date only few studies present data on the photocatalytic activity of these materials,279,280 the preliminary results of Gascon et al., who reported the first evidence of the selective photo-oxidation of propylene on a MOF surface, are promising.2777. Alternative photocatalysts for CO2reduction
In 1921, the artificial photosynthesis of hydrocarbons from CO2 and water was already matter of research. Baly et al. studied the production of formaldehyde under visible light, using colloidal uranium and ferric hydroxides as catalysts.281,282 Nowadays, the environmental problems derived from the rising concentration of CO2 in the atmosphere have renewed the interest in this process. Since this gas is one of the major contributors to the greenhouse effect, the reduction of the thermodynamically stable CO2 molecule into useful hydrocarbon products has turned into a research priority. Photocatalysis provides an interesting route for CO2 fixation which mimics photosynthesis in green plants, although instead of sugar like glucose it produces partially reduced compounds, mainly C1 molecules such as methanol. This is a highly endothermic process (ΔH0 = +715 kJ mol−1) and consequently it constitutes a potential route for storing solar energy in the form of convenient liquid fuels. The majority of the references for CO2 photoreduction deal with the use of TiO2-based photocatalysts, but this is not the only material that is able to catalyze CO2 photoreduction processes. Fujishima, Honda and co-workers first reported the photocatalytic reduction of CO2 with water in the liquid phase in several semiconductors,283 showing that the yield of methanol increased as the conduction band became more negative with respect to the redox potential of H2CO3/CH3OH. Thus, a higher yield was obtained on semiconductors such as SiC compared to TiO2.Fujiwara et al. investigated the selective photoreduction of CO2 into HCOO− catalyzed by colloidal ZnS nanocrystallites in the presence of triethylamine as an electron donor.284 The photoreduction of gaseous CO2 to yield CO over MgO, using hydrogen as reductant, was reported by Yoshida and co-workers. It was observed that the CO molecules were produced from the surface formate species formed as intermediates during the irradiation, which convert the adsorbed CO2 into CO (see Fig. 11).285 It is worth mentioning that MgO is not a semiconductor but an insulator. Previously, the authors found that the band gap excitation of the photocatalyst was not necessary for the photoreduction of CO2 to CO over ZrO2. The CO2− radicals formed under irradiation were considered as the active species responsible for the photoreduction of gaseous CO2.286 The authors studied the kinetic H–D isotope effect and analyzed the reaction between hydrogen and photoactivated CO2, concluding that hydrogen could be activated in the dark to react with the CO2− radical formed under illumination to yield the surface formate. The photoreduction of CO2 by methane was also conducted on ZrO2.287,288 As in the reaction with H2, the photogeneration of CO2− radicals and their interaction with CH4 was detected. Therefore, photocatalysis provided a way to conduct the highly exothermic CO2reforming of methane (ΔH0 = 247.3 kJ mol−1) at room temperature.
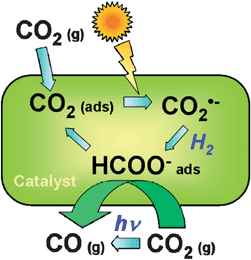 | ||
| Fig. 11 Pictorial diagram showing the proposed mechanism of CO2 photoreduction, using either H2 or CH4 as the reductant, over MgO or ZrO2. From ref. 286–288 | ||
Mesoporous materials have also been used to photocatalyze CO2reduction reactions. For instance, formic acid was detected as the primary product over a Ti silicalite molecular sieve (TS-1), under UV light and using methanol as electron donor, by Frei and co-workers, who studied the mechanism of the reaction by means of FTIR spectroscopy.289 They also proposed a mechanism for the CO2 photoreduction on a bimetallic ZrCu(I)–MCM-41 silicate sieve, implying that CO2 splits to CO and O2 at the excited metal-to-metal charge-transfer sites.290
Regarding the application of supported systems for this purpose, screening for the photoreduction of CO2 on different metal oxides on MgO, Al2O3 and SiO2 supports of an acid/basic nature, led to the conclusion that basic catalyst supports are more suitable for the selective conversion of CO2 into C1–C3 compounds.291
Methanol is the most valuable product from the CO2 photoreduction process because it can be directly used as an alternative fuel or as a building block by the chemical industry. For the photoconversion of CO2 into methanol in aqueous phase, higher yields were obtained over NiO and ZnO than over TiO2.292 CH3OH was also selectively produced over a NiO/InTaO4 photocatalyst under visible light irradiation.293 High efficiency and high selectivity for methanol was obtained in the photocatalytic reduction of CO2 with water, under UV illumination, over Ti-oxide/Y-zeolite catalysts containing highly dispersed isolated tetrahedral titanium oxide species. The charge-transfer excited state of these species is said to play a key role in the high selectivity for CH3OH, in contrast to the selectivity to CH4, obtained when bulk TiO2 was used as photocatalyst.294 Ti-incorporated mesoporous silicas also exhibited a much higher activity than bulk TiO2 in the photoreduction of CO2 with water to generate methanol and methane under UV irradiation.295 Besides the influence of the local structure of the Ti species, Anpo et al. investigated the effect of the hydrophilic–hydrophobic properties of the zeolite surface on the activity and selectivity of Ti-β zeolite photocatalysts in the photoreduction of CO2 with water.296 Although higher activity was observed on the catalyst with hydrophilic properties, enhanced selectivity to methanol was achieved in the hydrophobic system.
The effect of parameters such as temperature, pressure, light wavelength or type of reductant on the photocatalytic CO2reduction process was reviewed by Usubharatana et al., who also discussed different reactor configurations.297
Although it cannot be considered as a pure photocatalytic process, it is worth mentioning the work by Guan et al., who observed the formation of methanol in a composite catalyst consisting of a Pt–K2Ti6O13 photocatalyst combined with Cu/ZnO, as the CO2hydrogenation catalyst, using concentrated sunlight to increase the reaction temperature to 580 K.298 In this case, the H2 used as the reductant agent for the CO2reduction came from water decomposition on the hexa-titanate photocatalyst under solar illumination. No methanol formation was observed when a Xe- or Hg-lamp was used as the irradiation source.
Conclusions
The preponderant position of TiO2 as the archetypical photocatalyst relies on the favourable energetics of its band structure, and on its stability under irradiation. Besides, low cost and safety provide additional value for the large scale commercial exploitation of this material. Nevertheless, reduced quantum yield and null photoresponse under visible light illumination impose severe limits on the development of solar photocatalysis. Consequently there is increased interest in the development of alternative photocatalysts for the different fields of application of this technology.As it has been stated in this review, a number of novel photocatalysts have surpassed the performance of TiO2 for water splitting under different experimental constrains. Thus, the rate of H2 from pure water under UV irradiation over La-doped NaTaO3 is more than fifty times larger than when TiO2 is used. These results can be improved under visible light illumination using nanostructured CdS as photocatalyst and S2−/SO32− as sacrificial compound. However, if pure water is used as hydrogen source, the higher efficiency currently reported corresponds to (Ga0.88Zn0.12)(N0.88O0.12). Nevertheless, these results are still far from the quantum yield suggested as a threshold for the viability of this technology. Therefore further research is necessary not only to improve these achievements but also to ensure reproducibility, and discard the influence of additional variables not previously considered. On the other hand, if these semiconductors are used in the future for hydrogen generation, concerns about the cost and availability of elements such as Ga and Ta may arise. Similarly, the toxicity of Cd could require special precautions.
Regarding water detoxification, the existence of efficient photocatalyst alternatives to TiO2 are not evident, especially if UV-active materials are considered. As we have seen, ZnO shows high activity under UV light and is the subject of numerous studies, but the occurrence of anodic photocorrosion makes its use unviable in practice. Interesting results have been obtained with BiOX (X = halogen) photocatalysts, but few studies are available, and further investigations are needed to assess parameters like mineralization efficiency. In addition, the scarcity and higher cost of bismuth compared to titanium should be also considered. Regarding visible light activated catalysts, it is important to note that in order for a catalyst to be considered more active than TiO2, this activity must exceed that obtained with TiO2 under solar light owing to the UV contribution to the solar spectrum. In this sense, some metallates, considering their activities and stability, appear promising as materials for solar photocatalysis. However limitations with these materials arise from low mineralization yields. In summary, the limitations of TiO2 for this application have not been overcome up-to-date, but there are promising alternative materials that may lead to better utilization of solar light in the near future. Concerning air treatment, promising results are obtaining with oxides, mixed oxides and siliceous materials under UV or vis irradiation. β-Ga2O3 and SrTiO3 are some of the few semiconductors that show higher photocatalytic performance than TiO2 under UV irradiation. With respect to activation under visible light illumination, different oxynitrides and oxyfluorides as well as V-containing zeolites modified with alkaline metals have shown a significant conversion rate for VOCs and NOx abatement at relative high concentrations. Nevertheless, parameters such as stability and cost have to be further considered for commercial implementation. In contrast, photocatalytic disinfection with TiO2 alternatives are still in the initial stages of development, but preliminary results obtained with immobilised complexes and clays hold promise for the future design of novel and efficient photocatalysts for bacteria removal.
Due to the relatively high mineralization effectiveness of TiO2 low selectivity to partial oxidation products is usually achieved, and this provides significant opportunities for the use of alternative photocatalysts in organic synthesis. In this regard, interesting results have been obtained with dispersed oxides on high surface area solids like zeolites or mesoporous materials, but further advances in the design of photocatalysts for specific applications are envisaged in future research programs. In particular it is worth emphasising that, in spite of the high selectivities achieved in photocatalyzed synthetic reactions, product yields are generally too low to compete with methods currently applied in industry. In the case of CO2reduction although titania-based materials are still the preferred choice, higher selectivities have often been obtained when alternative photocatalysts were used. However, additional investigations are required in order to enhance the current yields of C1 products.
Acknowledgements
Financial support from the project MAT2008-01094/MAT from the Spanish Ministry of Science and Innovation is appreciated.References
- W. Doerffler and K. Hauffe, J. Catal., 1964, 3, 171 CrossRef CAS.
- S. R. Morrison and T. Freund, J. Chem. Phys., 1967, 47, 1543 CrossRef CAS.
- H. D. Muller and F. Steinbach, Nature, 1970, 225, 728 CrossRef CAS.
- F. Steinbach, Nature, 1969, 221, 657 CrossRef CAS.
- A. V. Pamfilov, Y. S. Mazurkev and S. Y. Mukhlya, Russ. J. Phys. Chem., 1970, 71(1–3), 14.
- M. Formenti, F. Juillet, P. Meriaudeau and S. J. Teichner, Chem. Technol., 1971, 1, 680.
- A. Fujishima and K. Honda, Nature, 1972, 238, 37 CAS.
- O. Carp, C. L. Huisman and A. Reller, Prog. Solid State Chem., 2004, 32, 33 CrossRef CAS.
- J.-M. Herrmann, Catal. Today, 1999, 53(1), 115 CrossRef CAS.
- M. R. Hoffmann, S. T. Martin, W. Choi and D. W. Bahnemann, Chem. Rev., 1995, 95(1), 69–96 CrossRef CAS.
- A. L. Linsebigler, G. Lu and J. T. Yates, Chem. Rev., 1995, 95(3), 735–758 CrossRef CAS.
- A. Mills and S. Le Hunte, J. Photochem. Photobiol., A, 1997, 108, 1–35 CrossRef CAS.
- A. Fujishima, T. N. Rao and D. A. Tryk, J. Photochem. Photobiol., C, 2000, 1(1), 1 CrossRef CAS.
- A. Fujishima and X. T. Zhang, C. R. Chim., 2006, 9, 750 CrossRef CAS.
- A. A. Adesina, Catal. Surv. Asia, 2004, 8(4), 265 CrossRef CAS.
- A. Mills and S.-K. Lee, J. Photochem. Photobiol., A, 2002, 152, 233 CrossRef CAS.
- S. Malato, J. Blanco, A. Vidal, D. Alarcón, M. I. Maldonado, J. Cáceres and W. Gernjak, Sol. Energy, 2003, 75(4), 329 CrossRef CAS.
- M. Romero, J. Blanco, B. Sánchez, A. Vidal, S. Malato, A. Cardona and E. García, Sol. Energy, 1999, 66(2), 169 CrossRef CAS.
- M. Kitano, M. Matsuoka, M. Ueshima and M. Anpo, Appl. Catal., A, 2007, 325(1), 1 CrossRef CAS.
- T. L. Thompson and J. T. Yates, Chem. Rev., 2006, 106, 4428 CrossRef CAS.
- F. Xu, P. Zhang, A. Navrotsky, Z.-Y. Yuan, T.-Z. Ren, M. Halasa and B.-L. Su, Chem. Mater., 2007, 19(23), 5680 CrossRef CAS.
- F. Zhao, X. Li, J.-G. Zheng, X. Yang, F. Zhao, K. Wong, W. Sing, L. Jing, W. Wenjiao, S. Mingmei and Q. Qiang, Chem. Mater., 2008, 20(4), 1197 CrossRef CAS.
- N. Bao, L. Shen, T. Takata and K. Domen, Chem. Mater., 2008, 20(1), 110 CrossRef CAS.
- D. Jing and L. Guo, J. Phys. Chem. B, 2006, 110(23), 11139 CrossRef CAS.
- K. Maeda and K. Domen, J. Phys. Chem. C, 2007, 111(22), 7851 CrossRef CAS.
- F. E. Osterloh, Chem. Mater., 2008, 20(1), 35 CrossRef CAS.
- A. Kudo and Y. Miseki, Chem. Soc. Rev., 2009, 38, 253 RSC.
- M. Anpo, T.-H. Kim and M. Matsuoka, Catal. Today, 2009, 142(3–4), 114 CrossRef CAS.
- H. Chen, A. Nambu, W. Wen, J. Graciani, Z. Zhong, J. C. Hanson, E. Fujita and J. A. Rodriguez, J. Phys. Chem. C, 2007, 111(3), 1366 CrossRef CAS.
- W. Gernjak, M. Fuerhacker, P. Fernández-Ibañez, J. Blanco and S. Malato, Appl. Catal., B, 2006, 64(1–2), 121 CrossRef CAS.
- H. J. Harmon, Chemosphere, 2006, 63(7), 1094 CrossRef CAS.
- E. Gkika, A. Troupis, A. Hiskia and E. Papaconstantinou, Appl. Catal., B, 2006, 62(1–2), 28 CrossRef CAS.
- C. Tanielian, Coord. Chem. Rev., 1998, 178–180, 1165 CrossRef CAS.
- A. Maldotti, A. Molinari and R. Amadelli, Chem. Rev., 2002, 102(10), 3811 CrossRef CAS.
- A. J. Bard and M. A. Fox, Acc. Chem. Res., 1995, 28(3), 141 CrossRef CAS.
- B. Ohtani, Chem. Lett., 2008, 37(3), 216 CrossRef.
- M. Matsuoka and M. Anpo, J. Photochem. Photobiol., C, 2003, 3(3), 225 CrossRef CAS.
- H. Z. Zhang and J. F. Banfield, J. Phys. Chem. B, 2000, 104, 3481 CrossRef CAS.
- X. Chen and S. S. Mao, Chem. Rev., 2007, 107(7), 2891 CrossRef CAS.
- A. J. Maira, K. L. Yeung, C. Y. Lee, P. L. Yue and C. K. Chan, J. Catal., 2000, 192(1), 185 CrossRef CAS.
- N. Serpone, D. Lawless and R. Khairutdinov, J. Phys. Chem., 1995, 99, 16646 CrossRef CAS.
- C. Wang, J. Rabani, D. W. Bahnemann and J. K. Dohrmann, J. Photochem. Photobiol., A, 2002, 148, 169 CrossRef CAS.
- J. M. Coronado, B. Sánchez, R. Portela and S. Suárez, J. Sol. Energy Eng., 2008, 113, 0110161.
- V. Svrcek, A. Slaoui and J.-C. Muller, Thin Solid Films, 2004, 451–452, 384 CAS.
- T. Lindgren, J. Lu, A. Hoel, C. G. Granqvist, G. R. Torres and S. E. Lindquist, Sol. Energy Mater. Sol. Cells, 2004, 84, 145 CrossRef CAS.
- J. Ryu and W. Choi, Environ. Sci. Technol., 2008, 42(1), 294 CrossRef CAS.
- R. I. Bickley, T. Gonzalez-Carreno, J. S. Lees, L. Palmisano and R. D. Tilley, J. Solid State Chem., 1991, 92, 178 CrossRef CAS.
- T. Ohno, K. Sarukawa, K. Tokieda and M. Matsumura, J. Catal., 2001, 203, 82–86 CrossRef CAS.
- M. A. Henderson, Langmuir, 1996, 12, 5093 CrossRef CAS.
- M. Nagao and Y. Suda, Langmuir, 1989, 5, 42 CrossRef CAS.
- W. C. Wu, C. C. Chuang and J. L. Lin, J. Phys. Chem. B, 2000, 104, 8719 CrossRef CAS.
- M.-C. Lu, W.-D. Roam, J. N. Chen and C.-P. Huan, Water Res., 1996, 30(7), 1670 CrossRef CAS.
- “Mineral Commodities Summaries 2009” US Geological Survey. http://minerals.usgs.gov/minerals/pubs/mcs/2009/mcs2009.pdf Search PubMed.
- J. X. Wang, G. Q. Zhou, C. Y. Chen, H. W. Yu, T. C. Wang, Y. M. Ma, G. Jia, Y. X. Gao, B. Li, J. Sun, Y. F. Li, F. Jiao, Y. L. Zhao and Z. F. Chai ZF, Toxicol. Lett., 2007, 168(2), 176 CrossRef CAS.
- V. H. Grassian, A. Adamcakova-Dodd, J. M. Pettibone, P. T. O'Shaughnessy and P. S. Thorne, Nanotoxicology, 2007, 1(3), 211 CrossRef CAS.
- C.-M. Liao, Y.-H. Chiang and C.-P. Chio, J. Hazard. Mater., 2009, 162(1), 57 CrossRef CAS.
- S. Y. Chae, M. K. Park, S. K. Lee, T. Y. Kim, S. K. Kim and W. I. Lee, Chem. Mater., 2003, 15(17), 3326 CrossRef CAS.
- T. Kasuga, M. Hiramatsu, A. Hoson, T. Sekino and K. Niihara, Langmuir, 1998, 14(12), 3160 CrossRef CAS.
- A. Yamamoto and H. Imai, J. Catal., 2004, 226, 462 CrossRef CAS.
- H. Yi, T. Peng, D. Ke, D. Ke, L. Ling and C. Yan, Int. J. Hydrogen Energy, 2008, 33(2), 672 CrossRef CAS.
- J. C. Yu, X. Wang and X. Fu, Chem. Mater., 2004, 16(8), 1523 CrossRef CAS.
- C. Adán, A. Bahamonde, M. Fernández-García and A. Martínez-Arias, Appl. Catal., B, 2007, 72(1–2), 11 CrossRef CAS.
- J. Lukáč, M. Klementová, P. Bezdička, S. Bakardjieva, J. Šubrt, L. Szatmáry, Z. Bastl and J. Jirkovský, Appl. Catal., B, 2007, 74(1–2), 83 CAS.
- D. Robert, Catal. Today, 2007, 122, 20 CrossRef CAS.
- Q. Liu, X. Wu, B. Wang and Q. Liu, Mater. Res. Bull., 2002, 37, 2255 CrossRef CAS.
- R. Asahi, T. Morikawa, T. Ohwaki, K. Aoki and Y. Taga, Science, 2001, 293(5528), 269 CrossRef CAS.
- M.-C. Yang, T.-S. Yang and M.-S. Wong, Thin Solid Films, 2004, 469–470, 1 CrossRef CAS.
- J. Yuan, M. Chen, J. Shi and W. Shangguan, Int. J. Hydrogen Energy, 2006, 31, 1326 CrossRef CAS.
- J. M. Coronado, B. Sánchez, F. Fresno, S. Suárez and R. Portela, J. Sol. Energy Eng., 2008, 130(4), 041012 CrossRef.
- N. Serpone, D. Lawless and R. Terzian, Sol. Energy, 1992, 49, 221 CrossRef CAS.
- T. Kodama and N. Gokon, Chem. Rev., 2007, 107, 4048 CrossRef CAS.
- M. G. Gonzalez, E. Oliveros, M. Worner and A. M. Braun, J. Photochem. Photobiol., C, 2004, 5(3), 225 CrossRef CAS.
- A. Nozik, Appl. Phys. Lett., 1977, 30(11), 567 CrossRef CAS.
- R. M. Navarro, M. C. Sánchez-Sánchez, M. C. Alvarez-Galván, F. del Valle and J. L. G. Fierro, Energy Environ. Sci., 2009, 2, 35 RSC.
- Data obtained by integration of the standardised solar spectrum provided by NREL available at http://rredc.nrel.gov/solar/spectra/am1.5/%23about.
- H. Kato, K. Asakura and A. Kudo, J. Am. Chem. Soc., 2003, 125(10), 3082 CrossRef CAS.
- K. Maeda, K. Teramura and K. Domen, J. Catal., 2008, 254, 198 CrossRef CAS.
- K. Domen, J. N. Kondo, M. Hara and T. Tanaka, Bull. Chem. Soc. Jpn., 2000, 73, 1307 CrossRef CAS.
- K. Domen, J. Yoshimura, T. Sekine, A. Tanaka and T. Onishi, Catal. Lett., 1990, 4(4–6), 339 CrossRef CAS.
- K. Yamaguti and S. Sato, J. Chem. Soc., Faraday Trans. 1, 1985, 81, 1237 RSC.
- Y. Ebina, A. Tanaka, J. N. Kondo and K. Domen, Chem. Mater., 1996, 8(10), 2534 CrossRef CAS.
- Y. Ebina, T. Sasaki, M. Harada and M. Watanabe, Chem. Mater., 2002, 14(10), 4390 CrossRef CAS.
- J. F. Reber and K. Meier, J. Phys. Chem., 1984, 88(24), 5903 CrossRef CAS.
- A. Kudo, Int. J. Hydrogen Energy, 2006, 31(2), 197 CrossRef CAS.
- I. Tsuji, H. Kato and A. Kudo, Angew. Chem., Int. Ed., 2005, 44(23), 3565 CrossRef CAS.
- J. F. Reber and M. Rusek, J. Phys. Chem., 1986, 90, 824 CrossRef CAS.
- W. F. Shangguan and A. Yoshida, J. Phys. Chem. B, 2002, 106(47), 12227 CrossRef CAS.
- K. Domen, S. Naito, M. Soma, T. Onishi and K. Tamaru, J. Chem. Soc., Chem. Commun., 1980,(12), 543 RSC.
- J. Kim, D. W. Hwang, H. G. Kim, S. W. Bae, J. S. Lee, W. Li and S. H. Oh, Top. Catal., 2005, 35(3–4), 295 CrossRef CAS.
- D. W. Hwang, H. G. Kirn, J. S. Lee, J. Kim, W. Li and S. H. Oh, J. Phys. Chem. B, 2005, 109(6), 2093 CrossRef CAS.
- T. Takata, Y. Furumi, K. Shinohara, A. Tanaka, M. Hara, J. N. Kondo and K. Domen, Chem. Mater., 1997, 9(5), 1063 CrossRef CAS.
- S. Ikeda, M. Hara, J. N. Hondo, K. Komen, H. Takahashi, T. Okubo and M. Kakihana, Chem. Mater., 1998, 10, 72 CrossRef CAS.
- Y.-W. Tai, J.-S. Chen, C.-C. Yang and B.-Z. Wan, Catal. Today, 2004, 97, 95 CrossRef CAS.
- Y. Miseki, H. Kato and A. Kudo, Energy Environ. Sci., 2009, 2, 306 RSC.
- A. Furube, T. Shiozawa, A. Ishikawa, A. Wada, K. Domen and C. Hirose, J. Phys. Chem. B, 2002, 106(12), 3065 CrossRef CAS.
- Y. Noda, B. Lee, K. Domen and J. N. Kondo, Chem. Mater., 2008, 20(16), 5361 CrossRef CAS.
- H. Otsuka, K. Y. Kim, A. Kouzu, I. Takimoto, H. Fujimori, Y. Sakata, H. Imamura, T. Matsumoto and K. Toda, Chem. Lett., 2005, 34(6), 822 CrossRef CAS.
- H. Kato and A. Kudo, Chem. Phys. Lett., 1998, 295(5–6), 487 CrossRef CAS.
- A. Kudo, R. Niishiro, A. Iwase and H. Kato, Chem. Phys., 2007, 339(1–3), 104 CrossRef CAS.
- Y. Sakata, Y. Matsuda, T. Yanagida, K. Hirata, H. Imamura and K. Teramura, Catal. Lett., 2008, 125(1–2), 22 CrossRef CAS.
- K. Maeda, N. Saito, D. Lu, Y. Inoue and K. Domen, J. Phys. Chem. C, 2007, 111, 4749 CrossRef CAS.
- J. K. Reddy, G. Suresh, C. H. Hymavathi, V. D. Kumari and M. Subrahmanyam, Catal. Today, 2009, 141, 89 CrossRef.
- P. Ritterskamp, A. Kuklya, M. A. Wustkamp, K. Kerpen, C. Weidenthaler and M. Demuth, Angew. Chem., Int. Ed., 2007, 46, 7770 CrossRef CAS.
- J. R. Smith, T. H. Van Steenkiste and X. G. Wang, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 79, 041403 CrossRef.
- Z. Zou and H. Arakawa, J. Photochem. Photobiol., A, 2003, 158, 145 CrossRef CAS.
- Y. Lee, H. Terashima, Y. Shimodaira, K. Teramura, M. Hara, H. Kobayashi, K. Domen and M. Yashima, J. Phys. Chem. C, 2007, 111.
- M. Miyauchi, A. Nakajima, T. Watanabe and K. Hashimoto, Chem. Mater., 2002, 14, 2812–2816 CrossRef CAS.
- L. B. Khalil, W. E. Mourad and M. W. Rophael, Appl. Catal., B, 1998, 17, 267–273 CrossRef CAS.
- I. Poulios, M. Kositzi and A. Kouras, J. Photochem. Photobiol., A, 1998, 115, 175 CrossRef CAS.
- E. Evgenidou, I. Konstantinou, K. Fytianos, I. Poulios and T. Albanis, Catal. Today, 2007, 124, 156 CrossRef CAS.
- G. Colón, M. C. Hidalgo, J. A. Navío, E. Pulido Melián, O. González Díaz and J. M. Doña Rodríguez, Appl. Catal., B, 2008, 83, 30 CrossRef CAS.
- M. L. Zhang, G. Y. Sheng, J. M. Fu, T. C. An, X. M. Wang and X. H. Hu, Mater. Lett., 2005, 59, 3641 CrossRef CAS.
- C. C. Hsu and N. L. Wu, J. Photochem. Photobiol., A, 2005, 172, 269–274 CrossRef CAS.
- S. Sakthivel, S. U. Geissen, D. W. Bahnemann, V. Murugesan and A. Vogelpohl, J. Photochem. Photobiol., A, 2002, 148, 283 CrossRef CAS.
- H. F. Lin, S. C. Liao and S. W. Hung, J. Photochem. Photobiol., A, 2005, 174, 82 CrossRef CAS.
- B. Dindar and S. Içli, J. Photochem. Photobiol., A, 2001, 140, 263 CrossRef CAS.
- S. K. Pardeshi and A. B. Patil, Sol. Energy, 2008, 82, 700 CrossRef CAS.
- M. D. Hernández-Alonso, A. B. Hungría, A. Martínez-Arias, M. Fernández-García, J. M. Coronado, J. C. Conesa and J. Soria, Appl. Catal., B, 2004, 50, 167 CrossRef.
- P. F. Ji, J. L. Zhang, F. Chen and M. Anpo, J. Phys. Chem. C, 2008, 112, 17809 CrossRef CAS.
- P. Ji, J. Zhang, F. Chen and M. Anpo, Appl. Catal., B, 2009, 85, 148 CrossRef CAS.
- Y. Zhai, S. Zhang and H. Pang, Mater. Lett., 2007, 61, 1863 CrossRef CAS.
- C. Lettmann, H. Hinrichs and W. F. Maier, Angew. Chem., Int. Ed., 2001, 40, 3160 CrossRef CAS.
- A. Sclafani, L. Palmisano, G. Marcí and A. M. Venezia, Sol. Energy Mater. Sol. Cells, 1998, 51, 203 CrossRef CAS.
- R. Abe, H. Takami, N. Murakami and B. Ohtani, J. Am. Chem. Soc., 2008, 130, 7780 CrossRef CAS.
- A. Watcharenwong, W. Chanmanee, N. R. de Tacconi, C. R. Chenthamarakshan, P. Kajitvichyanukul and K. Rajeshwar, J. Electroanal. Chem., 2008, 612, 112 CrossRef CAS.
- O. Hamanoi and A. Kudo, Chem. Lett., 2002, 838–839 CrossRef CAS.
- S. Kohtani, Y. Ohama, Y. Ohno, I. Tsuji, A. Kudo and R. Nakagaki, Chem. Lett., 2005, 34, 1056–1057 CrossRef CAS.
- S. Kohtani, S. Makino, A. Kudo, K. Tokumura, Y. Ishigaki, T. Matsunaga, O. Nikaido, K. Hayakawa and R. Nakagaki, Chem. Lett., 2002, 660 CrossRef CAS.
- S. Kohtani, M. Koshiko, A. Kudo, K. Tokumura, Y. Ishigaki, A. Toriba, K. Hayakawa and R. Nakagaki, Appl. Catal., B, 2003, 46, 573 CrossRef CAS.
- X. Zhang, Z. H. Ai, F. L. Jia, L. Z. Zhang, X. X. Fan and Z. G. Zou, Mater. Chem. Phys., 2007, 103, 162 CrossRef CAS.
- Y. Zheng, J. Wu, F. Duan and Y. Xie, Chem. Lett., 2007, 36, 520 CrossRef CAS.
- L. Zhou, W. Z. Wang, L. Zhang, H. L. Xu and W. Zhu, J. Phys. Chem. C, 2007, 111, 13659 CrossRef CAS.
- S. Kohtani, J. Hiro, N. Yamamoto, A. Kudo, K. Tokumura and R. Nakagaki, Catal. Commun., 2005, 6, 185 CrossRef CAS.
- S. Kohtani, M. Tomohiro, K. Tokumura and R. Nakagaki, Appl. Catal., B, 2005, 58, 265 CrossRef CAS.
- L. Ge, Mater. Chem. Phys., 2008, 107, 465 CrossRef CAS.
- L. Ge, J. Inorg. Mater., 2008, 23, 449 Search PubMed.
- H. Xu, H. M. Li, C. D. Wu, J. Y. Chu, Y. S. Yan, H. M. Shu and Z. Gu, J. Hazard. Mater., 2008, 153, 877 CrossRef CAS.
- J. W. Tang, Z. G. Zou and J. H. Ye, Catal. Lett., 2004, 92, 53 CrossRef CAS.
- C. Zhang and Y. Zhu, Chem. Mater., 2005, 17, 3537 CrossRef CAS.
- H. Fu, C. Pan, W. Yao and Y. Zhu, J. Phys. Chem. B, 2005, 109, 22432 CrossRef CAS.
- S. Zhu, T. Xu, H. Fu, J. Zhao and Y. Zhu, Environ. Sci. Technol., 2007, 41, 6234 CrossRef CAS.
- H. Fu, S. Zhang, T. Xu, Y. Zhu and J. Chen, Environ. Sci. Technol., 2008, 42, 2085 CrossRef CAS.
- L. H. Zhang, W. Z. Wang, Z. G. Chen, L. Zhou, H. L. Xu and W. Zhu, J. Mater. Chem., 2007, 17, 2526 RSC.
- J. Wu, F. Duan, Y. Zheng and Y. Xie, J. Phys. Chem. C, 2007, 111, 12866 CrossRef CAS.
- F. Amano, K. Nogami, R. Abe and B. Ohtani, J. Phys. Chem. C, 2008, 112, 9320 CrossRef CAS.
- F. Amano, K. Nogami, R. Abe and B. Ohtani, Chem. Lett., 2007, 36, 1314 CrossRef CAS.
- H. Fu, C. Pan, L. Zhang and Y. Zhu, Mater. Res. Bull., 2007, 42, 696 CrossRef CAS.
- Ye, D. Z. Li, W. J. Zhang, M. Sun, Y. Hu, Y. F. Zhang and X. Z. Fu, J. Phys. Chem. C, 2008, 112, 17351 CrossRef.
- A. Martinez-de la Cruz, S. O. Alfaro, E. L. Cuellar and U. O. Mendez, Catal. Today, 2007, 129, 194 CrossRef.
- C. Belver, C. Adan and M. Fernández-García, Catal. Today, 2009, 143(3–4), 274 CrossRef CAS.
- J. W. Tang, Z. G. Zou, M. Katagiri, T. Kako and J. H. Ye, Catal. Today, 2004, 93–95, 885 CrossRef CAS.
- W. K. Chang, K. K. Rao, H. C. Kuo, J. F. Cai and M. S. Wong, Appl. Catal., A, 2007, 321, 1 CrossRef CAS.
- Y. Ku, L. C. Wang and C. M. Ma, Chem. Eng. Technol., 2007, 30, 895 CrossRef CAS.
- W. F. Yao, H. Wang, X. H. Xu, S. X. Shang, Y. Hou, Y. Zhang and M. Wang, Mater. Lett., 2003, 57, 1899 CrossRef CAS.
- K. W. Li, H. Wang and H. Yan, J. Mol. Catal. A: Chem., 2006, 249, 65 CrossRef CAS.
- W. Q. Meng, F. Li, D. G. Evans and X. Duan, J. Porous Mater., 2004, 11, 97 CrossRef CAS.
- D. F. Wang, T. Kako and J. H. Ye, J. Am. Chem. Soc., 2008, 130, 2724 CrossRef CAS.
- K. G. Keramidas, G. P. Voutsas and P. I. Rentzeperis, Z. Kristallogr., 1993, 205, 35 CAS.
- K.-L. Zhang, C.-M. Liu, F.-Q. Huang, C. Zheng and W.-D. Wang, Appl. Catal., B, 2006, 68, 125 CrossRef CAS.
- C. H. Wang, C. L. Shao, Y. C. Liu and L. Zhang, Scr. Mater., 2008, 59, 332 CrossRef CAS.
- J. Zhang, F. J. Shi, J. Lin, D. F. Chen, J. M. Gao, Z. X. Huang, X. X. Ding and C. C. Tang, Chem. Mater., 2008, 20, 2937 CrossRef CAS.
- K. Demeestere, J. Dewulf and H. Van Langenhove, Crit. Rev. Environ. Sci. Technol., 2007, 37, 489 CrossRef CAS.
- T. Long, K. Takabatake, S. Yin and T. Sato, J. Cryst. Growth, 2009, 311, 576 CrossRef CAS.
- J. Liqiang, W. Baiqi, X. Baifu, L. Shudan, S. Keying, C. Weimin and F. Honggang, J. Solid State Chem., 2004, 177(11), 4221 CrossRef.
- C. Wu, X. Zhao, Y. Ren, Y. Yue, W. Hua, Y. Cao, Y. Tang and Z. Gao, J. Mol. Catal. A: Chem., 2005, 229, 233 CrossRef CAS.
- Y. Hou, L. Wu, X. Wang, Z. Ding, Z. Li and X. Fu, J. Catal., 2007, 250, 12 CrossRef CAS.
- A. Kudo, K. Omori and H. Kato, J. Am. Chem. Soc., 1999, 121, 11459 CrossRef CAS.
- M. Wiegel, W. Middel and G. Blasse, J. Mater. Chem., 1995, 5, 981 RSC.
- X. Hu, C. Hu and J. Qu, Appl. Catal., B, 2006, 69, 17 CrossRef CAS.
- M. Futsuhara, K. Yoshioka and O. Takai, Thin Solid Films, 1998, 317, 322 CrossRef CAS.
- D. Li and H. Haneda, J. Photochem. Photobiol., A, 2003, 155, 171 CrossRef CAS.
- D. Li, H. Haneda, N. Ohashi, S. Hishita and Y. Yoshikawa, Catal. Today, 2004, 93–95, 895.
- D. Li, H. Haneda, N. Ohashi, N. Saito and S. Hishita, Thin Solid Films, 2005, 486, 20 CrossRef CAS.
- L. Chen, S. Zhang, L. Wang, D. Xue and S. Yin, J. Cryst. Growth, 2009, 311, 746 CrossRef CAS.
- J. Wang, S. Yin, M. Komatsu, Q. Zhang, F. Saito and T. Sato, Appl. Catal., B, 2004, 52, 11 CrossRef CAS.
- M. Miyauchi, M. Takashio and H. Tobimatsu, Langmuir, 2004, 20, 232 CrossRef CAS.
- R. Aguiar, A. Kalytta, A. Reller, A. Weidenkaff and S. G. Ebbinghaus, J. Mater. Chem., 2008, 18, 4260 RSC.
- S. B. Zhu, H. B. Fu, S. C. Zhang, L. W. Zhang and Y. F. Zhu, J. Photochem. Photobiol., A, 2008, 193(1), 33 CrossRef CAS.
- H. B. Fu, S. C. Zhang, L. W. Zhang and Y. F. Zhu, Mater. Res. Bull., 2008, 43(4), 864 CrossRef CAS.
- Y. He, Y. Zhu and N. Wu, J. Solid State Chem., 2004, 177, 3868 CrossRef CAS.
- J. Grannec, A. Yacoubi, J. Ravez and P. Hagenmuller, J. Solid State Chem., 1988, 75, 263 CrossRef CAS.
- T. Murase, H. Irie and K. Hashimoto, J. Phys. Chem. B, 2005, 109, 13420 CrossRef CAS.
- H. Kato, H. Kobayashi and A. Kudo, J. Phys. Chem. B, 2002, 106, 12441 CrossRef CAS.
- W. M. Zhou, L. J. Yan, Y. Wang and Y. F. Zhang, Appl. Phys. Lett., 2006, 89(1), 013105 CrossRef.
- F. Amano, T. Ito, S. Takenaka and T. Tanaka, J. Phys. Chem. B, 2005, 109, 10973 CrossRef CAS.
- T. Tanaka, S. Takenaka, T. Funabiki and S. Yoshida, Chem. Lett., 1994, 1585 CAS.
- M. Matsuoka, E. Matsuda, K. Tsuji, H. Yamashita and M. Anpo, J. Mol. Catal. A: Chem., 1996, 107, 399 CrossRef CAS.
- M. Anpo, Y. Shioya, H. Yamashita, E. Giamello, C. Morterra, M. Che, H. H. Patterson, S. Webber and S. Ouellette, J. Phys. Chem., 1994, 98, 5744 CrossRef CAS.
- M. Matsuoka, W. S. Ju, H. Yamashita and M. Anpo, J. Photochem. Photobiol., A, 2003, 160, 43 CrossRef CAS.
- W. S. Ju, M. Matsuoka, K. Iino, H. Yamashita and M. Anpo, J. Phys. Chem. B, 2004, 108, 2128 CrossRef CAS.
- S. Rodrigues, S. Uma, I. N. Martyanov and K. J. Klabunde, J. Photochem. Photobiol., A, 2004, 165, 51 CrossRef CAS.
- S. Rodrigues, K. T. Ranjit, S. Uma, I. N. Martyanov and K. J. Klabunde, J. Catal., 2005, 230, 158 CrossRef CAS.
- S. Rodrigues, S. Uma, I. N. Martyanov and K. J. Klabunde, J. Catal., 2005, 233, 405 CrossRef CAS.
- V. Krishna, V. S. Kamble, P. Selvam and N. M. Gupta, Catal. Lett., 2004, 98, 113 CrossRef CAS.
- H. Yamashita, K. Yoshizawa, M. Ariyuki, S. Higashimoto, M. Anpo and M. Che, Chem. Commun., 2001, 435 RSC.
- S. Malato, J. Blanco, D. C. Alarcón, M. I. Maldonado, P. Fernández-Ibáñez and W. Gernjak, Catal. Today, 2007, 122(1–2), 137 CrossRef CAS.
- T. Matsunaga, R. Tomoda, T. Nakajima, N. Nakamura and T. Komine, Appl. Environ. Microbiol., 1988, 54(6), 1330 CAS.
- T. Matsunaga, R. Tomoda, T. Nakajima and H. Wake, FEMS Microbiol. Lett., 1985, 29, 211 CrossRef CAS.
- O. Seven, B. Dindar, S. Aydemir, D. Metin, M. A. Ozinel and S. Icli, J. Photochem. Photobiol., A, 2004, 165(1–3), 103 CrossRef CAS.
- S. L. Kuo and C. J. Liao, Water Qual. Res. J. Can., 2006, 41(4), 365 Search PubMed.
- L. Villen, F. Manjon, D. Garcia-Fresnadillo and G. Orellana, Appl. Catal., B, 2006, 69(1–2), 1 CrossRef CAS.
- Y. Fueda, H. Suzuki, Y. Komiya, Y. Asakura, T. Shiragami, J. Matsumoto, H. Yokoi and M. Yasuda, Bull. Chem. Soc. Jpn., 2006, 79(9), 1420 CrossRef CAS.
- G. Palmisano, V. Augugliaro, M. Pagliaro and L. Palmisano, Chem. Commun., 2007, 3425–3437 RSC.
- J.-M. Herrmann, C. Duchamp, M. Karkmaz, B. T. Hoai, H. Lachheb, E. Puzenat and C. Guillard, J. Hazard. Mater., 2007, 146, 624–629 CrossRef CAS.
- M. Fagnoni, D. Donddi, D. Ravelli and A. Albini, Chem. Rev., 2007, 107, 2725–2756 CrossRef CAS.
- R. A. Sheldon and J. K. Kochi, Metal-catalyzed oxidation of organic compounds, Academic Press, New York, 1981 Search PubMed.
- P. Pichat, J. M. Herrmann, J. Didier and M. N. J. Mozzanega, J. Phys. Chem., 1979, 83, 3122–3126 CrossRef CAS.
- J.-M. Herrmann, Helv. Chim. Acta, 2001, 84, 2731–2750 CrossRef CAS.
- H. Harada, T. Sakata and T. Ueda, J. Am. Chem. Soc., 1985, 107, 1773–1774 CrossRef CAS.
- H. Harada, T. Ueda and T. Sakata, J. Phys. Chem., 1989, 93, 1542–1548 CrossRef CAS.
- T. Ohuchi, T. Miyatake, Y. Hitomi and T. Tanaka, Catal. Today, 2007, 120, 233–239 CrossRef CAS.
- H. Miyama, N. Fujii and Y. Nagae, Chem. Phys. Lett., 1980, 74, 523–524 CrossRef CAS.
- Q. Li, K. Domen, S. Naito, T. Onishi and K. Tamaru, Chem. Lett., 1983, 321–324 CrossRef CAS.
- K. Wada, K. Yoshida, Y. Watanabe and T. Suzuki, J. Chem. Soc., Chem. Commun., 1991, 726–727 RSC.
- K. Wada, K. Yoshida, T. Tsuyoshi and Y. Watanabe, Appl. Catal., A, 1993, 99, 21–36 CrossRef CAS.
- K. Wada, K. Yoshida, Y. Watanabe and T. Mitsudo, J. Chem. Soc., Faraday Trans., 1996, 92, 685–691 RSC.
- Z. Kang, C. H. A. Tsang, N.-B. Wong, Z. Zhang and S.-T. Lee, J. Am. Chem. Soc., 2007, 129, 12090–12091 CrossRef CAS.
- K. Schoumacker, C. Geantet, M. Lacroix, E. Puzenat, C. Guillard and J.-M. Herrmann, J. Photochem. Photobiol., A, 2002, 152, 147–153 CrossRef CAS.
- B. Ohtani, B. Pal and S. Ikeda, Catal. Surv. Asia, 2003, 7, 165–176 CrossRef CAS.
- P. Calza, V. Maurino, C. Minero, E. Pelizzetti, M. Sega and M. Vincenti, J. Photochem. Photobiol., A, 2005, 170, 61–67 CrossRef CAS.
- J. S. Hubbard, J. P. Hardy and N. H. Horowitz, Proc. Natl. Acad. Sci. U. S. A., 1971, 68, 574–578 CrossRef CAS.
- J. S. Hubbard, J. P. Hardy and G. E. Voecks, J. Mol. Evol., 1973, 2, 149–166 CrossRef CAS.
- J. S. Hubbard, G. E. Voecks, G. L. Hobby, J. P. ferris, E. A. Williams and D. E. Nicodem, J. Mol. Evol., 1975, 5, 223–241 CrossRef CAS.
- T. Suzuki, K. Wada, M. Shima and Y. Watanabe, J. Chem. Soc., Chem. Commun., 1990, 1059 RSC.
- K. Wada, K. Yoshida, Y. Watanabe and T. Suzuki, Appl. Catal., 1991, 74, L1–L4 Search PubMed.
- K. Wada, K. Yoshida and Y. Watanabe, J. Chem. Soc., Faraday Trans., 1995, 91, 1647–1654 RSC.
- K. Wada, H. Yamada, Y. Watanabe and T. Mitsudo, J. Chem. Soc., Faraday Trans., 1998, 94, 1771–1778 RSC.
- S. Takenaka, T. Tanaka, T. Funabiki and S. Yoshida, Catal. Lett., 1997, 44, 67–74 CrossRef CAS.
- W. B. Fisher, J. F. Van Pepper and M. Grayson (Eds.), Kirk-Othner Encyclopedia of Chemical Technology, 3rd ed., Wiley, New York, 1979, vol. 7, p. 411 Search PubMed.
- Y. Shiraishi, Y. Teshima and T. Hirai, Chem. Commun., 2005, 4569–4571 RSC.
- Y. Shiraishi, Y. Teshima and T. Hirai, J. Phys. Chem. B, 2006, 110, 6257–6263 CrossRef CAS.
- T. Tanaka, H. Nojima, H. Yoshida, H. Nakagawa, T. Funabiki and S. Yoshida, Catal. Today, 1993, 16, 297–307 CrossRef CAS.
- H. Yoshida, T. Tanaka, M. Yamamoto, T. Funabiki and S. Yoshida, Chem. Commun., 1996, 2125–2126 RSC.
- H. Yoshida, T. Tanaka, M. Yamamoto, T. Yoshida, T. Funabiki and S. Yoshida, J. Catal., 1997, 171, 351–357 CrossRef CAS.
- H. Yoshida, C. Murata and T. Hattori, Chem. Lett., 1999, 901–902 CrossRef CAS.
- H. Yoshida, C. Murata and T. Hattori, J. Catal., 2000, 194, 364–372 CrossRef CAS.
- C. Murata, H. Yoshida and T. Hattori, Chem. Commun., 2001, 2412–2413 RSC.
- H. Yoshida, T. Shimizu, C. Murata and T. Hattori, J. Catal., 2003, 220, 226–232 CrossRef CAS.
- H. Yoshida, Curr. Opin. Solid State Mater. Sci., 2003, 7, 435–442 CrossRef CAS.
- F. Amano and T. Tanaka, Catal. Commun., 2005, 6, 269–273 CrossRef CAS.
- F. Amano and T. Tanaka, Chem. Lett., 2006, 35, 468–473 CrossRef CAS.
- F. Amano, T. Yamaguchi and T. Tanaka, J. Phys. Chem. B, 2006, 110, 281–288 CrossRef CAS.
- H. Kanai, M. Nakao and S. Imamura, Catal. Commun., 2003, 4, 405–409 CrossRef CAS.
- T. Tanaka, S. Takenaka, T. Funabiki and S. Yoshida, Chem. Lett., 1994, 809–812 CAS.
- T. Tanaka, S. Takenaka, T. Funabiki and S. Yoshida, Acid–base Catalysis II: Proceedings of the International Symposium on Acid–base Catalysis II, Elsevier, 1994, 485–490 Search PubMed.
- S.-C. Moon, M. Fujino, H. Yamashita and M. Anpo, J. Phys. Chem. B, 1997, 101, 369–373 CrossRef CAS.
- N. Soggiu, H. Cardy, J. L. Habib Jiwan, I. Leray, J. P. Soumillion and S. Lacombe, J. Photochem. Photobiol., A, 1999, 124, 1–8 CrossRef CAS.
- H. Yamashita, S. Nishio, S. Imaoka, M. Shimada, K. Mori, T. Tanaka and N. Nishiyama, Top. Catal., 2008, 47, 116–121 CrossRef CAS.
- H. Yamashita, K. Mori, S. Shironita and Y. Horiuchi, Catal. Surv. Asia, 2008, 12, 88–100 CrossRef CAS.
- Y. Shiraishi, M. Morishita and T. Hirai, Chem. Commun., 2005, 5977–5979 RSC.
- M. Morishita, Y. Shiraishi and T. Hirai, J. Phys. Chem. B, 2006, 110, 17898–17905 CrossRef CAS.
- Y. Shiraishi, H. Ohara and T. Hirai, J. Catal., 2008, 254, 365–373 CrossRef CAS.
- K. Zama, A. Fukuoka, Y. Sasaki, Y. Fukushima and M. Ichika, Catal. Lett., 2000, 66, 251–253 CrossRef CAS.
- H. H. Lopez and A. Martinez, Catal. Lett., 2002, 83, 37–41 CrossRef CAS.
- Y. Hu, N. Wada, K. Tsujimaru and M. Anpo, Catal. Today, 2007, 120, 139–144 CrossRef CAS.
- Y. Hu, Y. Nagai, D. Rahmawaty, C. Wei and M. Anpo, Catal. Lett., 2008, 124, 80–84 CrossRef CAS.
- M. S. Hamdy, O. Berg, J. C. Jansen, T. Maschmeyer, J. A. Moulijn and G. Mul, Chem.–Eur. J., 2006, 12, 620–628 CrossRef.
- G. Mul, W. Wasylenko, M. S. Hamdy and H. Frei, Phys. Chem. Chem. Phys., 2008, 10, 3131–3137 RSC.
- O. Berg, M. S. hamdy, T. Maschmeyer, J. A. Moulijn, M. Bonn and G. Mul, J. Phys. Chem. C, 2008, 112, 5471–5475 CrossRef CAS.
- E. L. Clennan, W. Zhou and J. Chan, J. Org. Chem., 2002, 67, 9368–9378 CrossRef CAS.
- A. Corma and H. Garcia, Chem. Commun., 2004, 1443–1459 RSC.
- F. Blatter and H. Frei, J. Am. Chem. Soc., 1994, 116, 1812–1820 CrossRef CAS.
- H. Sun, F. Blatter and H. Frei, Catal. Lett., 1997, 44, 247–253 CrossRef CAS.
- F. Blatter, H. Sun, S. Vasenkov and H. Frei, Catal. Today, 1998, 41, 297–309 CrossRef CAS.
- H. Frei, Science, 2006, 313, 309–310 CrossRef CAS.
- K. B. Myli, S. C. Larsen and V. H. Grassian, Catal. Lett., 1997, 48, 199–202 CrossRef CAS.
- Y. Xiang, S. C. Larsen and V. H. Grassian, J. Am. Chem. Soc., 1999, 121, 5063–5072 CrossRef CAS.
- K. Shimizu, H. Akahane, T. Kodama and Y. Kitayama, Appl. Catal., A, 2004, 269, 75–80 CrossRef CAS.
- A. Hiskia and E. Papaconstantinou, Polyhedron, 1988, 7, 477–481 CrossRef CAS.
- E. Fornal and C. Giannotti, J. Photochem. Photobiol., A, 2007, 188, 279–286 CrossRef CAS.
- A. Maldotti, A. Molinari and F. Bigi, J. Catal., 2008, 253, 312–317 CrossRef CAS.
- S. Farhadi and M. Afshari, J. Chem. Res., 2006, 188–191 Search PubMed.
- S. Farhadi, M. Afshari, M. Maleki and Z. Babazadeh, Tetrahedron Lett., 2005, 46, 8483–8486 CrossRef CAS.
- S. Farhadi, Z. Babazadeh and M. Maleki, Acta Chim. Solv., 2006, 53, 72–76 Search PubMed.
- S. Farhadi and Z. Momeni, J. Mol. Catal. A: Chem., 2007, 277, 47–52 CrossRef CAS.
- M. D. Tzirakis, I. N. Lykakis, G. D. Panagiotou, K. Bourikas, A. Lycourghiotis, C. Kordulis and M. Orfanopoulos, J. Catal., 2007, 252, 178–189 CrossRef CAS.
- J. Gascon, M. D. Hernandez-Alonso, A. R. Almeida, G. P. M. van Klink, F. Kapteijn and G. Mul, ChemSusChem, 2008, 1, 981–983 CrossRef CAS.
- H. K. Chae, D. Y. Siberio-Perez, J. Kim, Y. B. Go, M. Eddaoudi, A. J. Matzger, M. O'Keeffe and O. M. Yaghi, Nature, 2004, 427, 523–527 CrossRef CAS.
- P. Mahata, G. Madras and S. Natarajan, J. Phys. Chem. B, 2006, 110, 13759–13768 CrossRef CAS.
- M. Alvaro, E. CArbonell, B. Ferrer, F. Xamena and H. Garcia, Chem.–Eur. J., 2007, 13, 5106–5112 CrossRef CAS.
- E. C. C. Baly, I. M. Heilbron and W. F. Barker, J. Chem. Soc., Trans., 1921, 119, 1025–1035 RSC.
- E. C. C. Baly, I. M. Heilbron and D. P. Hudson, J. Chem. Soc., Trans., 1922, 121, 1078–1088 RSC.
- T. Inoue, A. Fujishima, S. Konishi and K. Honda, Nature, 1979, 277, 637–638.
- H. Fujiwara, H. Hosokawa, K. Murakoshi, Y. Wada and S. Yanagida, Langmuir, 1998, 14, 5154–5159 CrossRef CAS.
- Y. Kohno, H. Ishikawa, T. Tanaka, T. Funabiki and S. Yoshida, Phys. Chem. Chem. Phys., 2001, 3, 1108–1113 RSC.
- Y. Kohno, T. Tanaka, T. Funabiki and S. Yoshida, Phys. Chem. Chem. Phys., 2000, 2, 2635–2639 RSC.
- Y. Kohno, T. Tanaka, T. Funabiki and S. Yoshida, Chem. Lett., 1997, 993 CrossRef CAS.
- Y. Kohno, T. Tanaka, T. Funabiki and S. Yoshida, Phys. Chem. Chem. Phys., 2000, 2, 5302–5307 RSC.
- N. Ulagappan and H. Frei, J. Phys. Chem. A, 2000, 104, 7834–7839 CrossRef CAS.
- W. Lin and H. Frei, J. Am. Chem. Soc., 2005, 127, 1610–1611 CrossRef CAS.
- M. Subrahnanyam, S. Kaneco and N. Alonso-Vante, Appl. Catal., B, 1999, 23, 169–174 CrossRef.
- A. H. Yahaya, M. A. Gondal and A. Hameed, Chem. Phys. Lett., 2004, 400, 206–212 CrossRef CAS.
- P.-W. Pan and Y.-W. Chen, Catal. Commun., 2007, 8, 1546–1549 CrossRef CAS.
- M. Anpo, H. Yamashita, Y. Ichihashi, Y. Fujii and M. Honda, J. Phys. Chem. B, 1997, 101, 2632–2636 CrossRef CAS.
- J.-S. Hwang, J.-S. Chang, S.-E. Park, K. Ikeue and M. Anpo, Top. Catal., 2005, 35, 311–319 CrossRef CAS.
- K. Ikeue, H. Yamashita and M. Anpo, J. Phys. Chem. B, 2001, 105, 8350–8355 CrossRef CAS.
- P. Usubharatana, D. McMartin, A. Veawab and P. Tontiwachwuthikul, Ind. Eng. Chem. Res., 2006, 45, 2558 CrossRef CAS.
- G. Guan, T. Kida, T. Harada, M. Isayama and A. Yoshida, Appl. Catal., A, 2003, 249, 11–18 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2009 |