Endohedral metallofullerenes in self-assembled monolayers†
Received
27th July 2009
, Accepted 6th October 2009
First published on
11th November 2009
Abstract
A method has been developed for the attachment of a dithiolane group to endohedral metallofullerenes via a 1,3-dipolar cycloaddition reaction. This sulfur-containing functional group serves as an anchor, enabling efficient immobilisation of endohedral fullerenes on Au(111) surfaces at room temperature, directly from the solution phase. The functionalised fullerenes form disordered monolayers that exhibit no long-range ordering, which is attributed to both the strong bonding of the dithiolane anchor to the surface and to the conformational flexibility of the functional group. Endohedral fullerenes Er3N@C80 and Sc3N@C80 have been used as models for functionalisation and subsequent surface deposition. Their chemical reactivity towards dithiolane functionalisation and their surface behaviour have been compared to that of C60. The endohedral fullerenes appear to be significantly less reactive towards the functionalisation than C60, however they bind in a similar manner to a gold surface as their dithiolane terminated C60 counterparts. The optical activity of Er3N@C80 molecules is preserved after attachment of the functional group. We report a splitting of the endohedral Er3+ emission lines due to the reduction in symmetry of the functionalised fullerene cage, as compared to the highly symmetrical icosahedral C80 cage of pristine Er3N@C80.
Introduction
Carbon is a unique element that can create hollow, polyhedral cages called fullerenes. The void space within the fullerene cage can be occupied by a small heteroatom, such as N, P, He or a metal atom, or by a small metallic cluster.1 Encapsulation of endohedral heteroatoms within fullerenes is a difficult task. These so-called “endohedral fullerenes” X@CN (where X is an atom or cluster incarcerated in the fullerene, and N is a number of carbon atoms in the fullerene cage) are usually formed in extremely low yields and require extensive purification, which hinders investigations of their properties and integration within functional materials.
The endohedral species in X@CN often possess useful magnetic or optical properties1 that could potentially be utilised in nano-electronic devices.2 In this case, the fullerene cage serves as a “nanocontainer” which facilitates the incorporation of individual endohedral atoms within supramolecular architectures, such as 1D molecular chains in carbon nanotubes2 or 2D molecular arrays on surfaces.3 However, fullerene cages tend to have relatively isotropic exteriors owing to their spheroidal shapes, and so precise control of their positions and orientations can be difficult to achieve. An attractive approach for solving this problem is through chemical functionalisation of fullerene cages. This would allow for control over the orientation of the molecules via well-defined chemical bonding or highly directional non-covalent interactions. For example, endohedral fullerenes functionalised with an appropriate chemical group could be able to form spontaneous molecular monolayers on surfaces or molecular chains inside carbon nanotubes, within which the distance between the endohedral atoms X could be precisely controlled through the chemical functionality of fullerene cage.4,5
To demonstrate this principle, we have selected the most abundant type of endohedral fullerenes–trimetallic nitride templated endohedral metallofullerenes (TNT EMFs). These molecules comprise three endohedral metal atoms arranged in a triangular fashion around a nitrogen atom incarcerated in a C80 cage (Fig. 1). It is generally accepted that TNT EMFs M3N@C80 are stabilized by six electrons transferred from the trimetallic nitride (M3N) cluster to the icosahedral (Ih) C80 carbon cage, resulting in a closed-shell electronic structure described as [M3N]6+@[C80]6−.6–8 The transparency of the [C80]6− cage for wavelengths longer that 1 μm, allows direct excitation of endohedral metal atoms, such as Er3+, whose 4f electronic transitions occur in the near-IR range, important for telecommunications. Future technological applications of TNT EMFs may require the fabrication of well-ordered arrays of these fullerenes on surfaces. We demonstrate that a dithiolane group, which has a strong affinity for metal surfaces,9 can be efficiently attached to Sc3N@C80 and Er3N@C80. We investigate the effects of the functional group on the optical properties of Er3N@C80 and the attachment of functionalised TNT EMFs to a gold surface.
Results and discussion
Synthesis and characterization of dithiolane functionalized M3N@C80 (M = Sc, Er)
Synthesis and purification.
We have explored the reactivity of TNT EMFs towards dithiolane functionalisation using the reaction of 1,3-dipolar cycloaddition of azomethine ylides (Fig. 1). The cycloaddition reactions are commonly used for functionalisation of fullerene cages, as they often yield isomerically pure products with high efficiency. However, one of the main drawbacks of cycloaddition reactions is the problem of addition of multiple functional groups to a fullerene cage.
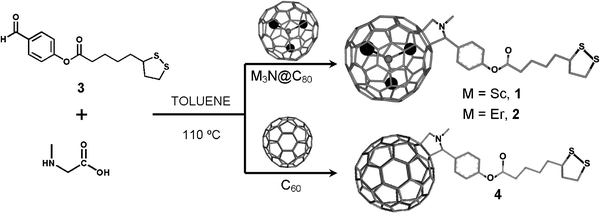
In order to address this problem, the reaction progress was carefully monitored by thin layer chromatography (TLC) and matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF MS, see Supporting Information file). Fullerene C60 appears to react smoothly with the dithiolane aldehyde 3 at 110 °C, forming a fullerene functionalised with one dithiolane group C60R (where R is a functional group), and subsequent products of bis-C60(R)2, tris-C60(R)3 and tetra-functionalised C60(R)4 fullerene (Fig. 2a). The reactivities of C70 and a higher fullerene C78 (Supporting Information file) appear to be significantly lower than C60 (Fig. 2b), which can be attributed to the fact that the proportion of reactive carbon–carbon bonds within the fullerene structure becomes lower as the fullerene cage becomes larger.10
![(a) Evolution of mono- (◆), bis- (■), tris- (●) and tetra-functionalised (▲) fullerenes in the reaction of 1,3-dipolar cycloaddition of dithiolane aldehyde 3 with C60 as a function of time. Irel is a relative conversion rate calculated as Irel (%) = [Iadduct/(∑Iadduct(i) + IC60)] × 100 measured by MALDI-TOF mass spectrometry. (b) Time required for bis-functionalised fullerene CN(R)2 to emerge in the reaction of 1,3-dipolar cycloaddition measured for different fullerenes.](/image/article/2010/CP/b915170b/b915170b-f2.gif) |
| | Fig. 2 (a) Evolution of mono- (◆), bis- (■), tris- (●) and tetra-functionalised (▲) fullerenes in the reaction of 1,3-dipolar cycloaddition of dithiolane aldehyde 3 with C60 as a function of time. Irel is a relative conversion rate calculated as Irel (%) = [Iadduct/(∑Iadduct(i) + IC60)] × 100 measured by MALDI-TOF mass spectrometry. (b) Time required for bis-functionalised fullerene CN(R)2 to emerge in the reaction of 1,3-dipolar cycloaddition measured for different fullerenes. | |
Since the cycloaddition takes place on the timescale of hours, the number of dithiolane groups appended to the fullerene cage can be conveniently controlled by the duration of the reaction. If the rate of formation of the mono-functionalised product is known, the yield of this product can be optimised by quenching the reaction before the bis-functionalised fullerene is formed in significant quantities. We adopted this strategy for the synthesis of pyrrolidinofullerene derivate N-methyl-2-(4-(liponyloxy)benzyl)-Sc3N@C80 (1), which was formed from Sc3N@C80, dithiolane aldehyde 3 and sarcosine at 110 °C (Fig. 1). The reactivity of the endohedral fullerene Sc3N@C80 towards 1,3-dipolar cycloaddition appears to be significantly lower than that of C60, and is comparable to that of higher fullerenes of similar size. However, Sc3N@C80 does not entirely support the trend (Fig. 2b), showing a slightly higher reactivity than a smaller fullerene C78, which may be related to the presence of the endohedral group Sc3N inside C80. The effects of Sc3N on the reactivity of the fullerene cage are difficult to evaluate directly, as the empty C80-Ih fullerene does not exist. Mono-adduct 1 was isolated with a 34% yield after purification by column chromatography. Negative mode MALDI-TOF mass spectrum of pure 1 (Fig. 3b) shows a pronounced M− peak at 1447.14 m/z and only a small degree of fragmentation leading to Sc3N@C80 at 1109.72 m/z.
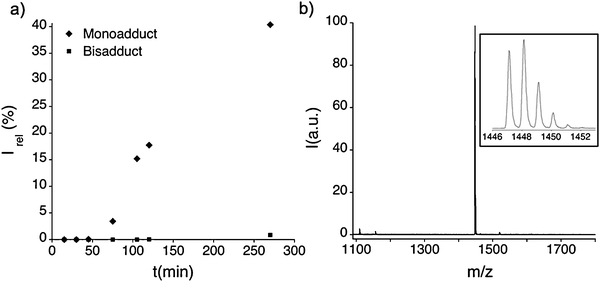 |
| | Fig. 3 (a) Evolution of the mono- and bis-functionalised Sc3N@C80 at 110 °C as a function of the reaction time. (b) MALDI-TOF mass spectrum of purified 1 (inset: isotopic distribution pattern confirming the composition of monoadduct 1). | |
TNT EMFs with the same structure of the fullerene cage are expected to have similar chemical reactivity. We have therefore used the optimised conditions found for the dithiolane addition reaction with Sc3N@C80 for functionalisation of Er3N@C80 without further modification. Mono-functionalised Er3N@C802 was isolated in a 28% yield following the same procedure as for fullerene 1. The product 2 was observed clearly in MALDI-TOF mass spectrum (negative ionisation mode) as a M− peak at 1815.53 m/z (Fig. 4a). It is interesting that the fullerene 2 undergoes a more extensive fragmentation in the mass spectrometer, using the same conditions, than fullerene 1; the peaks at 1682.51, 1658.45 and 1522.79 m/z emerge as a result the breakage of dithiolane functional group. The molecular peak of the mono-functionalised 2 is approximately twice as broad as that of 1. This is due to the wider isotopic distribution of erbium as compared to scandium, which is in a good agreement with the calculated distributions. High performance liquid chromatography (HPLC) through a 5PYE column (Nacalai Tesque) with toluene as eluent (flow rate 7 ml min−1) showed a retention time of 3.25 min for 2 (Fig. 4b) that is significantly shorter than the retention time observed for pristine Er3N@C80 under the same conditions (10.93 min, Fig. 4c). The dithiolane containing functional group attached to the carbon cage is quite polar and somewhat hydrophilic. This is expected to reduce the retention time in a reversed phase HPLC column. Moreover, the symmetry of the C80 cage is broken by the functional group, which could induce a substantial dipole moment on the cage. The purity of the product 2 was estimated to be at least 98% by HPLC. The absence of secondary peaks after 5 cycles of HPLC in a recycling mode confirm that 2 is formed as a single isomer.
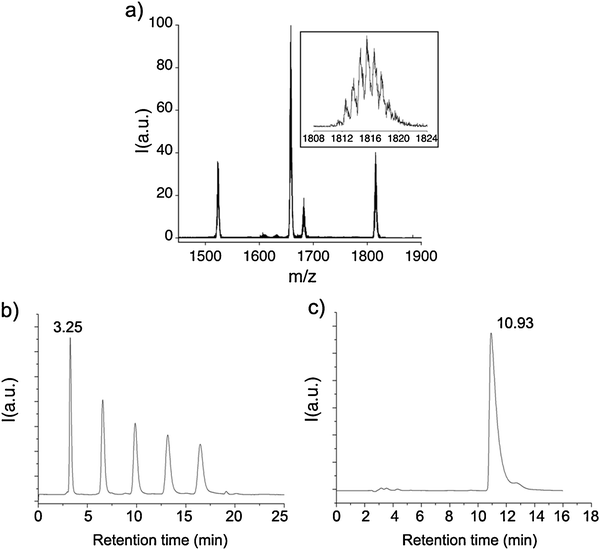 |
| | Fig. 4 MALDI-TOF mass spectrum (a) and recycling-HPLC trace (b) of the purified product 2. The retention time of 2 is 3.25 min. After 5 cycles no other peaks were detected. The retention time of 2 is significantly shorter than that of pristine Er3N@C80, under the same conditions (c). | |
NMR.
The five-member pyrrolidine ring is directly attached to the carbon cage (Fig. 5), rendering analysis of 1H and 13C NMR spectra of the pyrrolidine group very important for understanding the molecular structures of the functionalised metallofullerenes.11–18 The 1H NMR spectrum of 1 indicates the presence of a single regioisomer. The signals for the pyrrolidine ring geminal protons Ha and Hb in 1 are separated by 1.3 ppm and appear as two doublets at δ = 4.38 and 3.08 ppm (J = 9.7 Hz). The remaining pyrrolidine ring proton Hc gives a singlet peak at δ = 3.76 ppm, while for the N-methyl group Hd atoms a singlet is observed at δ = 3.15 ppm. The heteronuclear multiple quantum correlation (HMQC) spectrum (ESI)† enables correlation of the chemical shifts of the carbon atoms with corresponding hydrogen atoms attached to them, as summarised in Table 1. The relatively large difference in the chemical shifts of the geminal hydrogen atoms Ha and Hb, coupled with the presence of only one set of pyrrolidine carbon atoms, suggest the formation of a product where the pyrrolidine ring is attached to a [5,6]-bond of the C80 cage, (Fig. 5), similar to other examples of 1,3-dipolar cycloaddition reported for TNT EMFs.11–18 In the case of C60, where the cycloaddition reactions are known to take place at a [6,6]-bond, the mono-functionalised N-methyl-2-(4-(liponyloxy)benzyl)-[6,6]-C60 (4) has a much smaller difference between the chemical shifts of the geminal hydrogen atoms Ha and Hb of the pyrrolidine ring (Δδ = 0.7 ppm). This is because the Ha and Hb of the [6,6]-adduct 4 occupy more similar positions to those of the [5,6]-adduct 1 (Fig. 5).
![Structural diagrams of pyrrolidine ring attached to [5,6]-bond of Sc3N@C80 (a) and [6,6]-bond of C60 (b).](/image/article/2010/CP/b915170b/b915170b-f5.gif) |
| | Fig. 5 Structural diagrams of pyrrolidine ring attached to [5,6]-bond of Sc3N@C80 (a) and [6,6]-bond of C60 (b). | |
Table 1 Chemical shifts of H and C atoms of pyrrolidine rings in Sc3N@C80 (1) and C60 (4)
| Part of pyrrolidine ring |
Chemical shifts in 1/ppm |
Chemical shifts in 4/ppm |
| CH2 group |
Ha 4.4, Hb 3.1, C1 72.5 |
Ha 5.0, Hb 4.3, C1 70.1 |
| CH group |
Hc 3.8, C2 85.0 |
Hc 4.9, C2 83.0 |
| CH3 group |
Hd 3.1, C3 41.4 |
Hd 2.8, C3 40.0 |
Raman spectroscopy.
The Raman spectrum of 2 under a 532 nm laser excitation (Fig. 6) shows several peaks in the range of 450–750 cm−1, typical for the CS-SC moiety (Table 2). The frequencies of these modes are known to vary substantially depending on the precise structure of a ‘CS–SC’-containing compound,19 making a complete peak assignment difficult in this case. Nevertheless, a well-defined peak observed at 498 cm−1 is within the known range of the S–S bond vibration at 470–530 cm−1. The Raman peaks at 653 cm−1 and 659 cm−1 (Fig. 6) can be attributed to the C–S bonds of 2. A further proof of successful Er3N@C80 functionalisation is provided by Raman peaks at ∼3000 cm−1 (see inset in Fig. 6), indicating the presence of C–H bonds associated with the pyrrolidine, the dithiolane groups and the linking alkyl chain (–CH2–)4.
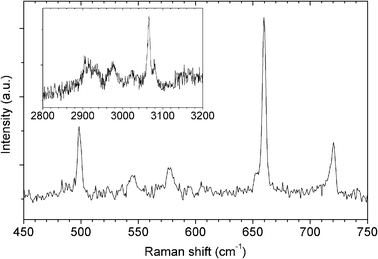 |
| | Fig. 6 Raman spectrum of 2 under 532 nm laser excitation, confirming the functionalisation. Raman shifts in the ranges typical for CS–SC (main plot), and C–H (inset) bond associated vibrations are shown. | |
Table 2 Comparison of Raman peak positions in the range of 450–750 cm−1 for 2 and compounds containing similar CS–SC groups19 (peaks associated with S–S bond are highlighted in bold)
|
2/cm−1 |
D,L-6,8-thioctic acid amide/cm−1 |
D,L-6,8-thioctic acid/cm−1 |
|
498
|
496
|
456 |
| 546 |
504 |
∼501 |
| 578 |
533 |
511 |
| 653 |
585 |
559 |
| 659 |
664 |
634 |
| 720 |
675 |
682 |
|
|
708 |
|
Photoluminescence of dithiolane functionalized Er3N@C80 (2) in solution
In order to gain an insight into the influence of the functional group on the inherent properties of the incarcerated species, comparative photoluminescence measurements of Er3N@C80 or 2 in solution have been performed. Optical excitation of the fullerene cage leads to population in the Er3+4I13/2 state via a series of incoherent non-radiative relaxation processes.20 Subsequent radiative relaxation occurs to the 4I15/2 manifold. The emitted photons are of wavelengths beyond the window in which the fullerene cage is absorbing, thus permitting their detection. The photoluminescence spectra of Er3N@C80 and 2 (Fig. 7) comprise a series of sharp lines corresponding to the emission from the lowest sublevel of 4I13/2, the only sublevel populated at 5 K, down to the different sublevels of the 4I15/2 ground state. Those sublevels arising from the crystal field splitting of the 4I13/2 and 4I15/2 Er3+ states are relatively sensitive to the Er3+ local environment in comparison to other sublevels.21 The emission lines of Er3N@C80 are monocomponent in nature (Fig. 7), whereas those of 2 are further split. In Er3N@C80, all Er3+ of the Er3 cluster occupy equivalent positions inside the C80 cage.22 In the case of 2, the functional group reduces the C80 cage symmetry and therefore perturbs the local environment of each Er3+ inside the cage. Although the presence of the functional group does not quench the luminescence of endohedral atoms, it appears to make the endohedral metal atoms in the trigonal Er3N cluster non-equivalent, which is manifested in the observed splitting of their emission peaks.
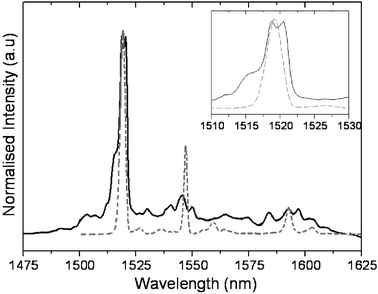 |
| | Fig. 7 Photoluminescence spectra of 2 (solid) and Er3N@C80 (dashed) in CS2 solutions at 5 K under 532 nm excitation. The inset shows the most intense emission line at ∼1520 nm in greater detail, highlighting the splitting of 2. | |
Deposition of ditholane functionalised Er3N@C80 (2) onto a gold surface
Deposition method.
To evaluate the affinity of the functionalised TNT EMFs to metal surfaces and their ability to form thin films, we have studied the deposition of 2 and 4 on a gold surface by immersing freshly prepared Au/mica substrates in dilute solutions of fullerenes.
Surface topography.
We have assessed the topography of the dithiolane functionalised Er3N@C802 self-assembled monolayers on Au(111) using scanning tunnelling microscopy (STM) (Fig. 8(a) and (b)). Spherical features are observed across the sample surface. The apparent diameters of these features range from 1.1 nm to 2.6 nm. Typically, the diameters of unfunctionalised TNT EMFs in close packed arrays are ∼1.15 nm.23,24 However, in our case the molecules are not densely packed and so tip convolution is likely to broaden their appearance. In addition, the functional group is conformationally flexible and so the fullerene cages are likely to move due to interactions between the STM tip and the fullerenes. This movement will significantly affect the measured fullerene diameter and increase the range of measured values. Our observations are consistent with these expectations, and therefore we attribute each spherical feature in Fig. 8(a) and (b) as a carbon cage of functionalised Er3N@C80.
 |
| | Fig. 8 Scanning tunnelling microscopy images of dithiolane functionalised Er3N@C802 on Au(111). Spherical features are observed and these are attributed to the fullerene cages of the molecules. (a) A large scan area image (70 × 70 nm) showing that the layer is almost complete. (b) A more detailed image (40 × 40 nm) in which the positions of individual molecules can be identified. A small close-packed cluster is highlighted by a white square and is shown in the inset. | |
Large areas of the surface are reasonably uniformly covered with a monolayer or sub-monolayer of 2 (Fig. 8). Unfunctionalised TNT EMFs deposited on Au-substrates from the solution phase under similar conditions show much less uniform coverage of the substrate, typically aggregating and forming multilayered islands on Au(111). As expected, the dithiolane group attached to the fullerene cage increases the affinity of fullerene to the metal surface and facilitates the formation of a fullerene monolayer. However, no long-range ordering of the molecules has been observed for monolayers of functionalised fullerenes. We attribute this observation to the strong bonding between the dithiolane group and the Au(111) surface. It is likely that two S–Au bonds per each molecule of 2 largely immobilise the functionalised fullerenes and so they remain at or close to their initial adsorption sites, preventing the surface migration of the molecules and formation of an ordered array. However, isolated patches comprising close packed fullerenes are occasionally seen illustrating the possibility of short-range molecular ordering (Fig. 8b, inset).
Bonding to the surface.
To explore further the mechanisms of the surface bonding for functionalised fullerenes, we have taken advantage of the high surface sensitivity and detailed chemical information that are offered by X-ray photoelectron spectroscopy (XPS). In general, sulfur-terminated functionalised fullerenes can bind to an Au(111) surface via either the functionalised group or the fullerene cage, leading to a range of possible geometries.25 XPS can be used to determine the orientation of the dithiolane functionalised fullerene monolayers with respect to the Au(111) surface. In particular, the S2p peaks provide excellent indicators of whether or not the functional group is bound to Au.
High resolution XPS spectra of functionalised fullerenes 2 and 4 show a good agreement in lineshape of the core level S2p peaks (Fig. 9), suggesting that the S atoms of dithiolane group have similar chemical environment in both cases (the tail towards higher energies for fullerene 2 is due to the onset of the Er 4d peak). Spectral fitting shows the presence of S2p3/2 (S2p1/2) peaks at 161.9 eV (163.1 eV) and 161.7 eV (162.9 eV) for the dithiolane functionalised Er3N@C80 and C60 monolayers, respectively. These values are in good agreement with the known spectral lines for S-atoms covalently bound to Au,25 and unequivocally demonstrate that the dithiolane group forms two S–Au bonds with the surface (Fig. 10). This binding mode provides a very efficient interaction of functionalised fullerenes with the surface, that is expected to be twice as strong as the standard Au–thiol interaction typically utilised for self-assembled monolayers. This confirms that the lack of long-range order in the monolayers of 2 and 4 is related to the strong interaction of the dithiolane group with the metal surface.
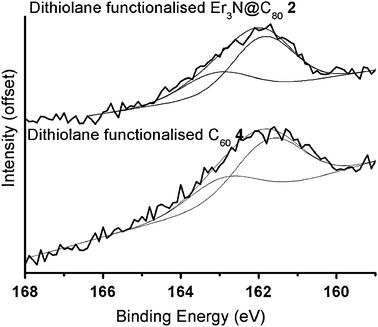 |
| | Fig. 9 S2p core level peak in XPS spectra of self-assembled monolayers of dithiolane functionalised Er3N@C80 (2) and dithiolane functionalised C60 (4) on Au(111). The peak intensity is offset for clarity. Gaussian peak fits are included. | |
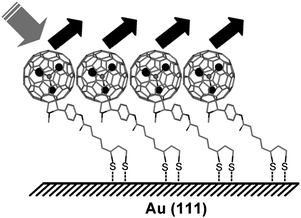 |
| | Fig. 10 Schematic representation of bonding of the dithiolane group to the Au-surface. | |
Surface photoluminescence of dithiolane functionalized Er3N@C80 (2) on Au(111)
The photoluminescence spectrum of dithiolane functionalised Er3N@C80 deposited on Au(111) surface was compared to that in solution (Fig. 11). An excellent agreement between the peak positions is observed, verifying that the optical functionality and chemical integrity of this fullerene have been retained. Variations in the relative intensities of the peaks are observed (this is particularly apparent for the ∼1519 nm emission line). These differences could arise from the preferential molecular orientation with respect to the substrate imposed by the Au–S bonds. We note that spatial mapping has highlighted inhomogeneities in the surface coverage, with photoluminescence signal only obtained from isolated regions of approximately 10 μm in diameter (an example of which is shown in the inset of Fig. 11). These patches are likely to arise from the regions of higher surface concentration of 2, possibly attracted to defect sites of the Au film.
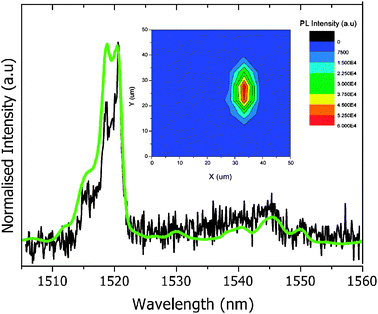 |
| | Fig. 11 Photoluminescence of 2 deposited on a Au(111) surface under a 800 nm laser excitation (black). The spectrum of 2 in CS2 as shown in Fig. 7 is included for comparison (green). A typical fluorescence map (50 × 50 μm) of 2 on Au(111) is shown in the inset. | |
Conclusions
Endohedral metallofullerenes have been functionalised with sulfur-containing groups for the first time and their interactions with gold surfaces have been explored. We have developed a method of attachment of dithiolane groups to Er3N@C80 and Sc3N@C80 TNT fullerenes, and have isolated isomerically pure, mono-functionalised endohedral fullerenes. The presence of the sulfur-containing group on TNT fullerenes has a significant effect on the interactions of these fullerenes with gold surfaces. The functionalised fullerenes deposited from solution give a more complete coverage of Au(111) surfaces than unfunctionalised TNT fullerenes, due to strong bonding between the dithiolane group and the metal surface. The effects of the exohedral functional group on the photoluminescence properties of the endohedral atoms have been demonstrated for the first time using Er3N@C80 as a model. The addition of chemical functionality to the highly symmetrical icosahedral C80 cage lowers the symmetry of the fullerene and results in splitting the major PL peaks of the endohedral Er-atoms. This may give a potential mechanism for controlling the functional properties of EMFs via the exohedral chemical functionality. The exohedral functionality does not quench the luminescence of the EMFs, so that the functionalised Er3N@C80 retains its optical properties within a monolayer assembled on the surface. The methodology for chemical functionalisation and surface deposition of EMFs described in this study can be further extended to electron spin active fullerenes, and in the long term could enable incorporation of endohedral fullerenes in functional electronic devices, harnessing their unique physicochemical properties for future technological applications.
Experimental
Raw erbium and scandium TNTs were supplied by Luna Innovations. Erbium TNTs were further purified by high performance liquid chromatography (HPLC) using a 5PYE column (Nacalai Tesque). Only one isomer of Er3N@C80 was detected. All other reagents and solvents were purchased from Aldrich and were used without further purification. All reactions were carried out under an argon atmosphere. Elemental analyses (C, H, N) were performed by the Elemental Analysis Service of London Metropolitan University. Infrared spectra were measured as either KBr discs or in solution on a Nicolet Avatar 380 FT-IR spectrometer over the range 400–4000 cm−1. 1H and 13C NMR spectra were obtained on a Bruker DPX300, 400, and AV(III)500 spectrometers. Coupling constants (J) are denoted in Hz, and chemical shifts (δ), in ppm. Multiplicities are denoted as follows: s = singlet, d = doublet, m = multiplet. Mass spectrometry was carried out on a Bruker Ultraflex III MALDI-TOF spectrometer using DCTB as matrix (355 nm) and on a Bruker MicroTOF with electrospray ionization (ESI). Analytical thin-layer chromatography (TLC) was performed using aluminium-coated Merck Kieselgel 60 F254 plates.
Dithiolane aldehyde precursor.
4-(Liponyloxy)benzaldehyde 3 was synthesized according with the procedure shown in the Supporting Information.
N-Methyl-2-(4-(liponyloxy)benzyl)-Sc3N@C80 fulleropyrrolidine (1).
0.8 mg of Sc3N@C80 (7.6 × 10−4 mmol), 1.1 mg of sarcosine (0.013 mmol) and 13.0 mg of 4-(liponyloxy)benzaldehyde (0.04 mmol) were dissolved in 15 mL of dry toluene in a 50 mL two-neck Schlenk flask equipped with a magnetic stirrer under argon. The mixture was heated and stirred for 270 min at 110 °C using an oil bath. After cooling to room temperature, the solvent was evaporated under reduced pressure until the volume was approximately 10 mL The reaction mixture was purified using silica gel column and toluene as eluent (Rf 0.13). After evaporation, 0.45 mg of a black powder was obtained (34% yield). MALDI-MS 1447.14 m/z [M]−.
N-methyl-2-(4-(liponyloxy)benzyl)-Er3N@C80 fulleropyrrolidine (2).
Following the procedure for the synthesis of 1, 1.8 mg of Er3N@C80 (1.2 × 10−3 mmol), 1.6 mg of sarcosine (0.018 mmol) and 18.5 mg of 4-(liponyloxy)benzaldehyde (0.06 mmol) were refluxed. The crude mixture was purified by column chromatography (silica, toluene) (Rf 0.14) to give 2 (0.55 mg) as a dark solid in 28% yield. MALDI-MS 1815.53 m/z [M]−. HPLC (5PYE column, 10 mm × 250 mm, 7 mL min−1 toluene, λ = 312 nm): 3.25 min.
N-methyl-2-(4-(liponyloxy)benzyl)-[6,6]-C60 fulleropyrrolidine (4).
A solution of 4-(liponyloxy)benzaldehyde (51.6 mg, 0.16 mmol) dissolved in dry toluene (5 mL) was added dropwise with stirring to a solution of C60 (100 mg, 0.14 mmol) and sarcosine (61.8 mg, 0.7 mmol) in dry toluene (35 mL). The resultant solution was refluxed at 110 °C under argon for 14 h. After cooling to room temperature, the solvent was evaporated, and the crude mixture was purified by column chromatography (silica, toluene) (Rf 0.22). Further purification was accomplished by subsequent precipitation with methanol to give 3 as a brown solid in 37% yield. 1H NMR {400 MHz, CDCl3, 300 K} δH 7.17 (m, 4H, aromatic H), 5.03 (d, J = 9.3 Hz, 1H, –CH2 pyrrolidine), 4.99 (s, 1H, –CH pyrrolidine), 4.33 (d, J = 9.3 Hz, 1H, –CH2 pyrrolidine), 3.57 (m, 1H, –CH), 3.20–3.10 (m, 2H, –CH2), 2.59 (t, J = 7.0 Hz, 2H, –CH2– alkyl chain), 2.49 (m, 1H, –CH2), 1.96 (m, 1H, –CH2), 1.76 (m, 2H, –CH2– alkyl chain), 1.61 (m, 2H, –CH2– alkyl chain) ppm. 13C NMR {500 MHz, CDCl3, 300 K} δC 170.70, 150.21, 148.41, 147.37, 146.63, 146.45, 146.37, 146.30, 146.22, 146.10, 146.0, 145.81, 145.34, 144.70, 144.47, 143.23, 142.72, 142.24, 142.32,142.14, 82.91, 70.10, 68.95, 68.41, 40.04, 34.02, 33.40, 28.15, 26.60, 24.75 ppm. MALDI-MS 1057.61 m/z [M]−. Elemental analysis found (expected)%: C 87.32 (87.32), H 2.16 (2.28), N 1.28 (1.32). IR (KBr disk) 2922 m (–C–H alkyl chain), 1740 m (–COO–), 1458 m (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C (s) phenyl group), 1384 (–O–CO–CH2–), 1458 m (CC (b) phenyl group). UV–vis (CHCl3) λmax: 431.66, 700. HPLC (SiO2 FORTIS HILIC (5μ) column, 250 mm × 21 mm, 5 mL min−1 3% ethylacetate in toluene, λ = 254 nm): 3.15 min (ESI).‡
C (s) phenyl group), 1384 (–O–CO–CH2–), 1458 m (CC (b) phenyl group). UV–vis (CHCl3) λmax: 431.66, 700. HPLC (SiO2 FORTIS HILIC (5μ) column, 250 mm × 21 mm, 5 mL min−1 3% ethylacetate in toluene, λ = 254 nm): 3.15 min (ESI).‡
Functionalised fullerenes were dissolved in CS2. The solution was drop coated on to a glass optical slide and dried in air. The Raman spectra were collected using a Horiba Jobin-Yvon Lab Aramis Confocal Raman Microscope in backscattering geometry, with an x100SLW objective (300 μm working distance). Measurements were performed at room temperature, under 532 nm excitation (solid state laser), on relatively thick (multi-layered film) sample areas.
Photoluminescence measurements
PL in solution.
Functionalised fullerenes were dissolved in CS2. The solution was placed in a quartz tube, degassed and sealed. For comparison, an Er3N@C80 solution was prepared in a similar manner. The concentration of each solution was not determined but was kept relatively low to avoid any clustering. Photoluminescence measurements were performed under a 532 nm excitation (15 mW), at 5 K using a He Oxford Instruments CF204 continuous flow cryostat. The detection was done through a monochromator (600 lines/nm grating) equipped with an InGaAs array detector.
PL on surface.
Samples comprising Er3N@C802 on Au(111) were prepared as outlined in the section “deposition of functionalised fullerenes on Au-substrates” below. PL measurements on these thin film samples were performed using a confocal microscope with an ×100 objective (Mitutoyo, 0.5 NA), which was mounted on a piezoelectric XYZ-stage 1 nm resolution. The samples were placed inside a continuous-flow liquid He microscope cryostat (Janis ST-500) in order to do the measurements at 5 K. The excitation was performed using a 800 nm Ti:Sapphire laser (Spectra-physics Mai-Tai). The detection was carried out through a monochromator (1200 lines/nm grating) equipped with the same InGaAs detector.
Surface deposition and characterisation
Deposition of functionalised fullerenes on Au-subsrtates.
Au(111) films (thickness 150 nm) grown on mica were used for the surface studies. These were prepared by flame annealing the substrate, a procedure well-known to produce the characteristic herringbone reconstruction of this surface. Thin film samples were prepared by immersing freshly prepared Au/mica substrates in dilute solutions of dithiolane functionalised C604 and Er3N@C802 in toluene and drying in a nitrogen gas stream.
STM.
STM images were obtained from an ultra high vacuum (UHV) JEOL JSTM4500S scanning tunnelling microscope (base pressure ∼10−9 mbar). The samples were prepared using the aforementioned ex situ procedure, immediately before transferring to the UHV chamber. Images were taken at room temperature at +2.3 V (bias applied to sample) and 0.2 nA using electrochemically etched tungsten tips.
XPS.
X-ray photoelectron spectroscopy measurements were performed using a VG Michrotech CLAM 4 MCD analyser system with a pass energy of 20 eV and slit size of 5 mm. X-rays were provided from a Mg Kα X-ray source operated at 200 W (base pressure ∼10−10 mbar), whilst data was obtained using SPECTRA version 8 operating system. A Shirley background subtraction was applied to all peaks prior to fitting. Binding energy scales were referenced using standard values for the Au4f7/2 (84.0 eV), Au4f5/2 (87.7 eV) and C1s (284.5 eV) peaks of gold and fullerenes, respectively. S2p1/2 and S2p3/2 peak fitting was performed whilst constraining the binding energy separation and relative peak abundances to values expected from spin–orbit coupling (1.2 eV and 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2, respectively).
:2, respectively).
Acknowledgements
This work was supported by the Engineering and Physical Sciences Research Council (EPSRC grant EP/D048761/01), the European Science Foundation (ESF), and the Royal Society. Raw samples of Er3N@C80 and Sc3N@C80 were supplied by Luna Innovations, Blacksburg, VA, USA.
References
- H. Shinohara, Rep. Prog. Phys., 2000, 63, 843–892 CrossRef CAS.
- A. N. Khlobystov, D. A. Britz and G. A. D. Briggs, Acc. Chem. Res., 2005, 38, 901–909 CrossRef CAS.
- J. A. Theobald, N. S. Oxtoby, M. A. Phillips, N. R. Champness and P. H. Beton, Nature, 2003, 424, 1029–1031 CrossRef CAS.
- T. W. Chamberlain, A. Camenisch, N. R. Champness, G. A. D. Briggs, S. C. Benjamin, A. Ardavan and A. N. Khlobystov, J. Am. Chem. Soc., 2007, 129, 8609–8614 CrossRef.
- T. W. Chamberlain, R. Pfeiffer, H. Peterlik, H. Kuzmany, F. Zerbetto, M. Melle-Franco, L. Staddon, N. R. Champness, G. A. D. Briggs and A. N. Khlobystov, Small, 2008, 4, 2262–2270 CrossRef CAS.
- M. M. Olmstead, A. de Bettencourt-Dias, J. C. Duchamp, S. Stevenson, D. Marciu, H. C. Dorn and A. L. Balch, Angew. Chem., Int. Ed., 2001, 40, 1223–1225 CrossRef CAS.
- K. Kobayashi, Y. Sano and S. Nagase, J. Comput. Chem., 2001, 22, 1353–1358 CrossRef CAS.
- L. Alvarez, T. Pichler, P. Georgi, T. Schwieger, H. Peisert, L. Dunsch, Z. Hu, M. Knupfer, J. Fink, P. Bressler, M. Mast and M. S. Golden, Phys. Rev. B: Condens. Matter Mater. Phys., 2002, 66 Search PubMed 035107/1–035107/7.
- T. Weider, F. Bretthauer, N. Ballav, H. Motschmann, H. Orendi, C. Brufn, U. Siemeling and M. Zharnikov, Langmuir, 2008, 24, 11691–11700 CrossRef CAS.
- T. Oshima, H. Kitamura, T. Higashi, K. Kokubo and N. Seike, J. Org. Chem., 2006, 71, 2995–3000 CrossRef CAS.
- E. B. Iezzi, K. H. Duchamp, T. E. Glass, H. M. Lee, M. M. Olmstead, A. L. Balch and H. C. Dorn, J. Am. Chem. Soc., 2002, 124, 524–525 CrossRef CAS.
- H. M. Lee, M. M. Olmstead, E. Iezzi, J. C. Duchamp, H. C. Dorn and A. L. Balch, J. Am. Chem. Soc., 2002, 124, 3494–3495 CrossRef CAS.
- C. M. Cardona, A. Kitaygorodskiy, A. Ortiz, M. A. Herranz and L. Echegoyen, J. Org. Chem., 2005, 70, 5092–5097 CrossRef CAS.
- C. M. Cardona, A. Kitaygorodskiy and L. Echegoyen, J. Am. Chem. Soc., 2005, 127, 10448–10453 CrossRef CAS.
- T. Cai, Z. Ge, E. B. Lezzi, T. E. Glass, K. Harich, H. W. Gibson and H. C. Dorn, Chem. Commun., 2005, 3594–3596 RSC.
- T. Cai, C. Slebodnick, L. Xu, K. Harich, T. E. Glass, C. Chancellor, J. C. Fettinger, M. M. Olmstead, A. L. Balch, H. W. Gibson and H. C. Dorn, J. Am. Chem. Soc., 2006, 128, 6486–6492 CrossRef CAS.
- C. M. Cardona, E. Bevan and L. Echegoyen, J. Am. Chem. Soc., 2006, 128, 6480–6485 CrossRef CAS.
- J. R. Pinzon, M. E. Plonska-Brzezinska, C. M. Cardona, A. J. Athans, S. Shankara Gayathri, D. M. Guldi, M. A. Herranz, N. Martin, T. Torres and L. Echegoyen, Angew. Chem., Int. Ed., 2008, 47, 4173–6 CrossRef CAS.
- H. E. Van Wart, A. Lewis, H. A. Scheraga and F. D. Saevat, Proc. Natl. Acad. Sci. U. S. A., 1973, 70, 2619 CAS.
- M. A. G. Jones, R. A. Taylor, A. Ardavan, K. Porfyrakis and G. A. D. Briggs, Chem. Phys. Lett., 2006, 428, 303–306 CrossRef CAS.
- A. Tiwari, G. Dantelle, K. Porfyrakis, R. A. Taylor, A. A. R. Watt, A. Ardavan and G. A. D. Briggs, J. Chem. Phys., 2007, 127, 194504 CrossRef.
- C. Norenberg, D. F. Leigh, D. Cattaneo, K. Porfyrakis, A. L. Bassi, C. S. Casari, M. Passoni, J. H. G. Owen and G. A. D. Briggs, J. Phys.: Conf. Ser., 2008, 100, 052080 CrossRef.
- D. F. Leigh, C. Norenberg, D. Cattaneo, J. H. G. Owen, K. Porfyrakis, A. Li Bassi, A. Ardavan and G. A. D. Briggs, Surf. Sci., 2007, 601, 2750–2755 CrossRef CAS.
- Y. Shirai, L. Cheng, B. Chen and J. M. Tour, J. Am. Chem. Soc., 2006, 128, 13479–13489 CrossRef CAS.
- T. M. Willey, A. L. Vance, C. Bostedt, T. van Buuren, R. W. Meulenberg, L. J. Terminello and C. S. Fadley, Langmuir, 2004, 20, 4939–4944 CrossRef CAS.
|
| This journal is © the Owner Societies 2010 |
Click here to see how this site uses Cookies. View our privacy policy here.