Large-scale synthesis of defined cobalt nanoparticles and magnetic metal–polymer composites
Justus
Schällibaum
a,
Florian H.
Dalla Torre
a,
Walter R.
Caseri
*b and
Jörg F.
Löffler
a
aLaboratory of Metal Physics and Technology, Department of Materials, ETH Zurich, Wolfgang-Pauli-Str. 10, 8093 Zurich, Switzerland
bLaboratory of Polymer Technology, Department of Materials, ETH Zurich, Wolfgang-Pauli-Str. 10, 8093 Zurich, Switzerland
First published on 28th September 2009
Abstract
We present a method where ε-cobalt nanoparticles with an average diameter of 4.5 nm can be synthesized in a controlled process and in significantly larger quantities than previously reported in the literature, based on the thermal decomposition of dicobaltoctacarbonyl in the presence of oleic acid and trioctylphosphine oxide (TOPO). Moreover, since the resulting particles are coated with an oleate layer, as shown by infrared (IR) spectroscopy, the colloids can be re-dispersed in organic solvents. These dispersions are suitable for the preparation of nanocomposites by a simple procedure, i.e. mixing of the cobalt dispersion with a polymer solution followed by casting and solvent evaporation. Magnetization measurements confirm the expected superparamagnetic behavior for both the cobalt nanoparticles and the metal–polymer nanocomposites.
1. Introduction
The magnetic properties of nanoparticles can differ from those of their bulk material counterparts, as noted e.g. in refs. 1 and 2. In particular, superparamagnetism can emerge for particles with diameters below the critical size of magnetic domain-wall formation, provided that most particles are well separated from each other.3 A convenient representative of a metal which shows superparamagnetism in the colloidal state is cobalt;4 however, the synthesis of large quantities of such particles which are present in a defined state is difficult and has been barely achieved so far. Accordingly, this report deals with the reproducible synthesis of large quantities of cobalt nanoparticles and, moreover, their use in the manufacture of superparamagnetic nancomposites.Previously, magnetic cobalt nanoparticles were synthesized, for instance, by reducing cobalt(II) chloride5 or cobalt(II) acetate6 or by thermolysis of dicobaltoctacarbonyl.7–10 The latter method has found particular attention, since it offers the advantage that the reaction byproducts can be easily removed, as only volatile byproducts (carbon monoxide) arise upon nanoparticle formation. The procedures reported so far are based on the rapid injection of dissolved dicobaltoctacarbonyl into a hot solvent, wherein the general mechanism of cobalt particle formation is often deduced from the La Mer11 method: When the cobalt precursor (CoCl2, (CH3COO)2Co or Co2(CO)8) is injected into a hot (typically 180–200 °C) solvent, metal atoms are supposed to coalesce spontaneously until a critical concentration is reached where the metal atoms rapidly form small clusters. These clusters subsequently grow by diffusion, which is believed to proceed under the conditions commonly applied during the cobalt precursor compound injection period. The temporal separation of the particle nucleation and growth processes triggers the formation of nanoscale particles with a relatively narrow size distribution. However, upon injection of larger amounts of dicobaltoctacarbonyl (1 g or more), the resulting cobalt particles become, according to our observations, rather inhomogeneous in size when the usual methods are employed. This probably occurs because the limited injection rate prolongs the injection period, interfering with nucleation and particle formation.
The size and shape of the cobalt particles are usually controlled by the interface between the particles and the adsorbed species.6,12 In the absence of polymers, which might act as stabilizing agents, this control can be supported by adding coordinating agents such as oleic acid,13 which has also been used in combination with trioctylphosphine oxide (TOPO)5,7 or triphenylphosphine (TPP).8 Remarkably, the presence of TOPO or TPP during the preparation of cobalt nanoparticles yields the metastable ε-cobalt phase, which shows a crystal symmetry like that in β-manganese.5,7,8,10 Such particles can in principle be isolated by precipitation with ethanol; however, it is hardly evident if such particles could be re-dispersed, which would allow the preparation of metal–polymer nanocomposites with randomly dispersed particles. In fact, especially in the preparation of nanocomposites, the cobalt particles have mostly been synthesized only in situ. For this purpose dicobaltoctacarbonyl was decomposed in the presence of a dissolved polymer,14–19 which stabilized the emerging cobalt particles, a strategy which was also transferred to the preparation of nanocomposites with iron or iron oxide by decomposing iron pentacarbonyl(Fe(CO)5).20–22 However, in those routes the average particle size depends strongly on the applied polymer and the metal content in the reaction system, which complicates the preparation of defined composites. Accordingly, the average particle sizes ranged from 5 nm to 100 nm depending on the polymer and the ratio between metal carbonyl complex and polymer. Thus the type of magnetism could not be specifired a priori, i.e. superparamagnetic or ferromagnetic properties were obtained at room temperature, depending on the particle size. In the case of larger particles (>20 nm), one-dimensional self-arrangements of particles into chains were also observed during solvent evaporation because of the particle dipole moment.23
The above considerations imply that there is a demand for the synthesis of larger quantities of cobalt nanoparticles with controlled sizes, which are also suitable for preparing nanocomposites of particles whose sizes are independent of the polymer matrix and the final metal content, and where the superparamagnetic behavior of the nanoparticles is also assured in a polymer matrix. The following study is devoted to this subject.
2. Results
2.1 Cobalt nanoparticles
Large quantities of cobalt particles (>1 g) were prepared by thermolysis of Co2(CO)8, as described in detail in the Experimental Section. Importantly, in contrast to related procedures described in the literature (see the Introduction), the Co2(CO)8 was not injected into the hot solvent but dissolved at room temperature. Only after adding oleic acid and TOPO was the reaction mixture rapidly heated to fast reflux. Therefore the solvent's boiling temperature defined the temperature during the formation of the cobalt particles, which varied between 111 °C and 196 °C for the applied solvents (toluene, chlorobenzene, p-xylene, 1,2,3-trichloropropane, σ-dichlorobenzene, and decaline, i.e. a mixture of cis + transdecahydronaphthalene). Only the reaction in toluene led to a homogeneous, magnetic dispersion of cobalt particles (the magnetic properties of the reaction mixtures were qualitatively evaluated by exposure to an external magnet). In contrast, when a typical procedure described in the literature7 was applied for large-scale synthesis in toluene (injection of Co2(CO)8 (dissolved in toluene) through a septum in the hot, refluxing reaction mixture (containing the same quantities of oleic acid and TOPO)), black precipitates emerged at the bottom of the reaction vessel.The cobalt particles were precipitated from toluene dispersions by adding ethanol, followed by washing of the filtered particles with ethanol in order to remove non-adsorbed TOPO and oleic acid, as described in detail in the Experimental Section. X-ray diffraction (XRD) measurements and electron diffraction studies were performed to identify the crystalline structure of the precipitated particles. The peaks in the X-ray pattern (see Fig. 1(a)) were broad and relatively weak due to the small particle size and the large quantity of adsorbed organic matter (oleate; see below). All XRD peaks agreed with those of metastable ε-cobalt. This structure was also found in several reports on the preparation of cobalt in the presence of TOPO.5,7,10 No distinct peaks which could correspond to cobalt oxide were detected, even after storing samples at ambient conditions for three months. This indicates that excessive oxidation is strongly retarded by the coordination of oleate on the particle surface, although naturally a certain degree of oxidation not manifested in XRD cannot be excluded. After removing the organic surface layer (oleate, see below) by annealing the cobalt powders at 500 °C for 3 h under argon, the particles became very sensitive to air. When such particles were exposed to ambient atmosphere, excessive cobalt oxide formed within as little as two days.
 | ||
| Fig. 1 (a) XRD pattern of precipitated cobalt particles; (b) electron diffraction pattern of cobalt particles (21.9% w/w) embedded in polychloroprene; and (c) radially-integrated intensities from (b), in which the sharp lines represent the position and relative intensities of ε-cobalt (d-spacing taken from ref. 5). | ||
Fig. 2 shows a typical TEM micrograph for as-prepared cobalt nanoparticles in the absence of a polymer matrix with a mean particle size of 4.5 nm. The darker contrast in the middle of Fig. 2 is due to particle agglomeration. Nevertheless, the particles are rather uniform in size and shape.
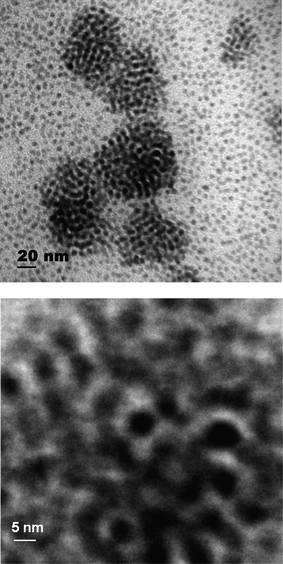 | ||
| Fig. 2 TEM micrograph for as-prepared cobalt nanoparticles in the absence of a polymer matrix, the lower image is a magnification of the darker contrast area in the middle of the upper image. | ||
The infrared spectra of the precipitated and washed particles and, for comparison, of oleic acid and TOPO are displayed in Fig. 3. Signals in the region of the C≡O stretching vibrations of the carbonyl groups of Co2(CO)8 (which appear at 2150–1750 cm−1) are absent in the spectrum of the precipitated cobalt particles (Fig. 3a), implying that the cobalt carbonyl precursor has decomposed completely. The spectrum of neat oleic acid shows an intense and sharp peak at 1710 cm−1, which is due to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration of the carboxylic acid24 (Fig. 3b), and that of pure TOPO shows a characteristic band at 1144 cm−1, which is derived from the PO group24 (Fig. 3c). These bands were missing in the spectrum of the Co nanoparticles, indicating that the excess of oleic acid and TOPO was efficiently removed by washing the particles with ethanol. Instead two new bands arose at 1557 cm−1 and 1417 cm−1 which are characteristic of the asymmetric and symmetric COO− stretching vibration of oleate on cobalt.13 Hence it appears that the cobalt surfaces were covered by a strongly adsorbed oleate layer.
O stretching vibration of the carboxylic acid24 (Fig. 3b), and that of pure TOPO shows a characteristic band at 1144 cm−1, which is derived from the PO group24 (Fig. 3c). These bands were missing in the spectrum of the Co nanoparticles, indicating that the excess of oleic acid and TOPO was efficiently removed by washing the particles with ethanol. Instead two new bands arose at 1557 cm−1 and 1417 cm−1 which are characteristic of the asymmetric and symmetric COO− stretching vibration of oleate on cobalt.13 Hence it appears that the cobalt surfaces were covered by a strongly adsorbed oleate layer.
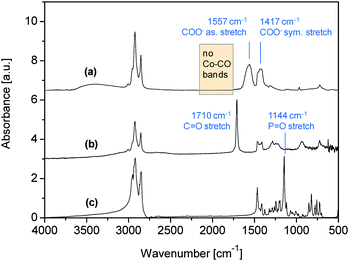 | ||
| Fig. 3 IR spectra of (a) Co nanoparticles; (b) oleic acid; and (c) TOPO. | ||
Thermogravimetric analysis of the oleate-capped cobalt nanoparticles (Fig. 4) reveals a decrease in mass starting at 286 °C and ceasing at 376 °C, with a maximum decomposition rate at 342 °C (not shown). The total mass loss at 376 °C was 70%, and was probably caused by the decomposition of oleate. This assumption is supported by elemental analyses, where the content of carbon amounted to 55.48% w/w and that of hydrogen to 8.54% w/w. From the carbon content and the stoichiometry of oleate an oxygen content of 8.21% w/w was calculated; this leads finally to an oleate fraction of 72% w/w. These values seem to be in reasonable agreement with the organic contents of dodecanethiol-coated silver particles of similar diameter,25 if we take into account the different lengths of dodecanethiol and oleate molecules.
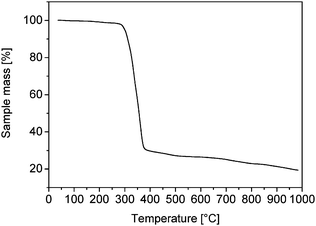 | ||
| Fig. 4 Thermogravimetric analysis of the oleat-capped Co nanoparticles revealing a total mass loss of 70%. | ||
2.2 Nanocomposites
Nanocomposites of cobalt particles and various polymers were prepared by mixing a polymer solution with re-dispersed cobalt nanoparticles. The polymers employed were low-density polyethylene, polycarbonate on the basis of bisphenol A (PC), poly(methyl methacrylate) (PMMA), polychloroprene, polyisobutylene (PIB) and an aliphatic polyurethane. The cobalt-containing polymer solutions were spread on a glass substrate to yield translucent films after solvent evaporation; these were investigated using an optical microscope. First the compatibility of the particles and the polymer was rated by eye. In the composites with PMMA, PC and PIB, macroscopic round-shaped inhomogeneities emerged during the evaporation of the solvent which were about 0.3–1 mm in size. All other polymer solutions yielded smooth, homogeneous films where no phase separation was observed, i.e. the above method can be applied in the preparation of homogeneous nanocomposites with various polymers.The surface-modified cobalt particles are most likely rather non-polar since the formation of carboxylate groups indicates their coordination to the particle surface, i.e. the hydrocarbon group of the oleic acid is anticipated to be exposed to the air. Hence, interaction of the particles is therefore expected to be with hydrocarbonpolymer segments, which are little pronounced in PMMA and PC. Therefore, particle agglomeration in those polymers is not surprising. On the other hand, particle agglomeration in the hydrocarbonpolymerPIB could indicate that kinetic effects might also play a role in related agglomeration processes as PIB is a quasi-liquid polymer at room temperature promoting higher mobility of the enclosed particles than in the other polymers.
In the following, polychloroprene was selected as a matrix for further studies because this polymer was the most favorable for processing from solution and film preparation. Films containing 9.9% w/w or 21.9% w/w cobalt were prepared. These films still displayed elastic properties, but became brittle after three months at ambient conditions. Samples comprising 9.9% w/w and 21.9% w/w cobalt embedded in a polychloroprene matrix were investigated by TEM, as shown in Fig. 5. A typical micrograph of a composite with 9.9% w/w cobalt (Fig. 5(a)) shows particles which are dispersed randomly within the polymer matrix. Upon incorporation of 21.9% w/w of cobalt, the particles arranged themselves partially into an ordered packing with a pattern resembling a cubic rather than a hexagonal grid. Fig. 5(b) reveals the self-assembling character of a more densily-populated area of particles. The inset to Fig. 5(b) depicts a high-resolution TEM image of a single particle, where the lattice planes are resolved. In the absence of polymers, agglomerates were found (see Fig. 2), which were disordered, i.e. they did not form superstructures. In the composite samples with polychloroprene, however, the particles were randomly distributed, i.e. agglomerates did not occur. The particle sizes were similar in all samples independent of their polymer content, i.e. the process of composite preparation did not generate a significant change in particle diameter or cause particle agglomeration.
 | ||
| Fig. 5 Cobalt particles in polychloroprene: (a) 9.9% w/w cobalt; (b) 21.9% w/w cobalt. Inset of (b): high-resolution TEM image of a single particle, and (c) size distribution of cobalt particles from the sample with 9.9% w/w cobalt. | ||
The particle size distribution (shown in Fig. 5(c)) which has been evaluated from the TEM micrograph (5(a)), and the mean particle diameter is approximately 4.5 nm. However, since the interface between metal core and adsorbed oleate may be blurred in the TEM images, the particles may in fact even be somewhat smaller.
Electron diffraction was performed with a sample containing 21.9% w/w cobalt embedded in a polychloroprene matrix. The diffraction patterns shown in Fig. 1(b) and 1(c) also show the presence of ε-cobalt in the polymer matrix, as the position and relative intensities of the previously5 reported d-spacing values of ε-cobalt agree well with the electron diffraction peaks. Hence, electron diffraction largely confirms the XRD results, although the weak and broad peak centered at ∼ 1.5 Å might be indicative of cobalt oxide (CoO).
2.3 Magnetic properties
The magnetic properties were studied in the precipitated cobalt particles in the absence of polymer and in samples with 9.9% w/w and 21.9% w/w cobalt in polychloroprene. Fig. 6(a) shows the magnetization curves for the precipitated cobalt nanoparticles at 300 K and 10 K. For an average particle size of 4.5 nm, most particles were expected to show superparamagnetic behavior at room temperature. Indeed, as predicted the coercivity increased from 15 Oe at 300 K to 550 Oe at 10 K (see inset of Fig. 6(a)), which is typical for a superparamagnetic-to-ferromagnetic transition; the coercivity of 15 Oe in the hysteresis loop at 300 K results from the testing method when using a superconducting solenoid as a magnetic source.26 The susceptibility at high fields in the hysteresis loop at 10 K probably results from a disordered spin structure at the surface of the nanoparticles.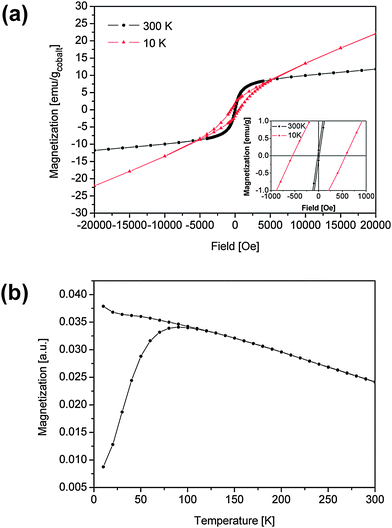 | ||
| Fig. 6 (a) Hysteresis loop of precipitated cobalt nanoparticles at temperatures of 10 K and 300 K (inset: hysteresis near zero field); (b) magnetization of precipitated cobalt nanoparticles after ZFC and FC in an external field of 400 Oe. | ||
Fig. 6(b) shows the magnetization as a function of temperature for the precipitated cobalt nanoparticles recorded at temperatures between 10 K and 300 K after zero-field-cooling (ZFC) and field-cooling (FC) in a magnetic field of 400 Oe. The zero-field-cooled measurements reveal a blocking temperature TB of 90 K. With 25kBTB ≈ KV,27 where K is the anisotropy constant and V is the average particle volume, K ≈ 6.4 × 106 erg cm−3. This value is even higher than the bulk value of (hcp) cobalt, which is 5.2 × 106 erg cm−3. This agrees, however, with other reports where higher values of the anisotropy have been observed for small nanoparticles.28,29 The broad transition from superparamagnetism to ferromagnetism is probably due to the particle size distribution (see Fig. 5(c)).
Hysteresis loops for the composites with 9.9% w/w and 21.9% w/w cobalt are shown in Fig. 7(a), together with the hysteresis loop for the precipitated particles for comparison (see also Fig. 6(a)). The magnetization curves at 300 K of the freshly prepared composite samples (Fig. 6(a)) show the same coercivity of 15 Oe as the precipitated particles, suggesting superparamagnetic behaviour (also taking into account the results of ref. 26). However, considerable changes in the saturation magnetization can be observed. The sample with 9.9% w/w shows a saturation magnetization of 50.6 emu/g (the mass unit refers to the mass of cobalt), while that with 21.9% w/w shows a value of 35.3 emu/g, compared to 9.9 emu/g for the precipitated particles (containing 30% w/w cobalt and 70% w/w oleate). This indicates a reduction in magnetization per cobalt particle with increasing cobalt fraction, indicating that the surface of the cobalt nanoparticles is partly oxidized, in particular in absence of the polymer. A reduction of the saturation magnetization has also been observed by Yang et al.8 upon dilution of cobalt nanoparticles in wax and was attributed there to a modification of the exchange coupling between the adjacent particles upon dilution of the nanoparticles.
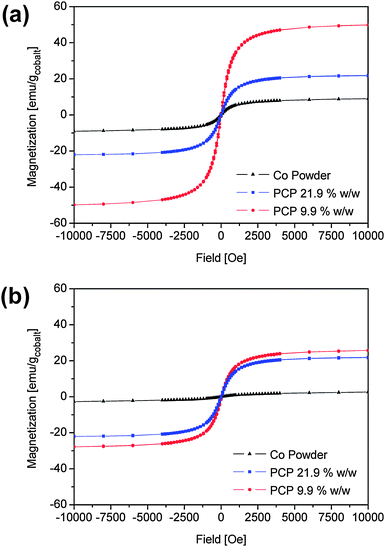 | ||
| Fig. 7 Magnetization curves for precipitated cobalt nanoparticles and composites containing polychloroprene and 9.9% w/w and 21.9% w/w cobalt nanoparticles. (a) Freshly-prepared samples; (b) samples after three months at ambient conditions. | ||
After three months in ambient conditions no changes in the coercivity were noted but the saturation magnetization decreased drastically (Fig. 7(b)). Although XRD and electron diffraction measurements did not reveal significant changes in the crystalline phases, the decrease in magnetization is probably due to further particle surface oxidation, which was, however, not pronounced enough to yield peaks in the related diffraction patterns (see Fig. 1(a) and 1(c)).
3. Discussion
3.1 Large-scale synthesis of nanoparticles
The results of this study show that the formation of large amounts of homogeneously-dispersed cobalt particles by decarbonylation of Co2(CO)8 in the presence of both oleic acid and TOPO is sensitive to the reaction temperature, the temperature profile (i.e. whether Co2(CO)8 is injected into the hot solvent, or whether or not a Co2(CO)8 solution is heated) and possibly also the nature of the solvent. Thus different mechanisms for the particle formation may occur, in particular if a Co2(CO)8 solution is heated or if the commonly-applied procedure is used (injection of Co2(CO)8 into a hot solvent). In the latter case, the composition of the reaction mixture is expected to be less uniform than in a Co2(CO)8 solution which has been heated, because the limited rate of Co2(CO)8 supply in the injection process causes an addition of Co2(CO)8 while nucleation and particle growth are already proceeding, limiting the quantity of Co2(CO)8 that can be injected. During heating of the reaction mixture, which contains Co2(CO)8, oleic acid and TOPO, nucleation and particle growth take place simultaneously because no temporal separation of those processes exists. This triggers the formation of large, polydisperse particles. Indeed, relatively high amounts of the stabilizing additives oleic acid and TOPO (mass ratio between oleic acid and cobalt of 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, molar ratio between oleic acid and TOPO of 5:1) were required to obtain homogeneous dispersions without agglomerated cobalt particles, i.e. quantities which exceeded those used in reports on the injection procedure.7 In addition, the presence of TOPO was necessary, as otherwise the reaction mixture did not respond to an external magnetic field, at least not within a reaction period of 24 h. Since TOPO is absent in the precipitated nanoparticles, it seems that TOPO is active only during particle formation. The oxygen atom in TOPO can basically coordinate to cobalt cations and therefore influence the ratio between nucleation and growth reaction rates or stabilize intermediate states on the way to particle formation.
:1, molar ratio between oleic acid and TOPO of 5:1) were required to obtain homogeneous dispersions without agglomerated cobalt particles, i.e. quantities which exceeded those used in reports on the injection procedure.7 In addition, the presence of TOPO was necessary, as otherwise the reaction mixture did not respond to an external magnetic field, at least not within a reaction period of 24 h. Since TOPO is absent in the precipitated nanoparticles, it seems that TOPO is active only during particle formation. The oxygen atom in TOPO can basically coordinate to cobalt cations and therefore influence the ratio between nucleation and growth reaction rates or stabilize intermediate states on the way to particle formation.
It should be emphasized, however, that in contrast to previous studies the procedure presented here makes possible the decomposition of larger quantities of Co2(CO)8 due to the absence of an injecting period. Therefore the volume of synthesized particles can be scaled-up readily. Hence the synthesis route described enables production of large quantities of cobalt nanoparticles in a straightforward, reproducible and easy-to-adapt manner.
3.2 Reproducibility of the synthesis method
The stability and handling of the starting material for the cobalt nanoparticle synthesis, i.e.Co2(CO)8, was never really addressed in previous studies. Here we used a common commercial product (see Experimental Section) which consisted of dark violet crystals moistened with hexane. When exposed to the environment, these crystals became desiccated and took on a whitish discoloration at their surfaces after only a few minutes. Upon exposure of those crystals to common solvents, such as toluene, p-xylene, decaline, hexane, σ-dichlorobenzene, or chlorobenzene, black, insoluble residues remained which were not present when the material from a freshly opened bottle was used. Re-moistening of Co2(CO)8 with hexane and storage of once-opened bottles under argon atmosphere showed a marginal retardation in these degradation effects, probably due to oxidation products. We also observed that thermolysis of Co2(CO)8 occurred at a slow rate even at room temperature. As a result of all these observations, Co2(CO)8 was stored at 5 °C, and once the bottle was opened, its entire contents (5 g) were employed at once by dissolution in toluene and the resulting solution was used immediately for the experiments.Care also has to be taken upon re-dispersion of the particles. When the precipitated particles were still wet they could be easily re-dispersed to translucent liquids in hexane or toluene. However, when the samples were completely dried only a part of the solids could be re-dispersed in hexane or toluene. Generally this behavior is typical of powders that contain agglomerated particles where the strong interactions between them impede their re-dispersion. Indeed, TEM micrographs revealed a pronounced tendency of the particles to form agglomerates (see Fig. 2) when the re-dispersed particles were dried (by evaporation on the TEM grid). In contrast, re-dispersion of wet precipitates led to a random particle distribution in the polymers (see Fig. 5(b)).
4. Conclusions
A large-scale synthesis of defined re-dispersable cobalt nanoparticles with average diameters of 4.5 nm was developed, based on the thermal decomposition of dicobaltoctacarbonyl in toluene containing dissolved oleic acid and trioctylphosphine oxide. In contrast to common synthetic routes where dicobaltoctacarbonyl is injected into the hot solvent, we dissolved the Co2(CO)8 together with the accessory agents oleic acid and TOPO in toluene at room temperature, followed by heating to fast reflux. The accessory agents must be present in large quantities to prevent (partial) precipitation of the particles. The cobalt nanoparticles are formed as ε-phase cobalt (possessing the β-manganese structure), which was also observed previously when TOPO was applied.7,10 The magnetic properties of the precipitated cobalt nanoparticles show typical superparamagnetic features.Precipitated cobalt particles which are in the wet state can be readily re-dispersed in organic solvents, which makes possible the preparation of metal–polymer nanocomposites by a convenient procedure, i.e. the mixing of particle dispersions with polymer solutions followed by casting and drying. TEM investigations revealed that the cobalt nanoparticles can be distributed randomly in a polymer matrix without considerable particle agglomeration. Magnetization measurements showed that the superparamagnetic properties of the cobalt nanoparticles were retained in the metal–polymer composites. Generally, composites with superparamagnetic particles have been proposed for applications such as electromagnetic interference shielding or biomedical sensing.
5. Experimental Section
5.1 Chemicals
Dicobaltoctacarbonyl [Co2(CO)8, 90–95%, moistened with hexane], toluene (99.7% anhydrous), decaline (mixture of cis+transdecahydronaphtalene, 98.0%), 1,2,3-trichloropropane (98.0%), hexane (97.0%) and a polyurethane (Tecoflex, an aliphatic polyurethane) were acquired from FLUKA, Buchs, Switzerland; ethanol (99.0% anhydrous) from Merck, Darmstadt, Germany; oleic acid (C18H34O2, 90%) and chlorobenzene (99.0%) from ABCR, Karlsruhe, Germany; σ-dichlorobenzene (99.0%), p-xylene (99.0%), trioctylphosphine oxide ([CH3(CH2)7]3PO, 99%), polyisobutylene (MW 4![[hair space]](https://www.rsc.org/images/entities/char_200a.gif) 700000, density 0.9180 g cm−3) and polycarbonate (MW 64000, density 1.20 g cm−3, on the basis of Bisphenol A) from ACROS organics, Geel, Belgium; poly(methyl methacrylate) (PMMA, MW 350000, density 1.17 g cm−3) and polychloroprene (85% trans, 10% cis, density 1.23 g cm−1) from Sigma Aldrich, Buchs, Switzerland; and low-density polyethylene (LDPE, Lupolen 1810 H, density 0.9190 g cm−3) from Basell, Wesseling, Germany. All chemicals were used without further purification.
700000, density 0.9180 g cm−3) and polycarbonate (MW 64000, density 1.20 g cm−3, on the basis of Bisphenol A) from ACROS organics, Geel, Belgium; poly(methyl methacrylate) (PMMA, MW 350000, density 1.17 g cm−3) and polychloroprene (85% trans, 10% cis, density 1.23 g cm−1) from Sigma Aldrich, Buchs, Switzerland; and low-density polyethylene (LDPE, Lupolen 1810 H, density 0.9190 g cm−3) from Basell, Wesseling, Germany. All chemicals were used without further purification.
5.2 Nanoparticle synthesis
In a typical experiment, 5 g (14.6 mmol) of dicobaltoctacarbonyl (content of cobalt atoms 1.7230 g, 29.2 mmol; we assume that the cobalt atoms are mostly present in cobalt nanoparticles) from a freshly opened bottle was dissolved in 60 mL of toluene. 8.6170 g (30.5 mmol) of oleic acid were added to this solution under stirring and subsequently a solution of 2.3590 g (6.1 mmol) of TOPO (trioctylphosphine oxide) in 20 mL of toluene. The mass ratio of oleic acid to cobalt amounted to 5:1 and the molar ratio of oleic acid to TOPO was 5:1. The resulting solution, which was kept in a two-necked round-bottomed flask equipped with a reflux condenser and a protecting gas inlet, was thoroughly flushed with either argon or nitrogen (no difference in the results was found between them). The flask was placed in a heating calotte (a calotte was favored over the conventional oil bath-heating/stirring plate system in order to avoid perturbations of the cobalt particles by the magnetic nature of the heating/stirring plate) and the solution was rapidly heated without stirring to a reflux of the condensing drops (approximately 4–5 drops per second). The argon flow was reduced but not halted. After 3 h the heating was stopped and the solution was allowed to cool down to room temperature. The reaction mixture appeared as a black, homogeneous solution. If a magnet was brought close to the reaction vessel, the reaction mixture responded vigorously to the magnetic field.
In order to remove excessive oleic acid and TOPO, a fourfold volume of ethanol (320 mL) was added to the reaction solution, whereupon the cobalt particles precipitated more or less completely as far as can be concluded from the color of the outstanding solution, which changed from deep black to a light brown and which no longer responded to a magnetic field (the drastic reduction in color intensity of the outstanding solution after particle precipitation suggests a yield of precipitated cobalt on the order of 99%). The supernatant was subsequently removed by decanting and the wet precipitates were immediately re-dispersed in hexane (80 mL). This procedure was repeated a second time (addition of 320 mL ethanol, precipitation and decanting, addition of 80 mL hexane), providing a stock solution for further experiments. When a part of the precipitates consisting of cobalt particles was dried in a vacuum oven (40 °C, 0.5 mbar) for 15 h, a waxy, granular material was obtained.
5.3 Preparation of nanocomposites
The particles were embedded in polychloroprene (PCP) using the above-described, freshly-prepared stock cobalt particle solution (containing 1.7230 g (29.2 mmol) cobalt atoms and 4.0203 g (14.3 mmol) adsorbed oleate, dispersed in 80 mL hexane). From this solution 8.220 mL (containing 3.0 mmol cobalt atoms) and 44.70 mL (containing 16.3 mmol cobalt atoms), respectively, were removed and added, under stirring, in each case to 1.2 g (13.7 mmol, referred to constitutional repeat units) of PCP dissolved in 12 mL toluene. The cobalt-containing polymer solutions were poured into round aluminium dishes (5 cm in diameter), and the solvent was allowed to evaporate for roughly 8 h under ambient conditions until a polymer film about 0.5 mm thick remained. Taking into account the quantities of the starting materials, the metal content in the films resulted in 9.9% w/w and 21.9% w/w, which roughly corresponds to 1.4% v/v and 3.1% v/v, using a density of 1.23 g cm−3 for polychloroprene (according to the supplier), 0.89 g cm−3 for oleate (corresponding to the density of oleic acid30) and 8.80 g cm−3 for cobalt.Films with several other polymers, which were soluble in organic solvents and expected to be suited for film formation, were also applied, in particular composites with polyisobutylene, a polycarbonate, poly(methyl metacrylate), low-density polyethylene, and an aliphatic polyurethane were prepared according to the method described above. 0.1010 g (1.8 mmol, referred to constitutional repeat units) polyisobutylene, 0.1320 g polycarbonate (0.52 mmol, referred to constitutional repeat units), 0.1290 g poly(methyl methacrylate) (1.3 mmol, referred to constitutional repeat units), and 0.1300 g (exact composition and therefore also molar mass unknown) polyurethane were dissolved in 5 mL THF while 0.1010 g polyethylene was dissolved in 5 mL p-xylene. Each polymer solution was mixed with 2.4 mL of the cobalt stock solution (containing 0.88 mmol cobalt) to yield a metal content of 17–19% w/w. Small amounts of these cobalt-containing polymer solutions were dropped on a glass substrate to yield translucent films after evaporation of the solvent. These films were examined with an optical microscope (Leica DM RX) in transmission.
5.4 Analyses
Transmission infrared spectra were obtained using a Bruker Vertex 70 FT-IR spectrometer. Spectra of pure TOPO were taken as a KBr disc. Spectra of neat oleic acid were recorded with the ATR technique (attenuated total reflection, device based on a Si-crystal), while the spectra of (oleate-capped) cobalt particles were obtained by placing ∼0.1 mL from the above-described stock solution in hexane on a KBr disc followed by evaporation of the solvent.Thermogravimetric analyses (TGA) were carried out on a TGA/SDTAe apparatus from Mettler-Toledo. Small amounts of the precipitated, waxy cobalt powder were heated to 1000 °C at a constant heating rate of 20 K min−1. The decomposition of Co2(CO)8 was followed at heating rates of 5 K min−1 to 600 °C.
Transmission electron microscopy (TEM) investigations were performed using a Phillips CM20 instrument operating at 200 kV, and high-resolution TEM was performed on a Phillips CM30 operating at 300 kV. Samples were prepared by drying solutions on a 300 mesh copper grid. In particular, the above-described stock solutions were diluted to a ratio of 1:7 with hexane, and 1 μL of the resulting solution was placed on a copper grid followed by evaporation of the solvent. In order to take images of the cobalt nanoparticles embedded in polychloroprene, 0.1 g of polychloroprene was dissolved in 4 mL and 0.72 mL of the stock solution to yield metal-to-polymer ratios of 21.9% w/w and 9.9% w/w, respectively. These solutions were also diluted to a ratio of 1:7 with hexane, and 1 μL of the resulting solutions was placed separately on a 300 mesh copper grid whereupon the solvent evaporated in ambient conditions.
X-ray diffraction (XRD) measurements were performed with a STOE STADI P diffractometer in reflection using Co Kα radiation (λ = 1.78896 Å and a 140° image-plate detector (STOE, IP-PSD)). The samples were prepared by placing small amounts of the above-described stock solution on a sample holder, followed by evaporation of the solvent in ambient conditions. This procedure was repeated five times, which yielded a thin film of cobalt particles of several micrometers in thickness.
The magnetic properties were characterized in the temperature range of 10–300 K using a Physical Property Measurement System (PPMS, Quantum Design), equipped with a 9 T magnet, using a dc magnetization option. For the zero-field-cooled (ZFC) and field-cooled (FC) procedure a field of 400 Oe was applied.
References
- D. L. Leslie-Pelecky and R. D. Rieke, Chem. Mater., 1996, 8, 1770 CrossRef CAS.
- R. H. Kodama, J. Magn. Magn. Mater., 1999, 200, 359 CrossRef CAS.
- J. F. Löffler, H. B. Braun and W. Wagner, Phys. Rev. Lett., 2000, 85, 1990 CrossRef CAS.
- J. P. Chen, C. M. Sorensen, K. J. Klabunde and G. C. Hadjipanayis, J. Appl. Phys., 1994, 76, 6316 CrossRef CAS.
- S. Sun and C. B. Murray, J. Appl. Phys., 1999, 85, 4325 CrossRef CAS.
- H. Shao and Y. Huang, J. Appl. Phys., 2006, 99, 08N702 CrossRef.
- V. F. Puntes, K. M. Krishnan and A. P. Alivisatos, Science, 2001, 291, 2115 CrossRef CAS.
- H. T. Yang, Y. K. Su, C. M. Shen, T. Z. Yang and H. J. Gao, Surf. Interface Anal., 2004, 36, 155 CrossRef CAS.
- Y. Bao, B. Pakhomov and K. M. Krishnan, J. Appl. Phys., 2005, 97, 10J317.
- D. P. Dinega and M. G. Bawendi, Angew. Chem., Int. Ed., 1999, 38, 1788 CrossRef CAS.
- V. K. LaMer and R. H. Dinegar, J. Am. Chem. Soc., 1950, 72, 4847 CrossRef CAS.
- N. Shukla, E. B. Svedberg, J. Ell and A. J. Roy, Mater. Lett., 2006, 60, 1950 CrossRef CAS.
- N. Wu, L. Fu, M. Su, M. Aslam, K. C. Wong and V. P. Dravid, Nano Lett., 2004, 4, 383 CrossRef CAS.
- J. R. Thomas, J. Appl. Phys., 1966, 37, 2914 CAS.
- P. H. Hess and P. H. Parker, Jr, J. Appl. Polym. Sci., 1966, 10, 1915 CrossRef CAS.
- R. Tannenbaum, C. L. Flenniken and E. P. Goldberg, J. Polym. Sci., Part B: Polym. Phys., 1987, 25, 1341 CrossRef CAS.
- J. P. Stevenson, M. Rutnakornpituk, M. Vadala, A. R. Esker, S. W. Charles, S. Wells, J. P. Dailey and J. S. Riffle, J. Magn. Magn. Mater., 2001, 225, 47 CrossRef CAS.
- J. I. Abes, R. E. Cohen and C. A. Ross, Chem. Mater., 2003, 15, 1125 CrossRef CAS.
- P. Y. Keng, I. Shim, B. D. Korth, J. F. Douglas and J. Pyun, ACS Nano, 2007, 1, 279 CrossRef CAS.
- T. W. Smith and D. Wychick, J. Phys. Chem., 1980, 84, 1621 CrossRef CAS.
- C. H. Griffiths, M. P. O'Horo and T. W. Smith, J. Appl. Phys., 1979, 50, 7108 CrossRef CAS.
- J. Lai, K. V. P. M. Shafi, A. Ulman, K. Loos, Y. Lee, T. Vogt, W. L. Lee and N. P. Ong, J. Phys. Chem. B, 2005, 109, 15 CrossRef CAS.
- K. Butter, P. H. H. Bomans, P. M. Frederik, G. J. Vroege and A. P. Philipse, Nat. Mater., 2003, 2, 88 CrossRef CAS.
- E. Pretsch, T. Clerc, J. Seibl, W. Simon, Strukturaufklärung organischer Verbindungen 3rd edn, Springer-Verlag, Berlin, 1990 Search PubMed.
- Y. Dirix, C. Bastiaansen, W. Caseri and P. Smith, J. Mater. Sci., 1999, 34, 3859 CrossRef CAS.
- G. Mastrogiacomo, J. F. Löffler and N. R. Dilley, Appl. Phys. Lett., 2008, 92, 082501 CrossRef.
- C. P. Bean and J. D. Livingston, J. Appl. Phys., 1959, 30, S120.
- J. P. Perez, V. Dupuis, J. Tuaillon, A. Perez, V. Paillard, P. Melinon, M. Treilleux, L. Thomas, B. Barbara and B. Bouche-Fabre, J. Magn. Magn. Mater., 1995, 145, 74 CrossRef CAS.
- J. F. Löffler, J. P. Meier, B. Doudin, J. P. Ansermet and W. Wagner, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 57, 2915 CrossRef CAS.
- J. Falbe, M. Regitz (Eds.), Römpp Lexikon Chemie, Georg Thieme, Stuttgart, 1998 Search PubMed.
| This journal is © The Royal Society of Chemistry 2009 |