Highly dispersible, superparamagnetic magnetite nanoflowers for magnetic resonance imaging†
Fengqin
Hu
,
Keith W.
MacRenaris
,
Emily
A. Waters
,
Elise A.
Schultz-Sikma
,
Amanda L.
Eckermann
and
Thomas J.
Meade
*
Department of Chemistry, Biochemistry and Molecular Biology and Cell Biology, Neurobiology and Physiology, and Radiology, Northwestern University, Evanston, IL 60208, USA. E-mail: tmeade@northwestern.edu; Fax: +1 847 491 3832; Tel: +1 847 491 2481
First published on 14th October 2009
Abstract
A one-pot reaction process was developed to synthesize highly dispersible, superparamagnetic Fe3O4 nanoflowers; the potential of these nanoflowers as MRI contrast agents was investigated.
Nanostructured magnetite (Fe3O4) is one of the most attractive nanomaterials for various biomedical applications such as magnetic separation, magnetic resonance imaging (MRI), targeted drug delivery, and hyperthermia treatment of cancer.1–8 In particular, magnetite nanoparticles are excellent MR contrast agents for noninvasive cellular and molecular imaging.4–7 The effectiveness of a contrast agent is determined by its relaxivity, or how much the relaxation rates of water protons are increased in the presence of the agent at a given concentration. Efforts to increase the relaxivity of magnetic nanoparticle contrast agents have focused on improvements in nanoparticle size,9 development of new magnetic core materials,10,11 or clustering.12–15 Increasing nanoparticle size increases the relaxivity, but also induces the superparamagnetic–ferromagnetic transition (at a domain size of ca. 30 nm for Fe3O4), so that nanoparticles are no longer dispersible in solution. The strategy of forming clusters of magnetic nanoparticles has the advantage of increasing the relaxivity while retaining the superparamagnetic properties.9,16
Magnetic nanoparticle clusters can be obtained either by assembly or through direct solution growth.12–17 Several methods for the direct synthesis of magnetic nanoparticle clusters have been developed, such as modified coprecipitation methods, high-temperature hydrolysis or decomposition of iron chloride in polyol.15–18 There are several barriers remaining in the preparation of high-quality magnetic nanoparticle clusters suitable for biological applications, including monodispersity, colloidal stability under physiological conditions and surface reactive moieties.
Herein, we describe an approach for the synthesis of highly dispersible, superparamagnetic Fe3O4 nanoparticle clusters via a one-pot reaction. By modification of literature procedures for the preparation of magnetic nanoparticles, a series of water-soluble Fe3O4 magnetic nanoflowers (NFs) were synthesized.4,19,20Fig. 1a–c shows representative TEM images of the Fe3O4 NFs with diameters of 42 ± 8 nm, 30 ± 5 nm and 19 ± 3 nm. The 30 nm Fe3O4 NFs were prepared by heating a mixture of 1.5 mmol Fe(acac)3, 6 g HOOC–PEG–COOH (600 g mol−1), 1.93 mL oleylamine, and 25 mL phenyl ether at 260 °C for 24 h. The 42 nm and 19 nm Fe3O4 NFs were synthesized by decreasing the quantity of HOOC–PEG–COOH and oleylamine and using shorter reaction times at 260 °C. The formation of NFs instead of individual nanoparticles may be the result of Fe3O4 nanoparticles spontaneously aggregating due to their high surface energy (see ESI†, Fig. S1). Oleylamine is necessary for the formation of high-quality Fe3O4 NFs (see ESI†, Fig. S2). The high resolution TEM image shown in Fig. 1d shows that the Fe3O4 NFs are made up of clusters of nanoparticles connected in three dimensions.
 | ||
| Fig. 1 (a–c) TEM images of (a) 42 nm, (b) 30 nm and (c) 19 nm Fe3O4 NFs. Insets are electron diffraction patterns. Scale bar: 100 nm. (d) High resolution TEM image of 30 nm Fe3O4 NFs. Scale bar: 10 nm. (e–f) Colloidal stability tests of 30 nm Fe3O4 NFs (e) in PBS and NaCl solutions, and (f) in several pH. | ||
Statistical results from high resolution TEM images of 42 nm, 30 nm and 19 nm Fe3O4 NFs show that the average diameters of the primary Fe3O4 nanoparticles in the NFs are approximately 4.3 nm (42 nm Fe3O4 NFs), 4.8 nm (30 nm Fe3O4 NFs), and 4.5 nm (19 nm Fe3O4 NFs), respectively (Fig. 1d; see ESI†, Fig. S3). Selected area electron diffraction patterns reveal that the Fe3O4 NFs are of high crystallinity (Fig. 1). The lattice spacings (calculated based on the diffraction patterns) are in agreement with those of bulk magnetite (JCPDS 19-0629).
The crystal structure of the Fe3O4 NFs was examined by powder X-ray diffraction (XRD). The position and relative intensity of all diffraction peaks match well with those from the JCPDS card (19-0629) for magnetite (see ESI†, Fig. S4). Calculations with the Debye–Scherrer formula for the strongest peak (311) gave grain size of 4.8 nm for 42 nm Fe3O4 NFs, 5.3 nm for 30 nm Fe3O4 NFs, and 5.0 nm for 19 nm Fe3O4 NFs. These sizes are comparable to the results from TEM.
The surface chemical structure of the Fe3O4 NFs was characterized by Fourier-transform infrared (FTIR) spectroscopy. As shown in Fig. S5 (see ESI†), 42 nm, 30 nm and 19 nm Fe3O4 NFs present similar spectra. The characteristic bands of HOOC–PEG–COOH at 1107 cm−1 (C–O–C stretch) and 1455 cm−1 (CH2 scissor) appear in the spectra of the Fe3O4 NFs, indicating that HOOC–PEG–COOH is present on the Fe3O4 nanocrystal surface. The distinct spectral difference between the spectrum of HOOC–PEG–COOH and that of the Fe3O4 NFs is that the carboxyl band at 1746 cm−1 for HOOC–PEG–COOH is shifted to a lower wavenumber, 1630 cm−1, in the spectra of the Fe3O4 NFs. This spectral change indicates binding between the carboxylate group and metal on Fe3O4, with one or two oxygen atoms interacting with the metal on the surface.21
These results demonstrate that the HOOC–PEG–COOH molecules are covalently bound to the Fe3O4 NFs through the carboxylic acid. In addition, all IR spectra of the Fe3O4 NFs exhibit characteristic peaks centered at 590 cm−1 that are attributed to lattice absorption of the magnetite.22
Colloidal stability under physiological conditions is one of the most important issues relating to the biomedical applications of nanomaterials. In the one-pot reaction process, PEG was selected as the surfactant because it can stabilize nanoparticles in a wide pH range due to hydrogen bonding between PEG and water. PEG layers have been shown to be non-toxic, non-immunogenic, non-antigenic and protein-resistant.23 Consequently, the Fe3O4 NFs synthesized by this procedure can be readily dispersed in H2O or PBS without further surface modification. Dynamic light scattering (DLS) measurements of 42 nm, 30 nm and 19 nm Fe3O4 NFs show that their hydrodynamic sizes (DH) are 74 ± 19 nm, 47 ± 9 nm, and 39 ± 6 nm, respectively (see ESI†, Fig. S6). To investigate the effects of pH and ionic strength on the colloidal stability of the Fe3O4 NFs, we exposed them to 0–1 M NaCl and pH 3–11 solutions, as shown in Fig. 1e and f. The Fe3O4 NFs are stable in NaCl solution at concentrations as high as 1 M. The NFs are stable over a pH range of 3–11, a remarkably broad tolerance.
The colloidal stability of the Fe3O4 NFs was investigated by measuring DH in PBS, NaCl solutions, DMEM (Dulbecco’s modified Eagle’s medium), EMEM (Eagle’s minimum essential medium) and at several pH levels. The DH results at 0 day and after placement for 21 days are shown in Fig. S7 (see ESI†). The DH values of the Fe3O4 NFs are similar under various conditions and present an excellent stability with DH being nearly unchanged for three weeks. To avoid the effects of serum on DH, the colloidal stability of the Fe3O4 NFs in fetal bovine serum (FBS), calf bovine serum (CBS), DMEM supplemented with 10% FBS, DMEM supplemented with 10% CBS and EMEM supplemented with 10% FBS was investigated through measuring the transverse relaxation time (T2) over time. The T2 results in PBS are shown for comparison. As shown in Fig. S8 (see ESI†), T2 values in various media are similar and have excellent stability during the three week measurement period. The DH and T2 results demonstrate that the Fe3O4 NFs prepared by the current procedure show excellent colloidal stability under physiological conditions, which is promising for in vitro and in vivo biomedical applications.
The zeta potentials of 42 nm, 30 nm and 19 nm Fe3O4 NFs at pH 7 are −26 mV, −28 mV and −12 mV, respectively, demonstrating that the Fe3O4 NFs are negatively charged at neutral pH. The zeta potentials of 30 nm Fe3O4 NFs under different pH conditions were investigated (see ESI†, Fig. S9). The zeta potential of the Fe3O4 NFs at pH 3 is 5.83 mV, i.e., the NFs are positively charged. At pH 5, the Fe3O4 NFs become negatively charged (−10.6 mV). Then a gradual increase of negative charge density was found at pH 7 (−27.6 mV) and pH 9 (−42.4 mV). As shown in Fig. S9 (see ESI†), the zeta potentials of the Fe3O4 NFs decreased linearly with increased pH from 3 to 9. Above pH 9, the zeta potentials are nearly unchanged. These results indicate that there are free carboxylic acid groups on the Fe3O4 NF surface. Further surface modification of the Fe3O4 NFs and conjugation with bio-targeting molecules are currently under investigation.
Magnetic properties of the Fe3O4 NFs were measured using a superconducting quantum interference device (SQUID). Magnetization vs. applied magnetic field was measured at 5 K and 250 K for each sample (Fig. 2). At 250 K, all samples exhibit superparamagnetic behavior without magnetic hysteresis and remanence, whereas ferromagnetic behavior with a coercivity of 401 Oe (42 nm Fe3O4 NFs), 532 Oe (30 nm Fe3O4 NFs), or 488 Oe (19 nm Fe3O4 NFs) is observed at 5 K since thermal energy is insufficient to induce moment randomization. This unique structure allows the Fe3O4 NFs to retain superparamagnetic behavior at 250 K even though the sizes exceed 30 nm. The saturation magnetizations of 42 nm, 30 nm and 19 nm Fe3O4 NFs at 250 K are 33, 45, 36 emu g−1 Fe, respectively. This trend is consistent with that of the primary Fe3O4 nanoparticle size in the Fe3O4 NFs.
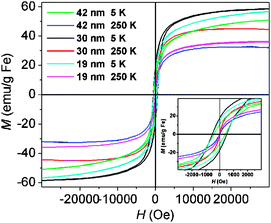 | ||
| Fig. 2 Magnetization (M) loops for 42 nm, 30 nm and 19 nm Fe3O4 NFs measured at 5 K and 250 K. | ||
To investigate the MR signal enhancement effects of the Fe3O4 NFs, aqueous solutions of various concentrations ([Fe] determined by ICP-AES) were measured on a 4.7 T horizontal-bore Bruker Biospec MR Imager. Fig. 3 shows the T2-weighted MR image of the Fe3O4 NFs at various Fe concentrations at 4.7 T (25 °C). Increasing concentrations of Fe3O4 NFs led to significant changes in signal intensity in the image. The transverse relaxivity (r2) values (determined by taking the slope of a linear fit of 1/T2versus Fe concentration) are 97 mM−1 s−1 (42 nm Fe3O4 NFs), 193 mM−1 s−1 (30 nm Fe3O4 NFs), and 88 mM−1 s−1 (19 nm Fe3O4 NFs), respectively. To show the MR signal enhancement effects of the Fe3O4 NFs at clinical field strength, the T2 of the Fe3O4 NF solutions at different Fe concentrations was measured on a 1.5 T (60 MHz) relaxometer at 37 °C (see ESI†, Fig. S10). The r2 values of 42 nm, 30 nm and 19 nm Fe3O4 NFs at 1.5 T are 148 mM−1 s−1, 238 mM−1 s−1, and 126 mM−1 s−1, respectively. The relaxivity differences between these Fe3O4 NFs are due to the synergistic effects of the primary Fe3O4 nanoparticle size and the number of nanoparticles per Fe3O4 NF (see ESI†).9,24
 | ||
| Fig. 3 T 2-weighted MR image of aqueous solutions of (a) 42 nm, (b) 30 nm and (c) 19 nm Fe3O4 NFs at various Fe concentrations (4.7 T, 25 °C). | ||
The r2 of 30 nm Fe3O4 NFs (238 mM−1 s−1, 1.5 T) is 2.3 times of that of the commercial Feridex (104 mM−1 s−1, 1.5 T) which has a similar primary particle size.10 Due to their excellent colloidal stability and high r2, the Fe3O4 NFs are promising candidates as high-efficiency T2 contrast agents for a variety of MR imaging applications. Initial experiments were conducted to determine the effects of these Fe3O4 NFs on cell viability using NIH/3T3 cells. As shown in Fig. 4, the Fe3O4 NFs of various concentrations (6.25, 12.5, 25.0, or 50.0 mg Fe L−1) have no effect on the NIH/3T3 cell viability after incubation with cells for 24 h.
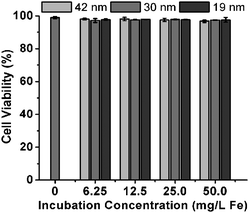 | ||
| Fig. 4 Cell viability of NIH/3T3 cells after 24 h incubation with 42 nm, 30 nm or 19 nm Fe3O4 NFs at various incubation iron concentrations. | ||
In conclusion, water-soluble, superparamagnetic Fe3O4 NFs were synthesized via a one-pot reaction. The Fe3O4 NFs show excellent colloidal stability in H2O, PBS, cell culture media and serum, and tolerate a high salt concentration (1 M NaCl) and a wide pH range from 3 to 11. Furthermore, these Fe3O4 NFs show high transverse relaxivity (238 mM−1 s−1) and have no effect on the viability of NIH/3T3 cells. This work describes a series of highly dispersible Fe3O4 NFs suitable for MR imaging applications and a facile one-pot reaction approach which can be extended to prepare other nanoclusters such as MFe2O4 (M = Mn, Zn, Co, Ni).
We thank Prof. Vinayak P. Dravid and Dr. George H. Chan for helpful discussions. This work was supported by the National Cancer Institute Center for Cancer Nanotechnology Excellence initiative at Northwestern University Award No. U54CA119341 and the National Institute of Biomedical Imaging and Bioengineering Award No. 1R01 EB005866-01. MRI was performed on instruments supported by NIH/NCRR Grant SIO RR 15685-01. We acknowledge NUANCE and IMSERC at Northwestern University for use of their instrumentation.
Notes and references
- U. Jeong, X. Teng, Y. Wang, H. Yang and Y. Xia, Adv. Mater., 2007, 19, 33 CrossRef CAS.
- H. Gu, K. Xu, C. Xu and B. Xu, Chem. Commun., 2006, 941 RSC.
-
R. A. Whitehead, M. S. Chagnon, E. V. Groman and L. Josephson, US Pat., 4
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 554088, 1985 Search PubMed.
554088, 1985 Search PubMed. - F. Hu, L. Wei, Z. Zhou, Y. Ran, Z. Li and M. Gao, Adv. Mater., 2006, 18, 2553 CrossRef CAS.
- R. Weissleder, A. Moore, U. Mahmood, R. Bhorade, H. Benveniste, E. A. Chiocca and J. P. Basilion, Nat. Med. (N. Y.), 2000, 6, 351 CrossRef CAS.
- O. Veiseh, C. Sun, J. Gunn, N. Kohler, P. Gabikian, D. Lee, N. Bhattarai, R. Ellenbogen, R. Sze, A. Hallahan, J. Olson and M. Zhang, Nano Lett., 2005, 5, 1003 CrossRef CAS.
- N. Nasongkla, E. Bey, J. Ren, H. Ai, C. Khemtong, J. S. Guthi, S. Chin, A. D. Sherry, D. A. Boothman and J. Gao, Nano Lett., 2006, 6, 2427 CrossRef CAS.
- M. Johannsen, A. Jordan, R. Scholz, M. Koch, M. Lein, S. Deger, J. Roigas, K. Jung and S. Loening, J. Endourol., 2004, 18, 495 CrossRef.
- J. Cheon and J. H. Lee, Acc. Chem. Res., 2008, 41, 1630 CrossRef CAS.
- W. S. Seo, J. H. Lee, X. M. Sun, Y. Suzuki, D. Mann, Z. Liu, M. Terashima, P. C. Yang, M. V. McConnell, D. G. Nishimura and H. J. Dai, Nat. Mater., 2006, 5, 971 CrossRef CAS.
- J. H. Lee, Y. M. Huh, Y. Jun, J. Seo, J. Jang, H. T. Song, S. Kim, E. J. Cho, H. G. Yoon, J. S. Suh and J. Cheon, Nat. Med. (N. Y.), 2007, 13, 95 CrossRef CAS.
- H. Ai, C. Flask, B. Weinberg, X. Shuai, M. D. Pagel, D. Farrell, J. Duerk and J. Gao, Adv. Mater., 2005, 17, 1949 CrossRef CAS.
- J. F. Berret, N. Schonbeck, F. Gazeau, D. E. Kharrat, O. Sandre, A. Vacher and M. Airiau, J. Am. Chem. Soc., 2006, 128, 1755 CrossRef CAS.
- J. Kim, J. E. Lee, S. H. Lee, J. H. Yu, J. H. Lee, T. G. Park and T. Hyeon, Adv. Mater., 2008, 20, 478 CrossRef CAS.
- J. H. Park, G. Maltzahn, L. Zhang, M. P. Schwartz, E. Ruoslahti, S. N. Bhatia and M. J. Sailor, Adv. Mater., 2008, 20, 1630 CrossRef CAS.
- J. Ge, Y. Hu, M. Biasini, W. P. Beyermann and Y. Yin, Angew. Chem., Int. Ed., 2007, 46, 4342 CrossRef CAS.
- K. C. Barick, M. Aslam, P. V. Prasad, V. P. Dravid and D. Bahadur, J. Magn. Magn. Mater., 2009, 321, 1529 CrossRef CAS.
- H. Deng, X. Li, Q. Peng, X. Wang, J. Chen and Y. Li, Angew. Chem., Int. Ed., 2005, 44, 2782 CrossRef CAS.
- Z. Li, L. Wei, M. Gao and H. Lei, Adv. Mater., 2005, 8, 1001 CrossRef.
- S. Liu, B. Jia, R. Qiao, Z. Yang, Z. Yu, Z. Liu, K. Liu, J. Shi, H. Ouyang, F. Wang and M. Gao, Mol. Pharm., 2009, 6, 1074 CrossRef CAS.
- Q. Liu and Z. Xu, Langmuir, 1995, 11, 4617 CrossRef CAS.
- F. Hu, Z. Li, C. Tu and M. Gao, J. Colloid Interface Sci., 2007, 311, 469 CrossRef CAS.
- J. Qin, S. Laurent, Y. S. Jo, A. Roch, M. Mikhaylova, Z. M. Bhujwalla, R. N. Muller and M. Muhammed, Adv. Mater., 2007, 19, 1874 CrossRef CAS.
- S. Laurent, D. Forge, M. Port, A. Roch, C. Robic, L. V. Elst and R. N. Muller, Chem. Rev., 2008, 108, 2064 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, TEM, XRD, IR, DLS, T2, zeta potential, relaxivity results. See DOI: 10.1039/b916562b |
| This journal is © The Royal Society of Chemistry 2010 |