A UV-Vis micro-spectroscopic study to rationalize the influence of Cl−(aq) on the formation of different Pd macro-distributions on γ-Al2O3 catalyst bodies†
L.
Espinosa-Alonso
,
K. P.
de Jong
and
B. M
Weckhuysen
*
Inorganic Chemistry and Catalysis group, Debye Institute for Nanomaterials Science, Utrecht University, Sorbonnelaan 16, 3584 CA Utrecht, The Netherlands. E-mail: b.m.weckhuysen@uu.nl
First published on 7th November 2009
Abstract
The influence of the Cl−(aq) concentration, solution pH and equilibration time on the PdCl42−(aq) dynamics and molecular structure after impregnation of γ-Al2O3 catalyst bodies has been studied using UV-Vis micro-spectroscopy. To do so, 0.2 wt% Pd catalysts have been prepared from acidic solutions (pH 1 and 5) of the Na2PdCl4 precursor salt with different amounts of NaCl. It was found that egg-shell catalysts are obtained when a less acidic pH (pH 5) is combined with [Cl−(aq)] < 0.6 M and less than 24 h of equilibration time are implemented, while to achieve egg-white catalysts the solution pH should be 1. Moreover, by increasing the equilibration time up to 96 h, the egg-shell profiles vanish to provide a uniform Pd distribution, while the egg-white distribution becomes egg-yolk. Additionally, Pd complexes appeared with different molecular structures depending on the solution pH, equilibration time and macro-distribution achieved. The protocol developed to create different Pd macro-distributions has been applied to prepare two 1 wt% Pd/γ-Al2O3 egg-shell and egg-white catalysts. The Pd dynamics and molecular structure have been followed after impregnation, drying and calcination, demonstrating that the profiles created after impregnation are retained.
Introduction
Heterogeneous catalysts with different shapes and sizes ranging from a few millimeters to hundreds of micrometers are generally used in the chemical industry. These catalysts are usually prepared via pore volume impregnation of the support bodies with a solution containing the precursor metal-ion, followed by drying, calcination and other activation step to obtain the catalytically active phase.1,2 In order to prepare an efficient industrial catalyst it is not only important to control the dispersion and the solid phase of the active species, but also its location in the support body. In this way, the macro-distribution of the active species can be uniform along the catalyst body as sometimes is desired, while for other applications non-uniform distributions are more efficient.3 Yet, it is not well understood how non-uniform distributions are created during the preparation process, since many different chemical and physical parameters are involved; e.g. the metal-ion precursor complex, pH, ionic strength or viscosity of the impregnation solution, and surface properties of the support material, such as the point of zero charge (pzc).4–7 All these parameters influence the adsorption properties of the metal-ion precursor on the support surface, and they also determine the transport rate of the active species towards the core of the support bodies. For many years numerous research groups have focused their attention on studying the first steps of catalyst preparation at a molecular level, and several adsorption models have been proposed.8–13 However, these studies have in general been performed in powdered catalysts, and when this knowledge is applied to prepare mm-sized heterogeneous catalysts, the understanding of the adsorption of the precursor metal-ion complexes on the support surface is not so straightforward, since a new variable is involved, namely the size of the support body. This new parameter can create, for instance, the speciation of the initial metal-ion precursor complex towards the core of the support material due to acid–base reactions between the alumina surface and the complex in the solution inside the pores.14 To understand the effect of the microscopic size of industrial support materials on the interactions between the active species and the support surface, the development of space resolved techniques is crucial, and large efforts are being made in developing spatiotemporal methods.14–20In this paper, we show how Cl−(aq) can affect the preparation of Pd/γ-Al2O3 pellets. We demonstrate that additional Cl−(aq) ions in the impregnation solution affect the Pd macro-distribution boosting more uniform profiles. Furthermore, we show how these ions influence the Pd speciation within catalyst bodies. It is well known that Cl−(aq) acts as a competitor in the adsorption of PdCl42− and PtCl62− on alumina supports favoring uniform distribution of the metals.21–24 However, only a few research groups have obtained an insight into the structural changes that PdII and PtVI undergo after contacting the support material.25–27 Even less is known about the changes that occur when Pd catalysts are prepared on support bodies, i.e. what is the influence of the size of the pellet on the adsorption and speciation of PdCl42− in microscopic support bodies. Based on this study, we have designed an experimental protocol to prepare different Pd catalysts that can be applied to prepare highly loaded catalysts.
Experimental
1 Catalyst preparation
Two series of impregnation solutions containing sodium tetrachloropalladate (Na2PdCl4, ca. 36.4 wt% Pd, Acros) in a final 20 mM Pd concentration were prepared in order to yield 0.2 wt% Pd on the catalysts. These series have been labeled as Series A and Series B. Series A was prepared by dissolving Na2PdCl4 in an aqueous 112 mM HCl solution (from conc. HCl, Acros). Six solutions were prepared from Series A containing increasing amounts of NaCl (Acros p.a.) to a final chloride concentration ranging from 112 to 1000 mM. The chloride concentration is referred to the sum of HCl and NaCl, and not to that from the precursor Na2PdCl4 salt. Seven solutions in Series B were prepared by dissolving Na2PdCl4 in water and adding increasing amounts of NaCl (Acros p.a.) to the same final concentration of Cl−(aq) as in Series A. The concentration of Cl−(aq) (as the sum of HCl and NaCl) and the experimental pH of the solutions are summarized in Table 1.Additionally, two solutions were prepared by dissolving the required amounts of Na2PdCl4 (ca. 36.4 wt% Pd, Acros) to a final 94 mM Pd concentration to yield 1 wt% Pd on the catalysts. The solution labeled as 1 wt%Pd-A1 was prepared by dissolving the Pd salt in a 112 mM HCl aqueous solution (from conc. HCl, Acros). The solution labeled as 1 wt%Pd-B4 was prepared by dissolving the Pd salt in water and adding NaCl (Acros p.a.) to a final NaCl concentration of 750 mM. The experimental pH of the solutions was 1.1 (solution 1 wt%Pd-A1) and 5.3 (solution 1 wt%Pd-B4).
Cylindrical γ-Al2O3 pellets (Engelhard, 3 mm in height and diameter) were used. The pellets were calcined at 450 °C for 8 h and stored at 120 °C until used. This material had a pore volume of 1.0 ml g−1 and a surface area of 200 m2 g−1. The point of zero charge (pzc) was determined to be 7.8 by mass titration.28 The γ-Al2O3 surface presented three different types of hydroxyl groups: basic, neutral and acidic, as determined in a previous work.14
The supported catalyst materials were prepared via pore volume impregnation on γ-Al2O3 pellets with all the Pd solutions prepared. The solutions were added drop-wise to the support bodies making sure that all the pellets in each batch (0.9 g) were wetted. The amount of solution added was equal to the pore volume plus 10%. After adding the impregnation solution, manual shaking was applied during 2 min to obtain an even distribution of the solution on the pellets and the extra solution added (10% of the pore volume) was removed with a pipette. After impregnation, the pellets were kept in a closed vessel with a wet tissue to avoid drying.
Two batches of pellets impregnated with solutions 1 wt%Pd-A1 and 1 wt%Pd-B4 were dried and calcined. The pellets were dried 3 h after impregnation (equilibration time). Drying was performed in a static air oven at 120 °C (5 °C min−1) during 4 h. For calcination, the temperature was increased up to 500 °C (5 °C min−1) and the pellets were held at this temperature for 4 h.
2 UV-Vis spectroscopic characterization
The impregnation solutions were characterized with UV-Vis spectroscopy. The UV-Vis spectra were recorded using a Cary 50 UV-Vis spectrophotometer in the range of 300 to 700 nm. Table 2 summarizes the position of the Pd absorption bands measured in this range together with the Pd species present in all the solutions, calculated from classical least squares (CLS) of the UV-Vis spectra measured. To do the CLS analysis, the UV-Vis spectra collected on solution B0 (PdCl42−) and B6 (PdCl3(H2O)−) were used as single components and linear combinations of the two spectra were made in order to achieve the best fit.| Series A | PdII d–d band/nm | % Species | Series B | PdII d–d band/nm | % Species | ||
|---|---|---|---|---|---|---|---|
| PdCl42− | PdCl3(H2O)− | PdCl42− | PdCl3(H2O)− | ||||
| a PdII speciation was calculated from the deconvolution of the PdII d–d transition band. | |||||||
| — | — | — | — | B0 | 430 | 0 | 100 |
| A1 | 455 | 56 | 44 | B1 | 449 | 53 | 47 |
| A2 | 471 | 79 | 21 | B2 | 469 | 82 | 18 |
| A3 | 474 | 90 | 10 | B3 | 471 | 89 | 11 |
| A4 | 474 | 96 | 4 | B4 | 473 | 98 | 2 |
| A5 | 474 | 95 | 5 | B5 | 473 | 96 | 4 |
| A6 | 474 | 100 | 0 | B6 | 474 | 100 | 0 |
The catalytic materials were measured after impregnation, drying or calcination with UV-Vis micro-spectroscopy, making use of a home-built set-up previously described in literature.20 All measurements on the catalyst bodies were performed applying a scan-line on the resulting surface of bisected pellets. The pellets were bisected by inserting them inside a silicon tube; the system tube-pellet was cut with a scalpel. Nine spectra were collected along the cross section of the bisected pellets with a spatial resolution of around 250 μm, for which 5 min were required. Sometimes, the UV-Vis spectra showed a spike at around 460 nm, this spike is due to interference from ambient light during the measurements.
3 Raman microscopy
Raman spectra at several positions on the calcined 1 wt%Pd pellets (after bisection) were collected using a Raman microscope (Renishaw) equipped with a 4.40 mW, 514 nm laser for excitation. The spot size was approximately 10 μm.Results and discussion
1 UV-Vis micro-spectroscopic study on the preparation of 0.2 wt% Pd catalysts
The electronic spectra of PdCl4−x(H2O)x(x−2) complexes in solution are very similar and are characterized by, among others, a spin-forbidden d–d transition (1A2g ← 1A1g) of square-planar PdII complexes in the region between 350–500 nm.29 All the impregnation solutions were very similar in color, this one being orange. This could also be inferred from the UV-Vis spectra, in which the only difference was the position of the Pd d–d spin-forbidden transition between 350–500 nm. The position of this band in the impregnation solutions is summarized in Table 2. The maximum absorption of the band shifts to shorter wavelengths when the chloride ligands are exchanged for water ligands (from solution A6 or B6 to solution A1 or B0), in agreement with the spectrochemical series of the ligands.30 The band centered at 475 nm has been assigned to the complex PdCl42− while the band centered at 431 nm has been assigned to the complex PdCl3(H2O)−.31 Thus, when the band is in between these two positions it indicates the presence of a mixture of both complexes. Further hydrolysis of PdCl3(H2O)− yields more hydrolyzed complexes, PdCl4−x(H2O)x(x−2)(x = 0–4). These complexes show a larger shift of the PdII d–d transition band to shorter wavelengths, and a change in color of the impregnation solutions is expected towards brown. Table 3 summarizes the position of the PdII d–d transition band, as reported by Elding et al.31| PdII chloride complex | λ/nm | ε/M−1 cm−1 |
|---|---|---|
| [PdCl4]2− | 474 | 161 |
| 345 | 200 | |
| [PdCl3(H2O)]− | 431 | 227 |
| [PdCl2(H2O)2] | 420 | 243 |
| [PdCl(H2O)3]+ | 406 | 175 |
| [Pd(H2O)4]2+ | 378 | 83 |
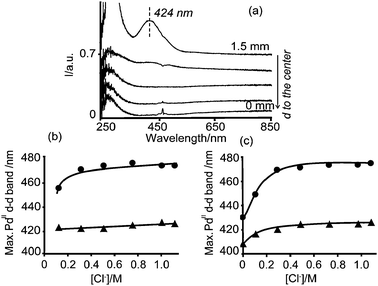 | ||
| Fig. 1 (a) Space resolved UV-Vis spectra measured on a γ-Al2O3 pellet, from the edge to the center, 5 min after impregnation with any of the prepared PdII impregnation solutions; (b) and (c) show the position of the PdII d–d transition band as a function of the Cl− (aq) concentration in the impregnation solutions (–●–) and measured in the edge of the impregnated pellets 5 min after impregnation (–▲–), from Series A (b) and Series B (c). | ||
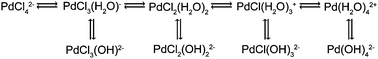 | ||
| Scheme 1 Equilibria of aqueous palladium chloride complexes.34,35 | ||
It is known that the PdCl42− complex undergoes the equilibria shown in Scheme 1 in solution in the absence of NaCl as a supporting electrolyte.34,35The formation of the aqueous species, top line in Scheme 1, is due to autohydrolysis, while the formation of the basic species (bottom line in the scheme) is generally due to alkaline hydrolysis. Moreover, autohydrolysis is dependent on the amount of additional Cl− ions in the solution.34 Hence, when Pd chloride complexes contact the alumina surface (represented as Al–OH), which acts as an alkaline medium due to its higher pzc compared to the solution pH, they suffer alkaline hydrolysis and they become PdCl2(OH)22−. Moreover, the basic OH groups (AlB–OH) from the alumina surface are most likely the easiest to become protonated in an acidic medium, as presented in eqn (1):14
| PdCl42− + 4AlB–OH + 2H3O+ → PdCl2(OH)22− + 4AlB–OH2+ + 2Cl− | (1) |
As an exception of the formation of PdCl2(OH)22− lies the impregnated pellet with solution B0, which did not contain any additional Cl− ions. The space resolved UV-Vis spectra of this pellet showed a band in the edge of the catalyst body with a larger shift to shorter wavelengths. The band appeared centered at 408 nm, which can be assigned to PdCl(OH)32−, Fig. 1b. The formation of a further hydrolyzed complex is due to the absence of additional Cl− ions, which are responsible for the stabilization of the chlorinated species.34 Moreover, this agreed with a brownish color of the pellet instead of the yellow color observed in the other samples.
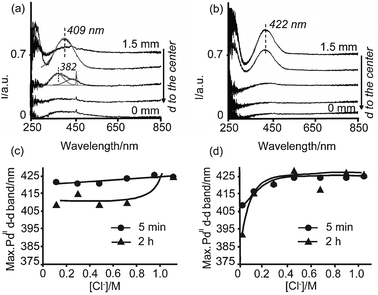 | ||
| Fig. 2 Space resolved UV-Vis spectra measured from the edge to the center of pellets impregnated with solution A3 (a) and B3 (b), 2 h after impregnation; (c, d) position of the PdII d–d transition band measured on pellets impregnated with Series A (c) and Series B (d) as a function of the Cl− (aq) concentration 5 min after impregnation at the edge (–●–), and 2 h after impregnation (–▲–) at the edge (Series B) or at 0.4 mm from the edge (Series A). | ||
On the other hand, the UV-Vis spectra of the impregnated pellet with solution A3 show a symmetrical band centered at 409 nm, measured at 1.1 mm from the center; i.e., not in the outer region but slightly deeper in the pellet in an egg-white distribution. Thus, with time after impregnation this band has shifted toward shorter wavelengths, which suggests the hydrolysis of PdCl2(OH)22− to form a structure similar to PdCl(OH)32− (Table 3).31 A Gaussian analysis of this band indicates indeed the presence of a single band at 409 nm. Moreover, the next spectrum close to the core (at 0.7 mm from the core) shows the maximum intensity at around 380 nm; i.e., at shorter wavelengths with respect to 409 nm. This band, additionally presents a strong interference with ambient light during the measurement (spike at around 460 nm). The deconvolution (Gaussian analysis) of this band yields a band centered at 382 nm due to Pd species and a broad contribution above 400 nm. This contribution has been partly assigned to the interference with ambient light. The band at 382 nm is close to the reference value for Pd(H2O)42+ at 378 nm (Table 3) and suggests the hydrolysis of PdCl(OH)32− while moving towards the core, forming a structure similar to Pd(OH)42− (which PdII d–d absorption band is expected at longer wavelengths than 378 nm, since OH− is a weaker ligand than H2O in the spectrochemical series). It is known, however, that the d–d band of Pd(H2O)42+ is nonsymmetrical in shape with a tail at longer wavelengths.31 This tail is probably part of the band above 400 nm obtained from the Gaussian analysis, which is also due to interference with ambient light. The hydrolysis of Pd chloride complexes when these are diffusing towards the core of the pellet is a direct consequence of the increase in pH of the solution inside the pores towards the core due to protonation of the alumina surface.
The space-resolved UV-Vis spectra in Fig. 2 indicate that the egg-shell distributions created 5 min after impregnation remained after 2 h when the pellets were impregnated with solution B3 (Fig. 2b), while impregnated pellets with solution A3 yielded Pd egg-white profiles (Fig. 2a). Moreover, Pd egg-shell profiles were obtained 2 h after impregnation of any solution from Series B, whereas Pd egg-white profiles were obtained 2 h after impregnation with any solution from Series A. Photographs taken 2 h after impregnation and bisection of the pellets clearly show these yellow egg-shell and egg-white profiles, as illustrated in Fig. 5.
The egg-white profile is formed due to the acidity of the impregnation solutions. As depicted in eqn (1), immediately after impregnation with an acidic solution the alumina surface (Al–OH) becomes protonated and PdCl2(OH)22− is formed. Protonation of the alumina surface occurs mainly on the basic hydroxyl groups (AlB–OH) and it yields an acid–base reaction between the protonated species AlB–OH2+ and Cl−, as depicted in eqn (2), and, consequently, alumina becomes more acidic (lower pzc):14,28,36
| AlB–OH + H+ + Cl− → AlB–OH2+ + Cl− ↔ AlB–OH2+⋯Cl− ↔ AlB–Cl + H2O | (2) |
| PdCl2(OH)22− + 2AlB–OH2+ + 2Cl− → PdCl2(OH)22− + 2AlB–OH2+⋯Cl− + 2 H2O | (3) |
It must be taken into account that alumina is not stable in acidic pH, and it can partially dissolve liberating Al3+ ions after impregnation of acidic solutions. These ions can ultimately become part of the active phase influencing the catalyst efficiency. In this work, no influence of the presence of Al3+ on the UV-Vis spectra of Pd chloride species could be observed. Therefore, even though the formation of Al3+ ions cannot be ignored, it will not be taken into account in the discussion regarding the UV-Vis micro-spectroscopic data.
Fig. 2c and d additionally show that the maximum absorption of the PdII d–d transition band shifted 2 h after impregnation of the pellets with Series A (Fig. 2c), while there was no shift of the position of this band 2 h after impregnation with Series B. When the pellets were impregnated with Series A, after 2 h, the PdII d–d transition band was measured at around 410 nm except for the pellets impregnated with solutions A6 and A7, which contained very high concentrations of Cl−(aq). In general, PdCl2(OH)22− has undergone further hydrolysis, forming a structure similar to PdCl(OH)32−. This is a consequence of the adsorption of Cl−(aq) on the alumina surface. Because of adsorption of Cl−(aq), the solution inside the pores lost Cl− ions, which no longer stabilized PdCl2(OH)22− complexes. When the pellets were impregnated with Series B, PdCl2(OH)22− complexes are stabilized (band at around 422 nm) because chloride adsorption does not occur to a large extent. Hence, the concentration of Cl− in solution is high and further hydrolysis of the PdCl2(OH)22− complex is avoided.
Fig. 3 shows the intensity of the PdII d–d transition band measured at different times after impregnation of solutions A6 and B6 and it indicates that longer equilibration times yield different distributions. This Figure illustrates the development of egg-yolk and uniform Pd macro-distributions as a function of time after impregnation.
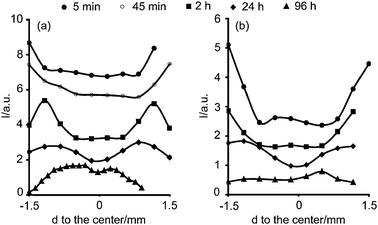 | ||
| Fig. 3 Maximum absorption of the PdII d–d transition band measured with UV-Vis micro-spectroscopy as a function of position inside the pellet and time after impregnation of solution (a) A6 and (b) B6. | ||
Impregnation with solution B6 yielded a uniform profile of Pd after 96 h, while impregnation with solution A6 gave rise to an egg-yolk Pd distribution. These Pd macro-distributions depended on the solution pH and a uniform profile was favorable for solutions with pH 5 (Series B), whereas solutions with pH 1 (Series A) yielded egg-yolk macro-distributions. Moreover, they also depended on the Cl− (aq) concentration, and they were only formed when the impregnation solutions had a very high concentration of Cl− ions (data not shown).
Additionally, the position of the maximum absorption of the PdII d–d transition band depended on the Cl− concentration and solution pH. Fig. 4 shows the position of the maximum absorption of the PdII d–d transition band in the catalyst bodies measured 2 h after impregnation at the point on the pellet where the intensity of the band was the highest, and in the center of the pellet 96 h after impregnation.
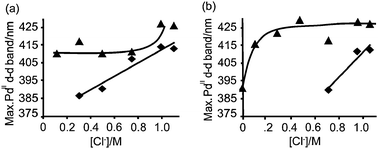 | ||
| Fig. 4 Position of the maximum absorption of the PdII d–d band as a function of Cl− (aq) concentration, 2 h close to the outer rim (▲) and 96 h in the center (◆) of the pellet after impregnation with Series A (a) and Series B (b). | ||
96 h after impregnation, the PdII d–d transition band was not observed in the center of the catalyst bodies for solutions with low Cl− (aq) concentration, for that reason some points do not appear in the Figure. Moreover, when PdII ions reached the core it was surrounded at maximum by one Cl− ligand as PdCl(OH)32−, as deduced from the position of the PdII d–d transition band measured between 385 and 412 nm (Table 2). As explained earlier in this work, the alkaline hydrolysis of Pd chloride complexes towards the core is a direct consequence of the buffering effect of the alumina support. Thus, the higher the concentration of Cl− (aq) in the impregnation solution the more chlorinated the PdII present in the core.
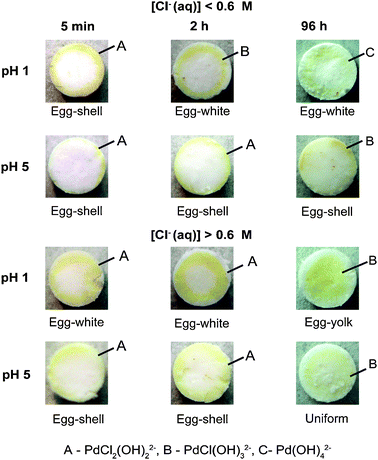 | ||
| Fig. 5 Photographs showing the different Pd macro-distributions developed as a function of the solution pH, Cl−(aq) concentration, and equilibration time; together with the Pd composition. | ||
Based on Fig. 5 we can conclude that in order to achieve uniform or egg-yolk Pd distributions, long equilibration times are required in combination with a [Cl−(aq)] ≥ 0.6 M.
As was previously described, Cl− act as a competitive anion in acidic environments. At low pH values, adsorption of Cl− on the alumina surface is preferential and PdCl2(OH)22− complexes can move forward in the support body while Cl− is retained in the outer rim occupying all the adsorption sites in that area. Consequently, an egg-yolk Pd distribution is created. On the other hand, when the solution pH is around 5, there is no competition between Cl− and PdCl2(OH)22− and, therefore, the probabilities of adsorption of one or the other anion are the same, yielding a uniform Pd macro-distribution. In any case, the concentration of active component is below the concentration required to saturate alumina surface. This, combined with the strong electrostatic interactions between the Pd complexes formed in the pores of the alumina, results in a slow transport rate of Pd towards the core of the support body, and long equilibration times are required to achieve uniform and egg-yolk Pd catalysts. If higher Pd loadings are required, the equilibration times could be reduced, as it is explained in the next section.
2 UV-Vis micro-spectroscopic study on the preparation of 1 wt% Pd catalysts
For some applications higher Pd loadings are required to have a better performance of the industrial process. For this reason, the developed methodology explained above to tune from egg-shell to egg-white, egg-yolk or uniform profiles was applied to prepare 1 wt% Pd/γ-Al2O3 catalyst bodies.2.1.1 5 min after impregnation. Fig. 6 illustrates the space resolved UV-Vis spectra of the impregnated pellets with 1 wt% Pd solutions, after 5 min of equilibration. Photographs of the bisected pellets after this time can also be found in Fig. B and C of the ESI.† A similar egg-shell distribution of Pd complexes for the two impregnated pellets was deduced. However, the position of the UV-Vis bands measured indicated that different Pd complexes were present depending on the impregnation solution.
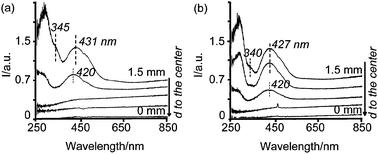 | ||
| Fig. 6 Space resolved UV-Vis spectra from the edge to the center of pellets 5 min after impregnation with solutions (a) 1 wt%Pd-A1, and (b) 1 wt%Pd-B4. | ||
The spectrum at the edge of the pellet impregnated with solution 1 wt%Pd-A1 (Fig. 6a) is characterized by a band centered at 431 nm and a shoulder at around 345 nm. However, 0.4 mm deeper in the extrudate the PdII d–d band at 431 nm shifted to 420 nm and the shoulder at around 345 nm disappeared. Even though a Gaussian analysis of these two spectra might be ambiguous due to the complexity of the system PdCl4−x(H2O)x(x−2) (x = 0–4); i.e., four different species with very similar spectra and the Pd d–d bands being very irregular in shape, further insight was attempted, as illustrated in Fig. D in the ESI.† The deconvolution of the spectrum close to the edge showed two important components at 343 and 425 nm. The band at 343 nm is assigned to PdCl42−, while the band at 425 nm is assigned to PdCl2(OH)22−, following the same reasoning described in Section 1.1. Moreover, the low intensity of the band at 343 nm compared to that at 425 nm (the absorption coefficient of these bands is known to be comparable and around 200 cm−1 M−1) indicates that the main component is PdCl2(OH)22−.29 A shift of the band centered at 431 nm to 420 nm when Pd moves towards the core, suggests the further hydrolysis of PdCl2(OH)22− species. Furthermore, the shoulder at 345 nm disappears. The Gaussian analysis presented in Fig. D of the ESI† clearly shows the shift of the component at 425 nm to 418 nm. This band is between that of PdCl2(H2O)2 and PdCl(H2O)3+ (Table 3) and confirms the partial hydrolysis of PdCl2(OH)22− species while moving towards the core. Additionally, a component at 338 nm is still measured with low intensity which suggests that PdCl42− is barely present and it has also undergone partial hydrolysis (indicated by the shift to shorter wavelengths). The UV-Vis spectrum at the edge of the pellet impregnated with solution 1 wt%Pd-B4 (Fig. 6b) also shows a shoulder at around 340 nm and a band centered at 427 nm. The Gaussian analysis of this spectrum, presented as Fig. E in the ESI†, yields a band at 339 nm, while the band centered at 427 nm can be fitted by using two components at 412 nm and at 467 nm. These bands do not match with any particular species, but they suggest the presence of several species: the band at 412 nm could indicate a mixture of PdCl2(H2O)2 and PdCl(H2O)3+, while the band at 467 indicates a mixture of PdCl42− and PdCl3(H2O)−, as compared to the values presented in Table 3. Moreover, the latter two species are known to have irregular band shapes.25,31 Hence, the only conclusions that can be drawn from this analysis is that small amounts of PdCl42− are present, indicated by the shoulder at 339 nm, and that there is a mixture of PdCl4−x(H2O)x(x−2) that in average gives a structure similar to PdCl2(OH)22− (following the same reasoning as for the 0.2 wt% Pd catalysts). Furthermore, the band centered at 427 nm appears at 426 nm in the second spectrum close to the edges (not indicated in the Figure). From the Gaussian analysis of this spectrum, presented in Fig. E in the ESI†, it can be concluded that less PdCl42− species are present in this position. Additionally, the two components that form the band at 426 nm appear at shorter wavelengths than in the spectrum close to the edges. This means that even though the average Pd structure in this point is also PdCl2(OH)22−, there are more hydrolyzed species present here when compared to closer to the edges. If one looks at the third spectrum close to the edges, it can be seen that the band at 427 nm has shifted further towards shorter wavelengths, 420 nm, which again indicates the hydrolysis of the initial Pd chlorinated species. In general, the presence of PdCl42− in the impregnated pellets with the 1 wt% Pd solutions suggests that part of the PdII ions in the pores of alumina do not interact with the surface of the alumina and remain in the bulk liquid of the pores. Hence, they do not suffer hydrolysis. Still, the dynamics and molecular structure of Pd, 5 min after impregnation with 1 wt% Pd solutions correlates rather well with the findings for the 0.2 wt% Pd catalysts described in this work.
2.1.2. 3 h after impregnation. During the equilibration time, Pd complexes moved deeper in the catalyst bodies, as can be deduced from the movement of the yellow color towards the core of the pellets. The photographs can be found in the ESI.†Fig. 7 shows the space resolved UV-Vis spectra from the edge to the center of the impregnated pellets after 3 h. When the pellet was impregnated with solution 1 wt%Pd-A1 and equilibrated for 3 h (Fig. 7a), the band appeared centered at 419 nm in a uniform distribution, except for the position close to the edge where the band appeared at around 429 nm. A Gaussian analysis presented in Fig. F in the ESI† indicates that the spectrum at the edge contains a small band at 340 nm, which is assigned to a transition band from PdCl42−, and the broad band at 429 nm composed by two others centered at 416 and 464 nm. The band at 416 nm suggests the presence of PdCl(OH)32− or a mixture of PdCl(OH)32− and PdCl2(OH)22−. On the other hand, the spectra measured towards the core are the same and show a band centered at 419 nm. The deconvolution of the spectrum in the center of the pellet, as presented in Fig. F in the ESI,† indicates the absence of PdCl42− species (no band at 340 nm is present) and the band at 419 nm was mainly constituted by a band centered at 416 nm that, as in the edge of the pellet, can be assigned to PdCl(OH)32− or a mixture of PdCl(OH)32− and PdCl2(OH)22−. Thus, the edge of the pellet impregnated with solution 1 wt%Pd-A1 contains more chlorinated species than the core, as indicated by the band at 340 nm.
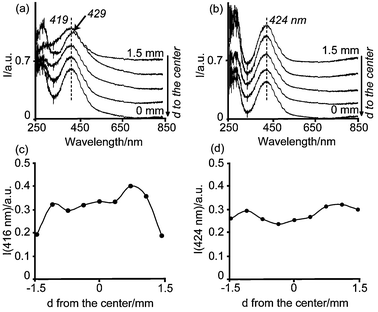 | ||
| Fig. 7 Space resolved UV-Vis spectra from the edge to the center of pellets 3 h after impregnation with solutions (a) 1 wt%Pd-A1 (b) 1 wt%Pd-B4. The intensity of the corresponding PdII d–d band is also represented as a function of the position in the catalyst body after impregnation with solution (c) 1 wt%Pd-A1, and (d) 1 wt%Pd-B4. | ||
The impregnated pellet with solution 1 wt%Pd-B4 showed the maximum absorption of the PdII d–d band 3 h after impregnation at around 424 nm. From the Gaussian analysis of these spectra it can be concluded that PdCl42− is not present any more. Hence, it has decomposed to form more hydrolyzed species. Moreover, the band at 424 nm can be fitted to two Gaussian curves at 418 nm and 476 nm. The 476-nm band cannot be assigned to PdCl42− species since the band at 340 nm is absent, and the latter has a higher absorption coefficient, as summarized in Table 3. Hence, the non-symmetrical band at 424 nm has been assigned to the complex PdCl2(OH)22−, in agreement to what was observed for the 0.2 wt% Pd system (sample B4 with the same concentration of Cl− ions). Thus, the position of the PdII d–d transition band shifted towards shorter wavelengths during the equilibration time, indicative of hydrolysis of the initial Pd chloride complexes.
Furthermore, the bands at 419 nm (1 wt% Pd-A1) and 424 nm (1 wt% Pd-B4) were measured along the cross section of the catalyst bodies, but with different intensities. The intensity of these bands as a function of the position in the catalyst bodies is also included in Fig. 7. After 3 h of equilibration time, Pd was somewhat depleted at the edges of the pellet when this was impregnated with the 1 wt%Pd-A1 solution. On the other hand, an almost uniform Pd distribution was noted if the impregnation was done with the 1 wt%Pd-B4 solution. These profiles were expected according to the solution pH, equilibration time and Cl− concentration (Fig. 5). However, these profiles were not as sharp as obtained for the 0.2 wt% Pd samples, and Pd was already detected in the core of the pellets.
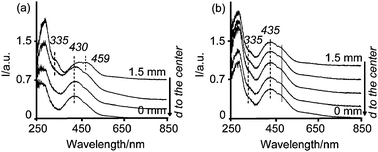 | ||
| Fig. 8 Space resolved UV-Vis spectra from the edge to the center of dried pellets impregnated with solutions (a) 1 wt%Pd-A1 (b) 1 wt%Pd-B4. | ||
The UV-Vis spectra of the dried pellet containing a 1 wt%Pd-B4 solution showed a shoulder at 335 nm and a band centered at 435 nm with a shoulder at around 460 nm homogeneously distributed (Fig. 8b). A Gaussian analysis of the spectra in the center and in the edge, presented as Fig. I in the ESI,† indicates that the spectra were almost identical regardless of the position along the catalyst body. All the spectra showed a band between 335–340 nm assigned to PdCl42− species and a band centered at 435 nm, which was deconvoluted in two components: at 425 and 490 nm. Considering that the species present in the pellets 3 h after impregnation are PdCl2(OH)22−, a shift towards longer wavelengths, to 435 nm, after drying can only be explained by an exchange of OH− groups with Cl− ligands forming PdCl3(O–Al)2− and PdCl4−. Therefore, we can conclude that after drying there is a mixture of PdCl2(O–Al)22− PdCl3(O–Al)2− and PdCl42− along the cross section of the catalyst body.
The formation of more chlorinated complexes after drying is in agreement with previous studies reported on the adsorption mechanism of Pd on alumina during catalyst preparation. Bozon-Verduraz et al.27 suggested that after impregnation of an acidic solution which contained PdCl2, the alumina surface becomes chlorinated and a deposit of PdCl2 is formed, while drying yielded the regeneration of PdCl42− as the chloride ions from the alumina surface enter the coordination sphere of Pd. In this study we have shown that indeed a regeneration of chlorinated Pd complexes after drying takes place. However, the regeneration capacity of PdCl42− depends on the effect that Cl− has on the alumina surface after impregnation. The chlorination of the alumina surface is pH dependent and is enhanced at more acidic pHs.36 Therefore, impregnation with a 1 wt%Pd-A1 solution yielded the exchange of alumina hydroxyl groups with Cl− groups, especially in the outer rim of the pellet. Thus, a ring of chlorinated alumina is formed close to the edges and the formation of a region richer in PdCl42− complexes in that area after drying is expected. On the other hand, alumina is less chlorinated in the core and Pd adsorbs on alumina forming a structure similar to PdCl3(O–Al)− or PdCl2(O–Al)2. This is also the case if the pellets are impregnated with a 1 wt%Pd-B4 solution.
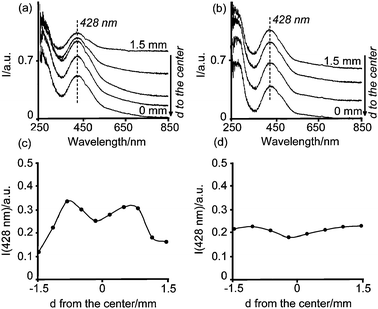 | ||
| Fig. 9 Space-resolved UV-Vis spectra from the edge to the center of calcined catalyst bodies after impregnation with solutions (a) 1 wt%Pd-A1, (b) 1 wt%Pd-B4. The intensity of the band at 428 nm as a function of the position in the catalyst body is also shown (c) 1 wt%Pd-A1, and (d) 1 wt%Pd-B4. | ||
The presence of the absorption band at 428 nm suggests that only partial loss of Cl− ligands from the molecular structure of Pd complexes during calcination took place yielding the formation of a structure similar to PdCl2(O–Al)22−, since if metallic Pd or PdO was formed during the calcination, this band would be non-existant or would appear at around 410 nm.37 The water ligands have been exchanged by the hydroxyl groups from the alumina surface, which are known to be weaker ligands in the spectrochemical series.33 The presence of Cl− can also be inferred from the absorption band at around 260 nm, which corresponds to a LMCT from Cl− to PdII. From the UV-Vis micro-spectroscopic data no speciation of PdCl4−x(H2O)x(x−2) complexes was measured along the catalyst body; i.e., the absorption band appeared centered at 428 nm regardless of the position within the pellet. However, this band showed a different intensity depending on its position in the catalyst body, as depicted in Fig. 9b and c. These Figures show that the Pd macro-distributions achieved 3 h after impregnation were retained after the drying and the calcination process. This was also confirmed by visual inspection, as can be observed in the photographs of bisected pellets in Fig. 10. It must be noted, however, that different drying conditions might enhance redistribution of Pd within the support bodies.
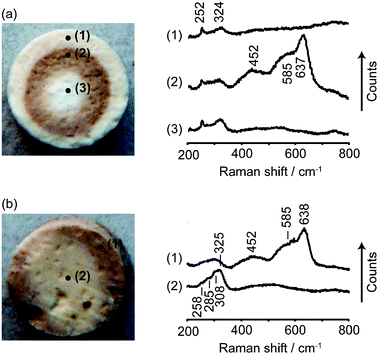 | ||
| Fig. 10 Photographs of bisected catalyst bodies after calcination and Raman spectra measured at several points on the bodies: (a) 1 wt%Pd-A1, and (b) 1 wt%Pd-B4. | ||
Fig. 10 shows the Raman spectra measured at different points of the bisected pellets after calcination. As compared to the UV-Vis micro-spectroscopic data, the space resolved Raman spectra indicate the presence of different Pd phases along the catalyst bodies. The Raman spectra measured close to the edge or in the center of the 1 wt%Pd-A1 pellet (Fig. 10a, positions (1) and (3)) show a sharp peak at 252 cm−1 and a broad peak at 324 cm−1. Both peaks appear at similar positions as for the stretching and bending vibrations (Cl–Pd–Cl) of PdCl2 or the ν(Pd–Cl) of PdCl42− or its hydrolysis products, suggesting that both the edge and the core of the pellets are rich in chlorinated Pd species.38 The higher intensity of these peaks in the core also indicates a higher concentration of these species in the core, which agrees with the higher intensity of the UV-Vis band at 428 nm in the center compared to the edge of the pellet illustrated in Fig. 9. On the other hand, the spectrum measured in position (2) of the calcined 1 wt%Pd-A1 pellet (Fig. 10a) shows, besides the two peaks at 252 and 324 cm−1, several broad peaks at 452, 585 and 637 cm−1 that can be assigned to PdO or PdOx/2Cl2−x.38,39 The presence of more oxidic Pd species in this inner ring also agrees with the browner color of PdO which is observed in the photograph.
Very similar spectra were measured on the calcined 1 wt%Pd-B4 pellet, as illustrated in Fig. 10b. A brown color was observed at the edge of this pellet which agreed with the presence of PdO or PdOx/2Cl2−x deduced from the Raman peaks at 452, 585 and 637 cm−1.38,39 Moreover, the core of the pellet showed a broad band centered at around 324 cm−1. The band at 258 cm−1 was also measured, but its intensity relative to the peak at 324 cm−1 was lower than in the 1 wt%Pd-A1 pellet. This suggests the presence of PdCl42− or its hydrolysis products.38,39 However, the Pd structure is slightly different for the two samples.
It must be noted that the spatial resolution of Raman microscopy is 25 times higher than that of UV-Vis micro-spectroscopy, which is the reason that the speciation of PdOx/2Cl2−x can be obtained from the Raman technique but not from UV-Vis micro-spectroscopy. Still, both techniques indicate that Cl− ions cannot be completely removed from the catalyst during the calcination step. Residual chlorine blocking the active sites of the Pd surface is known to have a detrimental effect on the catalyst performance.40–42 In order to remove the Cl− ions from the catalysts, higher temperatures or additional activation steps are required; and, consequently, a compromise between the desired particle size of Pd and the Cl− concentration in the catalytic materials should be reached.41,42
Conclusions
An experimental protocol to design Pd catalyst bodies with different Pd profiles (from egg-shell to uniform passing through egg-white and egg-yolk) has been developed thanks to the use of UV-Vis micro-spectroscopy. It has been demonstrated that, immediately after impregnation, PdCl42− is not stable in contact with alumina and it undergoes hydrolysis to form PdCl2(HO)22− species. Therefore, even though the main interactions between a Pd precursor species and an alumina surface have an electrostatic nature, Pd changes its molecular structure as soon as it contacts the alumina support. Moreover, the molecular structure of Pd, before drying, depended on the concentration of Cl−(aq), the solution pH and equilibration time. In this way, solutions with a high concentration of Cl−(aq) combined with a long equilibration time after impregnation gave rise to egg-yolk or uniform Pd distributions. The former profile was stabilized at acidic pH and the latter at slightly higher pH. On the other hand, a low concentration of Cl−(aq) created egg-shell Pd distributions if the solution was acidic or egg-white Pd profiles if the solution pH was around 5.In a second stage of this study the protocol developed for 0.2 wt% Pd catalysts was successfully applied to catalysts with higher loading. To do so, it has to be kept in mind that the higher the Pd concentration, the shorter the equilibration times required before drying. Moreover, UV-Vis micro-spectroscopy revealed that due to the strong interactions between Pd species and the alumina surface during the first step of catalyst preparation, i.e., pore volume impregnation, the profiles achieved during this step were retained during the subsequent preparation steps; namely, drying and calcination. However, after drying, chlorinated Pd is regenerated and retained after calcination. Thus, Cl− is not completely eliminated during calcination and it appears closely attached to the Pd active phase.
Acknowledgements
The authors thank Dr A. Iglesias-Juez (Utrecht University) for fruitful discussions and C. Harvey (Utrecht University) for her help carrying out the Raman microscopic measurements. The authors acknowledge financial support of ASPECT-ACTS.References
- Synthesis of Solid Catalysts, ed. K. P. de Jong, Wiley-VCH, Weinheim, 2009 Search PubMed.
- Catalyst Preparation: Science and Engineering, ed. J. R. Regalbuto, CRC Press, Boca Raton, 2007 Search PubMed.
- A. V. Neimark, L. I. Kheifets and V. B. Fenelonov, Ind. Eng. Chem. Prod. Res. Dev., 1981, 20, 439 CrossRef.
- M. S. Heise and J. A. Schwarz, J. Colloid Interface Sci., 1985, 107, 237 CrossRef CAS.
- M. S. Heise and J. A. Schwarz, J. Colloid Interface Sci., 1986, 113, 55 CrossRef CAS.
- M. S. Heise and J. A. Schwarz, J. Colloid Interface Sci., 1988, 123, 51 CrossRef CAS.
- J. A. Schwarz and M. S. Heise, J. Colloid Interface Sci., 1990, 135, 461 CrossRef CAS.
- K. Bourikas, C. Kordulis and A. Lycourghiotis, Catal. Rev. Sci. Eng., 2006, 48, 363 CrossRef CAS.
- A. Lekhal, B. J. Glasser and J. G. Khinast, Chem. Eng. Sci., 2004, 59, 1063 CrossRef CAS.
- X. Liu, J. G. Khinast and B. J. Glasser, Chem. Eng. Sci., 2008, 63, 4517 CrossRef CAS.
- J. R. Regalbuto, A. Navada, S. Shadid, M. L. Bricker and Q. Chen, J. Catal., 1999, 184, 335 CrossRef CAS.
- B. Shelimov, J.-F. Lambert, M. Che and B. Didillon, J. Am. Chem. Soc., 1999, 121, 545 CrossRef CAS.
- B. N. Shelimov, J. F. Lambert, M. Che and B. Didillon, J. Mol. Catal. A: Chem., 2000, 158, 91 CrossRef CAS.
- L. Espinosa-Alonso, K. P. de Jong and B. M. Weckhuysen, J. Phys. Chem. C, 2008, 112, 7201 CrossRef CAS.
- A. M. Beale, S. D. M. Jacques, J. A. Bergwerff, P. Barnes and B. M. Weckhuysen, Angew. Chem., Int. Ed., 2007, 46, 8832 CrossRef CAS.
- J. A. Bergwerff, A. A. Lysova, L. Espinosa Alonso, I. V. Koptyug and B. M. Weckhuysen, Angew. Chem., Int. Ed., 2007, 46, 7224 CrossRef CAS.
- I. V. Koptyug, A. V. Khomichev, A. A. Lysova and R. Z. Sagdeev, J. Am. Chem. Soc., 2008, 130, 10452 CrossRef CAS.
- B. M. Weckhuysen, Angew. Chem., Int. Ed., 2009, 48, 4910 CrossRef CAS.
- J. A. Bergwerff, T. Visser, G. Leliveld, B. D. Rossenaar, K. P. de Jong and B. M. Weckhuysen, J. Am. Chem. Soc., 2004, 126, 14548 CrossRef CAS.
- L. G. van de Water, J. A. Bergwerff, T. A. Nijhuis, K. P. de Jong and B. M. Weckhuysen, J. Am. Chem. Soc., 2005, 127, 5024 CrossRef CAS.
- R. Maatman, Ind. Eng. Chem., 1959, 51, 913 CrossRef CAS.
- R. W. Maatman and C. D. Prater, Ind. Eng. Chem., 1957, 49, 253 CrossRef CAS.
- T. Mang, B. Breitscheidel, P. Polanek and H. Knözinger, Appl. Catal., A, 1993, 106, 239 CrossRef CAS.
- Y.-S. Shyr and W. R. Ernst, J. Catal., 1980, 63, 425 CrossRef CAS.
- V. N. Ananin and A. I. Trokhimetz, React. Kinet. Catal. Lett., 1985, 27, 129 CrossRef CAS.
- K. Attyia and N. Fouad, J. Therm. Anal., 1994, 42, 1207 CrossRef CAS.
- F. Bozon-Verduraz, A. Omar, J. Escard and B. Pontvianne, J. Catal., 1978, 53, 126 CAS.
- A. R. McInroy, D. T. Lundie, J. M. Winfield, C. C. Dudman, P. Jones, S. F. Parker and D. Lennon, Catal. Today, 2006, 114, 403 CrossRef CAS.
- L. I. Elding and L. F. Olsson, J. Phys. Chem., 1978, 82, 69 CrossRef CAS.
- A. B. P. Lever, Inorganic Electronic Spectroscopy, Elsevier Science B.V., Amsterdam, 1987 Search PubMed.
- L. I. Elding, Inorg. Chim. Acta, 1972, 6, 647 CrossRef CAS.
- C. Lepetit and M. Che, J. Mol. Catal. A: Chem., 1995, 100, 147 CrossRef CAS.
- J.-F. Lambert, M. Hoogland and M. Che, J. Phys. Chem. B, 1997, 101, 10347 CrossRef CAS.
- N. B. Milić and Ž. D. Bugarčić, Transition Met. Chem., 1984, 9, 173 CrossRef CAS.
- S. Y. Troitskii, M. A. Fedotov and V. A. Likholobov, Russ. Chem. Bull., 1993, 42, 634 Search PubMed.
- J. A. Mieth, J. A. Schwarz, Y. J. Huang and S. C. Fung, J. Catal., 1990, 122, 202 CrossRef CAS.
- A. B. Gaspar and L. C. Dieguez, Appl. Catal., A, 2000, 201, 241 CrossRef CAS.
- W. Guihua, Y. Xibin, C. Xiaowei, L. Hexing and Z. Zongrang, J. Raman Spectrosc., 2000, 31, 1051 CrossRef.
- S. S. Chan and A. T. Bell, J. Catal., 1984, 89, 433 CAS.
- T. Lear, R. Marshall, J. A. Lopez-Sanchez, S. D. Jackson, T. M. Klapotke, M. Baumer, G. Rupprechter, H.-J. Freund and D. Lennon, J. Chem. Phys., 2005, 123, 174706 CrossRef.
- Q. Lin, Y. Ji, Z.-D. Jiang and W.-D. Xiao, Ind. Eng. Chem. Res., 2007, 46, 7950 CrossRef CAS.
- D. Roth, P. Gélin, M. Primet and E. Tena, Appl. Catal., A, 2000, 203, 37 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Gaussian deconvolution of UV-Vis spectra; digital photographs. See DOI: 10.1039/b915753k |
| This journal is © the Owner Societies 2010 |