Multilayer nanoparticles with a magnetite core and a polycation inner shell as pH-responsive carriers for drug delivery
Miao
Guo
a,
Yu
Yan
a,
Xiaozhou
Liu
a,
Husheng
Yan
*a,
Keliang
Liu
*b,
Hongkai
Zhang
c and
Youjia
Cao
c
aKey Laboratory of Functional Polymer Materials, Ministry of Education; and Institute of Polymer Chemistry, Nankai University, Tianjin 300071, China. E-mail: yanhs@nankai.edu.cn; Fax: (+86) 22 2350 3510
bBeijing Institute of Pharmacology and Toxicology, Beijing 100850, China. E-mail: keliangliu@yahoo.com; Fax: (+86) 10 6821 1656
cKey Laboratory of Bioactive Materials, Ministry of Education; and College of Life Sciences, Nankai University, Tianjin 300071, China
First published on 21st December 2009
Abstract
Nanocarriers with multilayer core–shell architecture were prepared by coating a superparamagnetic Fe3O4 core with a triblock copolymer. The first block of the copolymer formed the biocompatible outermost shell of the nanocarrier. The second block that contains amino groups and hydrophobic moiety formed the inner shell. The third block bound tightly onto the Fe3O4 core. Chlorambucil (an anticancer agent) and indomethacin (an anti-inflammation agent), each containing a carboxyl group and a hydrophobic moiety, were loaded into the amino-group-containing inner shell by a combination of ionic and hydrophobic interactions. The release rate of the loaded drugs was slow at pH 7.4, mimicking the blood environment, whereas the release rate increased significantly at acidic pH, mimicking the intracellular conditions in the endosome/lysosome. This can be attributed to the disruption of the ionic bond caused by protonation of the carboxylate anion of the drugs and the swelling of the inner shell caused by protonation of the amino groups.
Introduction
Many therapeutic agents, while pharmacologically effective in treatment, also exhibit side-effects because of their toxicities. For example, cytotoxic compounds used in cancer therapy can kill target cells, but also normal cells in the body resulting in undesired side-effects. Meanwhile, the biological levels of organization present many barriers to the delivery of therapeutic agents, including target-specific localization, enhanced clearance, and overexpressed membrane-associated multi-drug resistance.1–4 Therefore, many therapeutic agents are limited in their clinical application. Nanocarriers have been considered for target-specific delivery of drugs to different sites in the body. These engineered nanoparticulate carriers offer some advantages: passive targeting due to the enhanced permeability and retention (EPR) effect, functionalized surface features for target-specific localization, the opportunity to develop nanocarriers that respond to physiological stimuli, the combination of non-invasive imaging and drug therapy to monitor effects in real time, and the opportunity to combine drugs with energy (heat, light, and sound) delivery for synergistic therapeutic effects.1–4 Ideally, the drug-loaded carriers shoudld show longevity without clearance by the reticuloendothelial system (RES) and without significant release of the drugs in the blood circulation (pH 7.4), and eventually accumulate in the required organ or tissue and then get into target cells, where the drugs are released responsively due to the intracellular microenvironments such as lower pH in endosomes (pH 5–6) and lysosomes (pH 4–5).Therapeutic agents have been loaded into nanocarriers by acidic-pH-induced cleavable covalent bonds.5,6 Alternatively and more commonly, drugs are loaded into the inner core of polymeric micelles by non-covalent hydrophobic interactions.7–9 Generally, compared to chemical attachment, non-covalent entrapment is convenient and easy to prepare, but possibly causes a lower loading capacity due to the low solubility of the drug in the hydrophobic core, which depends on the structures of the polymer and drug used, and poor kinetics of release of the drug from the drug–core solid solution if the glass transition temperature of the polymer is higher than the release temperature.7,8
Polymeric nanocarriers with embedded superparamagnetic iron oxide nanoparticles are attractive platforms as multi-functional agents (therapy and imaging).8–13 The iron oxide nanoparticles allow the nanocarriers to be used as agents for magnetic resonance imaging (MRI), magnetic targeting with the assistance of external magnetic field gradients and hyperthermia treatment in an alternating magnetic field.14,15 The polymeric coatings serve as drug-loading sites in the nanocarriers, ideally, with a stimuli-sensitive interaction with the loaded drugs, and/or confer biocompatible surfaces ensuring nontoxicity under physiological conditions and preventing recognition by the RES.8,9,16–20 In this paper, multi-functional nanocarriers with a superparamagnetic magnetite core and multilayer polymer shells were prepared by a simple and environmentally friendly method. The nanoparticles contain a biocompatible outermost shell and an inner shell composed of polycations, which may act as a pH-sensitive place for drug loading. Model drugs with a carboxylic acid group and a hydrophobic moiety were loaded into the inner shell containing amino groups and hydrophobic moieties by the combination of ionic bonding and hydrophobic interactions. The combination of the ionic bonding and hydrophobic interactions led to high loading affinity and thus high loading capacity. In a lower pH environment, protonation of the carboxylate anion of the drugs, which would disrupt the ionic bonding, and protonation of the amino groups of the inner shell, which would cause swelling of the shell, led to release of the drug with good kinetics.
Experimental
Materials
Methoxypoly(ethylene glycol) (MPEG–OH, 2000 Da, Aldrich) was dehydrated by azeotropic distillation of water in toluene, precipitated with cold ethyl ether, filtered, washed with ether, and dried under vacuum. (N,N-dimethylamino)ethyl methacrylate (DMAEMA, Acros) and (N,N-diethylamino)ethyl methacrylate (DEAEMA, Acros) were distilled over CaH2 under reduced pressure before use. 1,1,4,7,7-Pentamethyldiethylenetriamine (PMDETA) was purchased from Acros. Solketal methacrylate (SMA) were prepared by reaction of solketal (2,2-dimethyl-1,3-dioxolane-4-methanol) with methacryloyl chloride, according to our previous reports.21,22Chlorambucil (CLB) was purchased from Heumann PCS GmbH (Germany). Indomethacin (IND) was the product of Nanjing Tian Zun Chemical Inc. (Nanjing, China). Macroinitiator, MPEG–Br, was synthesized by reacting MPEG–OH with 2-bromoisobutyryl bromide (Aldrich) as reported previously.21Preparation of copolymer-coated Fe3O4 nanocarriers
Block copolymers MPEG-b-PDEAEMA-b-PGMA, MPEG-b-PDMAEMA-b-PGMA, PDEAEMA-b-PGMA and MPEG-b-PGMA were synthesized by atom transfer radical polymerization (ATRP) using similar procedures reported previously.21,22 The synthesis of MPEG-b-PDEAEMA-b-PGMA, as an example shown in Scheme 1, is described below.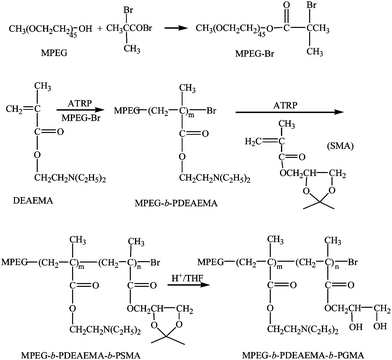 | ||
| Scheme 1 Synthesis of MPEG-b-PDEAEMA-b-PGMA. | ||
MPEG-Br (1.0 g, 0.5 mmol), CuBr (72 mg, 0.5 mmol) and cyclohexanone (2.5 mL) were added to a Schlenk flask and the system was evacuated and back-filled with N2 for 2 h. To this was added deoxygenated PMDETA (105 μL, 0.5 mmol) using a syringe and the mixture was stirred for 10 min. Then deoxygenated DEAEMA (3.6 g, 20 mmol) was added into the flask. After purging with N2 for another 2 h, the flask was sealed and immersed in an oil bath preheated to 70 °C. The polymerization was allowed to proceed for 12 h under stirring. The resulting viscous liquid was diluted with acetone (10 mL) and then filtered through a column packed with neutral alumina to remove the catalyst. The filtrate was concentrated by rotary evaporation under reduced pressure, and the polymer was precipitated into hexane and dried to give MPEG-b-PDEAEMA.
SMA (2.8 g, 15 mmol) and CuBr (36 mg, 0.25 mmol) were added to a Schlenk flask and the system was evacuated and back-filled with N2 for 2 h. To this was added deoxygenated PMDETA (52.3 μL, 0.25 mmol) using a syringe and the mixture was stirred for 10 min. This solution was then added to a Schlenk flask containing a deoxygenated solution of MPEG-b-PDEAEMA (2.0 g) in cyclohexanone (2 mL). After purging with N2 for another 2 h, the flask was sealed and immersed in an oil bath preheated to 90 °C. After stirring for 12 h, the reaction mixture was diluted with acetone (10 mL) and then filtered through a column packed with neutral alumina to remove the catalyst. The filtrate was concentrated by rotary evaporation under reduced pressure, and the polymer was precipitated into hexane and dried to yield MPEG-b-PDEAEMA-b-PSMA. Gel permeation chromatography (GPC) gave: Mn = 9.56 × 103, PDI = 1.21. The number of repeat units of the first block MPEG was 45 according to the average molecular weight of the original material MPEG given by the supplier. The numbers of repeat units of PDEAEMA and PSMA, respectively, were determined to be 26 and 21 by 1H NMR using MPEG as the reference based on the integration ratios of the peak at 2.61 ppm for the N,N-diethylamino group of PDEAEMA and the peaks at 1.39 and 1.45 ppm for the ketal group of PSMA to the peak at 3.60 ppm for the ethylene group of MPEG.
MPEG-b-PDEAEMA-b-PSMA (0.5 g) was dissolved in tetrahydrofuran (THF, 6 mL), followed by addition of 6 M HCl (1 mL). The mixture was stirred at ambient temperature for 2 h to remove the ketal protection group of PSMA. After most of the THF and HCl were removed by rotary evaporation under reduced pressure, the residue was dissolved in water and the solution was dialyzed (molecular weight cut-off, Mcut-off = 3500) against water and then was lyophilized to give MPEG-b-PDEAEMA-b-PGMA.
Magnetite nanoparticles were prepared by the coprecipitation of ferric and ferrous salts in anaerobic conditions at ambient temperature. A solution of FeCl2·4H2O (2.0 g, 10 mmol) in 25 mL of 1.0 M HCl and a solution of FeCl3·6H2O (5.4 g, 20 mmol) in 25 mL of double-distilled water were combined into a three-neck, round-bottom flask with magnetic stirrer and N2 protection. To this was added slowly 160 mL of 1.5 M ammonium hydroxide solution by a drop funnel and the color of the suspension turned black immediately. After stirring for 24 h under N2 protection, the resulting black precipitate was collected with a strong magnet and the supernatant was decanted. The resulting powder was washed with deoxygenated double-distilled water four times. The precipitate was peptized with 50 mL of 1.0 M HClO4 and then the mixture was dialyzed against water for 2–3 days, giving aqueous dispersion of magnetite nanoparticles stabilized with HClO4. The iron oxide content of the dispersion was 22.9 mg mL−1 determined by evaporating 1 mL of the dispersion to dryness.
An aqueous solution (10 mL) of MPEG-b-PDEAEMA-b-PGMA (0.25 g) was added to an aqueous dispersion of the magnetite nanoparticles stabilized with HClO4 (1.5 mL). The mixture was stirred overnight and dialyzed (Mcut-off = 14![[hair space]](https://www.rsc.org/images/entities/char_200a.gif) 000) against water to remove the unbound polymer, giving MPEG-b-PDEAEMA-b-PGMA-coated magnetite nanoparticles. MPEG-b-PDMAEMA-b-PGMA- and PDEAEMA-b-PGMA-coated Fe3O4nanoparticles were obtained with a similar method. The iron content of the nanoparticles was determined by spectrophotometric measurements at 340 nm using a TU1810PC UV–Vis spectrometer (Beijing Purkinje General Instrument Co. Ltd., China) after a 2 h digestion of the nanoparticles in 30% v/v HCl at elevated temperatures (50–60 °C) according to a literature method.17
000) against water to remove the unbound polymer, giving MPEG-b-PDEAEMA-b-PGMA-coated magnetite nanoparticles. MPEG-b-PDMAEMA-b-PGMA- and PDEAEMA-b-PGMA-coated Fe3O4nanoparticles were obtained with a similar method. The iron content of the nanoparticles was determined by spectrophotometric measurements at 340 nm using a TU1810PC UV–Vis spectrometer (Beijing Purkinje General Instrument Co. Ltd., China) after a 2 h digestion of the nanoparticles in 30% v/v HCl at elevated temperatures (50–60 °C) according to a literature method.17
The average particle size, size distribution and morphology of the polymer-coated nanoparticles were studied by transmission electron microscopy (TEM) and dynamic light scattering (DLS). TEM measurements were performed on a JEM-2010F transmission electron microscope (JEOL, Japan). A drop of well dispersed nanoparticles dispersion was placed onto an amorphous carbon-coated 200-mesh copper grid, followed by drying the sample at ambient temperature before it was loaded into the microscope. DLS analysis of the samples dispersed in aqueous media was carried out on a BI-200SM (Brookhaven, NY, USA) equipped with a BI-900AT digital correlator at 636 nm. Magnetization of the samples was measured as a function of the applied magnetic field H with a 9600VSM (LDJ, USA) superconducting quantum interference device (SQUID) magnetometer. The hysteresis of the magnetization was obtained by changing H between +6000 and −6000 Oe at 300 K.
Stability of the nanocarriers in the presence of oxalic acid
The stability of the aqueous dispersion of the nanocarriers was inferred from optical absorbance as a function of oxalic acid solution. Small aliquots of the dispersion of the copolymer-coated iron oxide nanoparticles were added to oxalic acid aqueous solutions (10 mL each) with final oxalic acid concentrations ranging from 0.01 to 0.05%. The mixture was stirred for several hours and then centrifuged at 6000 rpm for 10 min. The UV absorptions of the supernatants at 380 nm were determined.Cell viability assay of the nanocarriers
Cell viability assays were carried out using perviously reported procedures.17,23 OCTY (osteoblast-like cells) mouse cells were plated at a density of 1 × 104 cells/well in a 96-well plate at 37 °C in 5% CO2 atmosphere. After 12 h of culture, the medium in the wells was replaced with fresh medium containing the nanoparticles in the iron concentrations ranging from 0 to 400 mg mL−1. After 24 h, the medium was removed and the cells were washed three times with PBS to remove the nanoparticles and imaged using a fluorescent microscope. Then 20 μL of combined 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS)/phenazine methosulfate (PMS) solution (MTS solution: PMS solution = 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v, Cell-Titer 96® AQueous kit) was added to each well of the 96-well assay plates containing 100 μL of cells in culture medium. After incubating the plate for 2 h at 37 °C in a humidified 5% CO2 atmosphere, the absorbance at 490 nm was recorded using an ELISA plate reader. The spectrophotometer was calibrated to zero absorbance using culture medium without cells. The relative cell viability related to control wells containing cell culture medium without the nanoparticles was calculated by [A]test/[A]control, where [A]test is the absorbance of the test sample and [A]control is the absorbance of control sample.
:1, v/v, Cell-Titer 96® AQueous kit) was added to each well of the 96-well assay plates containing 100 μL of cells in culture medium. After incubating the plate for 2 h at 37 °C in a humidified 5% CO2 atmosphere, the absorbance at 490 nm was recorded using an ELISA plate reader. The spectrophotometer was calibrated to zero absorbance using culture medium without cells. The relative cell viability related to control wells containing cell culture medium without the nanoparticles was calculated by [A]test/[A]control, where [A]test is the absorbance of the test sample and [A]control is the absorbance of control sample.
Drug loading and release
Drug loading was carried out as described below. An aqueous solution of indomethacin sodium (10 mL, 1 mg mL−1) was added dropwise into the nanocarrier dispersion (35.4 mg of nanoparticles in 8 mL volume). After stirring overnight, the suspension was dialyzed (Mcut-off 14000) against water changed 10–15 times during 3 days to remove the free indomethacin sodium. The loading capacity was estimated by subtraction of the amount of indomethacin sodium in the collected outer solutions determined by a spectrophotometric method (254 nm) from the total amount of indomethacin sodium added. Chlorambucil sodium was loaded into nanocarriers with the same method, and the loading capacity was also determined spectrophotometrically, at 302 nm.
For the determination of release of the loaded indomethacin, the indomethacin-loaded nanocarrier dispersion (3 mL, ∼ 1.5 mg mL−1) was dialyzed (Mcut-off 14000), respectively, against phosphate buffer (10 mM phosphate, 50 mL) with or without 0.9% NaCl at a pH ranging from 4.5 to 7.4. At dialysis times of 1, 3, 5, 7, 10 and 24 h, the outer solution was replaced with fresh buffer. The concentration of indomethacin in the removed outer solution was determined by a spectrophotometric method at a wavelength of 254 nm (the measured absorbance values ranging from 0.122 to 0.982). All release measurements were carried out in triplicate for each sample, and an average value was adopted. The release of the loaded chlorambucil was determined by the same method (the measured absorbance values ranging from 0.103 to 0.931).
Results and discussion
Preparation and characterization of the nanocarriers
Four block copolymers were synthesized by ATRP using similar procedures to those reported previously21,22 and the structure of the block copolymers is shown in Table 1. The block copolymer-coated Fe3O4nanoparticles were prepared by a ligand-exchange method by adding an aqueous solution of the copolymer to an aqueous dispersion of Fe3O4nanoparticles stabilized with HClO4. The released HClO4 and the excess polymer were removed by dialysis. All block copolymer-coated Fe3O4nanoparticles had a similar appearance in the TEM micrographs. Fig. 1 shows the TEM images of MPEG-b-PDMAEMA-b-PGMA-Fe3O4 and MPEG-b-PDEAEMA-b-PGMA-Fe3O4nanoparticles. The average diameters of the core Fe3O4nanoparticles were estimated to be 9–10 nm from the images, which could not show the outside copolymers clearly. The average hydrodynamic diameters of MPEG-b-PDMAEMA-b-PGMA-Fe3O4 and MPEG-b-PDEAEMA-b-PGMA-Fe3O4nanoparticles dispersed in pure water were 27 and 30 nm, respectively, with narrow size distribution determined by dynamic light scattering (DLS) (Fig. 2). The average hydrodynamic diameters of MPEG-b-PDEAEMA-b-PGMA-Fe3O4nanoparticles dispersed in physiological saline (pure water containing 0.9% NaCl), pH 7.4 buffer (10 mM phosphate with 0.9% NaCl) and pH 4.5 buffer (10 mM phosphate with 0.9% NaCl) were also determined by DLS and their values were 30, 23 and 35 nm, respectively. It seems that the effect of the ionic strength on the average hydrodynamic diameter is negligible, while the average hydrodynamic diameter depends on the pH. Upon the pH decreasing from 7.4 to 4.5, the average hydrodynamic diameter increased from 23 to 35 nm. This can be attributed to the more stretched polymer chains at acidic pH due to the protonation of the amino groups of the PDEAEMA block.| Copolymer | Number of repeat units of PDEAEMA or PDMAEMAa | Number of repeat units of PGMAa | PDIb |
|---|---|---|---|
| a The numbers were derived from the numbers of the corresponding repeat units of the precursor polymers (see Experimental), which were determined by 1H NMR. The molecular weight of MPEG was 2000 Da, as given by the supplier. b Polydispersity index of the precursor polymers, as determined by GPC. | |||
| MPEG-b-PDEAEMA-b-PGMA | 26 | 21 | 1.21 |
| MPEG-b-PDMAEMA-b-PGMA | 23 | 25 | 1.19 |
| PDEAEMA-b-PGMA | 31 | 27 | 1.16 |
| MPEG-b-PGMA | — | 40 | — |
 | ||
| Fig. 1 TEM images of (a) MPEG-b-PDEAEMA-b-PGMA-Fe3O4nanoparticles and (b) MPEG-b-PDMAEMA-b-PGMA-Fe3O4nanoparticles. | ||
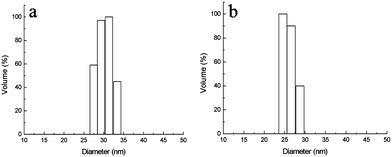 | ||
| Fig. 2 Size distributions of (a) MPEG-b-PDEAEMA-b-PGMA-Fe3O4nanoparticles and (b) MPEG-b-PDMAEMA-b-PGMA-Fe3O4nanoparticles dispersed in water, determined by DLS. Results are means (n = 3), and the SD values are (a) 0.5 nm and (b) 0.7 nm. | ||
We have previously shown that both MPEG and PDMAEMA have low affinities for Fe3O4nanoparticles: no stable magnetic fluid could be obtained when homopolymerPDMAEMA or MPEG was used as the stabilizer in aqueous media, whereas PGMA has a strong affinity for Fe3O4nanoparticles due to the cooperation of multi-dentate interactions of 1,2-diols on the polymer chain with iron atoms at the surface of the Fe3O4nanoparticles.21 Accordingly, MPEG-b-PDMAEMA-b-PGMA-Fe3O4 and MPEG-b-PDEAEMA-b-PGMA-Fe3O4nanoparticles dispersed in aqueous solution would be expected to possess a multi-layer core–shell architecture with a Fe3O4 core, to which the PGMA block was attached, while the PDMAEMA or PDEAEMA and MPEG blocks form the outside layer, extending in the aqueous matrix. This multi-layer core–shell architecture was further confirmed by the stability of dispersions of MPEG-b-PDEAEMA-b-PGMA-Fe3O4 and PDEAEMA-b-PGMA-Fe3O4nanoparticles in the presence of oxalic acid, as shown in Fig. 3. For the PDEAEMA-b-PGMA-Fe3O4 dispersion, the ionic interaction of oxalic acid with the outermost PDEAEMA shell of the particles led to inter-particle crosslinking and thus precipitate formation. In contrast, the ionic interaction of oxalic acid with the inner PDEAEMA shell of the MPEG-b-PDEAEMA-b-PGMA-Fe3O4nanoparticles could not lead to inter-particle crosslinking and thus no precipitate formed.
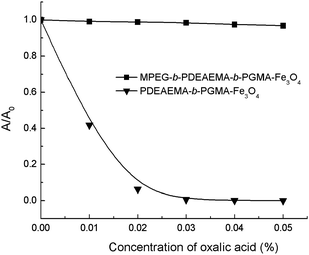 | ||
| Fig. 3 Stabilities of dispersions of MPEG-b-PDEAEMA-b-PGMA-Fe3O4 and PDEAEMA-b-PGMA-Fe3O4nanoparticles in the presence of oxalic acid. | ||
Magnetic measurement studies indicated that these polymer-coated iron oxide nanoparticles showed superparamagnetic behavior without magnetic hysteresis due to the small size of the Fe3O4 core. Fig. 4 shows the magnetization curves of MPEG-b-PDMAEMA-b-PGMA-Fe3O4 and MPEG-b-PDEAEMA-b-PGMA-Fe3O4nanoparticles. The saturation magnetization of these polymer-coated iron oxide nanoparticles expressed in pure iron oxide was lower than that of pure bulk magnetite (92 emu g−1), for example, the saturation magnetization for MPEG-b-PDEAEMA-b-PGMA-Fe3O4nanoparticles was 21.5 emu g−1 (Fig. 4), which is equivalent to 39.9 emu g−1 normalized to pure iron oxide. The decrease of the saturation magnetization is most likely attributed to the small particle size of the iron oxide nanoparticles and the disordered structure at the interface between the iron oxide nanoparticles and the coating.24
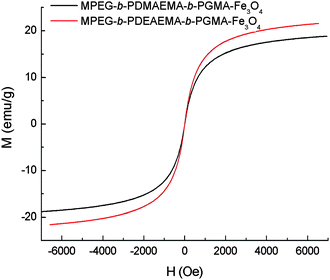 | ||
| Fig. 4 SQUID magnetization curves of MPEG-b-PDEAEMA-b-PGMA-Fe3O4 and MPEG-b-PDMAEMA-b-PGMA-Fe3O4nanoparticles at 300 K. | ||
Cytotoxicity of the nanocarriers
Nanoparticles with hydrophobic or charged surfaces could interact with proteins and cells upon contact with blood, showing cytotoxicity and recognition by the macrophages of the mononuclear phagocyte system (MPS).25 In particular, nanoparticles with positively charged surfaces show nonspecific cell sticking.26 In this paper, we used the PDMAEMA or PDEAEMA layer as the drug-loading sites in the particles. As expected, PDEAEMA-b-PGMA-Fe3O4nanoparticles with positive charges on their surfaces showed strong cytotoxicity, as shown in Fig. 5. Compared with the result published in our previous paper,17 positively charged PDEAEMA-b-PGMA-Fe3O4nanoparticles showed much higher cytotoxicity than the negatively charged PMAA-b-PGMA-Fe3O4 [PMAA = poly(methacrylic acid)] nanoparticles did. To circumvent this problem, Fe3O4nanoparticles were coated with MPEG-b-PDMAEMA-b-PGMA or MPEG-b-PDEAEMA-b-PGMA, of which the uncharged hydrophilic MPEG block formed the outermost layer, shielding the inner layer of PDMAEMA or PDEAEMA. Aqueous dispersions of MPEG-b-PDMAEMA-b-PGMA-Fe3O4 and MPEG-b-PDEAEMA-b-PGMA-Fe3O4nanoparticles showed much lower cytotoxicities than dispersions of the corresponding nanoparticles without the MPEG layer. Fig. 5 shows that the viability of cells after incubation with MPEG-b-PDEAEMA-b-PGMA-Fe3O4nanoparticles with iron concentrations ranging from 25 to 400 μg mL−1 was close to that of the control cells and varied in the range from 99% to 95%, whereas only 20% of the cells remained viable after incubation with PDEAEMA-b-PGMA-Fe3O4nanoparticles with iron concentration of 400 μg mL−1. Therefore, MPEG-b-PDEAEMA-b-PGMA-Fe3O4nanoparticles have no distinct effect on the cell viability as the iron concentration increased in the range studied, but PDEAEMA-b-PGMA-Fe3O4nanoparticles could markedly reduce the cell viability.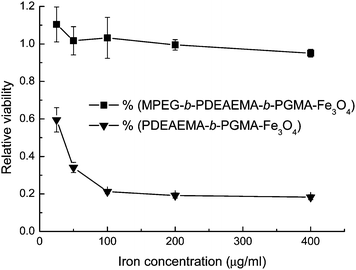 | ||
| Fig. 5 MTS assay viability of OCTY cells incubated with polymer-coated Fe3O4nanoparticles at 37 °C for 24 h. Results are means ± SD (n = 3). | ||
Drug loading studies
CLB, an anticancer agent, and IND, an anti-inflammation agent, each with a carboxylic acid group and a hydrophobic moiety, were used as model drugs for loading and controlled release. CLB and IND in salt form might interact with the nanocarriers by an ionic bond between the carboxylate anion of the drugs and the protonated amine cation of the carriers, meanwhile the hydrophobic moiety of the drugs might form hydrophobic interactions with the substituent groups of the amino groups and the main-chain of the polymer of the nanocarriers. Table 2 shows the drug loading capacities of the nanocarriers. The much higher loading capacities of MPEG-b-PDEAEMA-b-PGMA-Fe3O4 and MPEG-b-PDMAEMA-b-PGMA-Fe3O4 nanocarriers than that of the MPEG-b-PGMA-Fe3O4 nanocarrier (see Table 2) shows the ionic bond between the drugs and the former carriers existed. On the other hand, it was found that the carrier containing more hydrophobic PDEAEMA block had a higher loading capacity than that containing less hydrophobic PDMAEMA block by comparing the loading capacities of MPEG-b-PDEAEMA-b-PGMA-Fe3O4 and MPEG-b-PDMAEMA-b-PGMA-Fe3O4 nanocarriers. This result indicated that, in addition to the ionic bond mentioned above, hydrophobic interactions between the carriers and drugs also existed. From the data in Table 2, we also noticed that the MPEG layer led to a high loading capacity (comparing the loading capacities of MPEG-b-PDEAEMA-b-PGMA-Fe3O4 and PDEAEMA-b-PGMA-Fe3O4 nanocarriers in Table 2). This may be attributed to the MPEG layer creating a hydrophobic microenvironment within the carriers, leading to stronger hydrophobic interactions between the carriers and drugs. A similar interaction was observed in our previous paper.17| Copolymers | Loading of CLB | Loading of IND | ||
|---|---|---|---|---|
| mg mg−1 | mg mmol−1a | mg mg−1 | mg mmol−1 | |
| a mg of drugs per mmol of amino groups in the carriers. | ||||
| MPEG-b-PDEAEMA-b-PGMA | 0.132 ± 0.016 | 87.6 | 0.134 ± 0.009 | 88.9 |
| MPEG-b-PDMAEMA-b-PGMA | 0.110 ± 0.005 | 56.0 | 0.136 ± 0.015 | 69.2 |
| PDEAEMA-b-PGMA | 0.043 ± 0.007 | 16.1 | 0.058 ± 0.011 | 21.7 |
| MPEG-b-PGMA | 0.029 ± 0.003 | — | 0.031 ± 0.004 | — |
The above results show that the loading capacity was low if the loading was driven by the ionic bond alone (e.g., loading in PDEAEMA-b-PGMA-Fe3O4 nanocarriers) or the hydrophobic interactions alone (e.g., loading in MPEG-b-PGMA-Fe3O4 nanocarriers). However, if the loading was driven by the combination of these two interactions, the synergistic effect of them greatly enhanced the loading affinity and thus the loading capacity increased significantly (e.g., the loading in MPEG-b-PDEAEMA-b-PGMA-Fe3O4 nanocarriers).
Drug release studies
As shown above, the drugs were loaded into the carriers by the combination of an ionic bond and hydrophobic interactions. Once the pH of the medium decreases, the carboxylate anion of the drugs would be protonated to form uncharged free carboxylic acid and thus the ionic bond would disappear, while the hydrophobic interactions alone are weak, as shown above, due to the moderate hydrophobicity of both of the carriers and drugs. Therefore, the affinity of the drugs for the carriers at acidic pH would be low. Figs. 6 and 7 show the pH-responsive releases of CLB and IND from the drug-loaded MPEG-b-PDEAEMA-b-PGMA-Fe3O4 and MPEG-b-PDMAEMA-b-PGMA-Fe3O4 nanocarriers. It can be seen from the figures that the release of the drugs from both MPEG-b-PDEAEMA-b-PGMA-Fe3O4 and MPEG-b-PDMAEMA-b-PGMA-Fe3O4 nanocarriers was correlated with the pKa values of the drugs (the pKa values of carboxyl groups of CLB and IND were 5.8 and 4.5, respectively27,28). At pH values above the pKa, the release was slow due to the existence of both of the ionic bond and hydrophobic interactions between the drugs and carriers. For example, only 26% CLB was released from MPEG-b-PDEAEMA-b-PGMA-Fe3O4 carriers at pH 7.4 after 24 h. When the pH value of the medium decreased to the pKa value or below it, the carboxylate anion of the drugs would be protonated gradually and become uncharged free carboxylic acid, leading to the breaking of the ionic bond between the polymer chain and drugs, so the drugs were released rapidly. As shown in Fig. 6, 55% CLB or 40% IND was released from MPEG-b-PDEAEMA-b-PGMA-Fe3O4 carriers at pH 5.5 after 24 h, and 90% CLB or 75% IND was released from the carriers at pH 4.5 after 24 h. Similar results for MPEG-b-PDMAEMA-b-PGMA-Fe3O4 carrier can be observed from Fig. 7. The much higher release rate of CLB than IND at lower pH values can be mainly attributed to their different pKa values. CLB is also a derivative of aniline. The amino group of CLB may be protonated at acidic pH (e.g., pH 4.5) and this would also promote the release.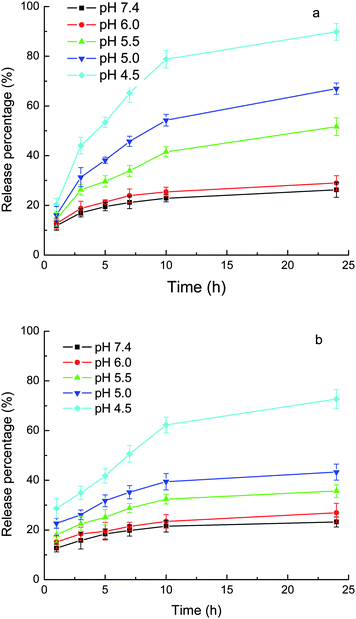 | ||
| Fig. 6 Release of loaded drugs from MPEG-b-PDEAEMA-b-PGMA-Fe3O4 nanocarriers in phosphate (10 mM) buffer with 0.9% NaCl. (a) CLB and (b) IND. Results are means ± SD (n = 3). | ||
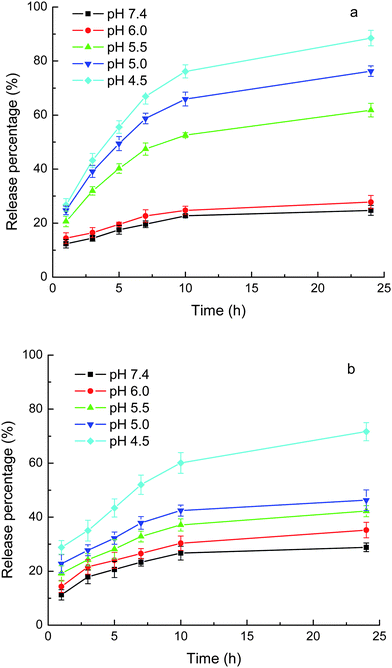 | ||
| Fig. 7 Release of loaded drugs from MPEG-b-PDMAEMA-b-PGMA-Fe3O4 nanocarriers in phosphate (10 mM) buffer with 0.9% NaCl. (a) CLB and (b) IND. Results are means ± SD (n = 3). | ||
Figs. 6 and 7 show that at a pH around pKa, the release rate of IND (pKa = 4.5) was much greater than that of CLB (pKa = 5.8) over the same release durations. The loaded IND was released at a significant magnitude even at pH values above the pKa but still acidic, e.g., more than 40% of the loaded IND was released from MPEG-b-PDEAEMA-b-PGMA-Fe3O4 carrier at pH 5.0 after 24 h. These results can be attributed to the high swelling degree of the drug-loading layer at acidic pH due to the protonation of the amino groups of the carriers, as shown above by the DLS results. The drugs can diffuse out more freely in the swollen nanocarriers. There have been several publications that report the pH-responsive release of drugs from micelles composed of PDMAEMA- or PDEAEMA-containing block copolymers due to the swelling of the micelles caused by the protonation of the PDMAEMA or PDEAEMA block.29–32 It is clear from this study that the influence of the decrease in pH on the release caused by the disruption of the ionic bond due to the protonation of the carboxylate anion of the drugs and the swelling of the carriers due to the protonation of the amines, have the same tendency.
The PDEAEMA layer in the MPEG-b-PDEAEMA-b-PGMA-Fe3O4 carriers should be more hydrophobic than the PDMAEMA layer in the MPEG-b-PDMAEMA-b-PGMA-Fe3O4 carriers. This led to the higher release rate from the MPEG-b-PDMAEMA-b-PGMA-Fe3O4 carriers than from the MPEG-b-PDEAEMA-b-PGMA-Fe3O4 carriers for the same drug under the same conditions (pH and release time), as shown in Figs. 6 and 7, due to the hydrophobic interactions between the drugs and carriers. Figs. 6 and 7 also show that the release rate of IND was greater than that of CLB from both of the carriers at pH 6–7.4, at which the strength of the ionic bonds for both of the drugs may be considered to be the same. The release rate difference may be attributed to the weaker hydrophobic interactions between the carriers and IND than that between the carriers and CLB due to the lower hydrophobicity of IND than that of CLB. Consequently, the release rate may be controlled by the hydrophobicity of the substituents at the N atom of the amine-containing drug-loading layer of the carriers according to the hydrophobicity of the drug used.
As mentioned above, the loading of the drugs into the carriers included ionic bonding. Therefore, the ionic strength should influence the release. The release medium shown in Figs. 6 and 7 was phosphate buffer containing 0.9% NaCl, which mimics the physiological environment. Fig. 8 shows the release of CLB and IND from MPEG-b-PDEAEMA-b-PGMA-Fe3O4 carriers in phosphate buffer without the addition of NaCl. Compared with the release in phosphate buffer containing 0.9% NaCl (see Fig. 6), the release rate of the same drug at the same pH and release duration without the addition of NaCl was lower.
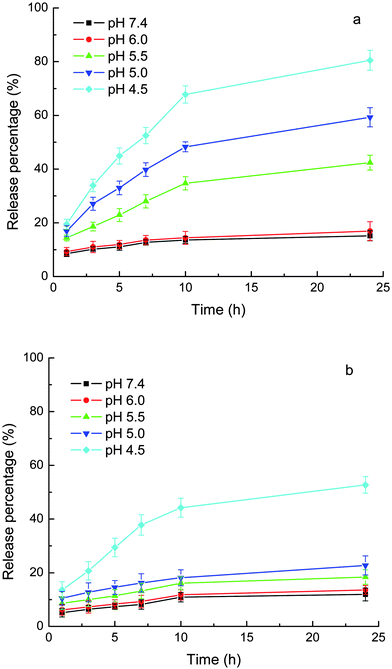 | ||
| Fig. 8 Release of loaded drugs from MPEG-b-PDEAEMA-b-PGMA-Fe3O4 nanocarriers in phosphate (10 mM) without added NaCl. (a) CLB and (b) IND. Results are means ± SD (n = 3). | ||
Conclusions
This article reports a type of novel nanocarrier with a Fe3O4nanoparticle as the core and a triblock copolymer as the shell. The triblock copolymer (viz. MPEG-b-PDEAEMA-b-PGMA or MPEG-b-PDMAEMA-b-PGMA) was attached tightly onto the surface of the Fe3O4nanoparticles by a PGMA block. Carboxyl- and hydrophobic moiety-containing drugs, CLB and IND, were loaded into the amine-containing inner shell composed of PDEAEMA/PDMAEMA block by a combination of an ionic bond and hydrophobic interactions. The biocompatible MPEG block formed the outermost shell of the carriers. It was also found that the MPEG layer created a hydrophobic microenvironment, which enhanced the loading capacity. The release of the loaded drugs from the nanocarriers was pH-responsive. In phosphate buffer containing 0.9% NaCl (pH 7.4), which mimicks the blood environment, the release rate of the loaded drugs was slow. Upon a decrease in the pH of the release medium, the release rate increased due to the influence of two factors. The first factor was that, at acidic pH, protonation of the carboxylate anion of the drugs broke the ionic bond between the carrier and drug. The second factor was the swelling of the PDEAEMA or PDMAEMA layer caused by protonation of the amino groups at acidic pH.Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant No. 20974052), the National Key Technologies R & D Program for New Drugs of China (Grant No. 2009ZX09301-002), and the Natural Science Foundation of Tianjin Municipality (Grant No. 09JCZDJC22900).References
- R. Langer, Science, 1990, 249, 1527–1533 CrossRef CAS.
- S. Ganta, H. Devalapally, A. Shahiwala and M. Amiji, J. Controlled Release, 2008, 126, 187–204 CrossRef CAS.
- X. Wang, L. Yang, Z. Chen and D. M. Shin, Ca–Cancer J. Clin., 2008, 58, 97–110 Search PubMed.
- V. P. Torchilin, AAPS J., 2007, 9, E128–E147 Search PubMed.
- Y. Bae, S. Fukushima, A. Harada and K. Kataoka, Angew. Chem., Int. Ed., 2003, 42, 4640–4643 CrossRef CAS.
- J. Ko, K. Park, Y. S. Kim, M. S. Kim, J. K. Han, K. Kim, R. W. Park, I. S. Kim, H. K. Song, D. S. Lee and I. C. Kwon, J. Controlled Release, 2007, 123, 109–115 CrossRef CAS.
- X. Shuai, H. Ai, N. Nasongkla, S. Kim and J. Gao, J. Controlled Release, 2004, 98, 415–426 CrossRef CAS.
- N. Nasongkla, E. Bey, J. Ren, H. Ai, C. Khemtong, J. S. Guthi, S. F. Chin, A. D. Sherry, D. A. Boothman and J. Gao, Nano Lett., 2006, 6, 2427–2430 CrossRef CAS.
- J. Yang, C. H. Lee, H. J. Ko, J. S. Suh, H. G. Yoon, K. Lee, Y. M. Huh and S. Haam, Angew. Chem., Int. Ed., 2007, 46, 8836–8839 CrossRef CAS.
- J. R. McCarthy and R. Weissleder, Adv. Drug Delivery Rev., 2008, 60, 1241–1251 CrossRef CAS.
- C. Sun, J. S. H. Lee and M. Zhang, Adv. Drug Delivery Rev., 2008, 60, 1252–1265 CrossRef CAS.
- Q. Yuan, R. Venkatasubramanian, S. Hein and R. D. K. Misra, Acta Biomater., 2008, 4, 1024–1037 CrossRef CAS.
- T. K. Jain, J. Richey, M. Strand, D. L. Leslie-Pelecky, C. A. Flask and V. Labhasetwar, Biomaterials, 2008, 29, 4012–4021 CrossRef CAS.
- S. Mornet, S. Vasseur, F. Grasset and E. Duguet, J. Mater. Chem., 2004, 14, 2161–2175 RSC.
- W. J. M. Mulder, A. W. Griffioen, G. J. Strijkers, D. P. Cormode, K. Nicolay and Z. A. Fayad, Nanomedicine, 2007, 2, 307–324 CrossRef CAS.
- F. Hu, L. Wei, Z. Zhou, Y. Ran, Z. Li and M. Gao, Adv. Mater., 2006, 18, 2553–2556 CrossRef CAS.
- M. Guo, Y. Yan, H. Zhang, H. Yan, Y. Cao, K. Liu, S. Wan, J. Huang and W. Yue, J. Mater. Chem., 2008, 18, 5104–5112 RSC.
- B. Wang, C. Xu, J. Xie, Z. Yang and S. Sun, J. Am. Chem. Soc., 2008, 130, 14436–14437 CrossRef CAS.
- J. H. Park, G. von Maltzahn, E. Ruoslahti, S. N. Bhatia and M. J. Sailor, Angew. Chem., Int. Ed., 2008, 47, 7284–7288 CrossRef CAS.
- X. Yang, Y. Chen, R. Yuan, G. Chen, E. Blanco, J. Gao and X. Shuai, Polymer, 2008, 49, 3477–3485 CrossRef CAS.
- S. Wan, Y. Zheng, Y. Liu, H. Yan and K. Liu, J. Mater. Chem., 2005, 15, 3424–3430 RSC.
- Y. Zhen, S. Wan, Y. Liu, H. Yan, R. Shi and C. Wang, Macromol. Chem. Phys., 2005, 206, 607–612 CrossRef CAS.
- S. Wan, J. Huang, M. Guo, H. Zhang, Y. Cao, H. Yan and K. Liu, J. Biomed. Mater. Res. A, 2007, 80, 946–954 CrossRef.
- S. H. Huang, M. H. Liao and D. H. Chen, Biotechnol. Prog., 2003, 19, 1095–1100 CrossRef CAS.
- R. H. Muller, Colloidal Carriers for Controlled Drug Delivery and Targeting: Modification, Characterization and In Vivo Distribution, CRC Press, Boca Raton, 1991 Search PubMed.
- T. Fujita, M. Nishikawa, Y. Ohtsubo, J. Ohno, Y. Takakura, H. Sezaki and M. Hashida, J. Drug Targeting, 1994, 2, 157–165 Search PubMed.
- R. B. Mikkelsen, C. Asher and T. Hicks, Biochem. Pharmacol., 1985, 34, 2531–2534 CrossRef CAS.
- A. Nokhodchi, Y. Javadzadeh, M. R. Siahi-Shadbad and M. Barzegar-Jalali, J. Pharm. Pharmaceut. Sci., 2005, 8, 18–25 CAS.
- Y. Shen, Y. Zhan, J. Tang, P. Xu, P. A. Johnson, M. Radosz, E. A. Van Kirk and W. J. Murdoch, AIChE J., 2008, 54, 2979–2989 CrossRef CAS.
- J. O. You and D. T. Auguste, Biomaterials, 2008, 29, 1950–1957 CrossRef CAS.
- M. Oishi, H. Hayashi, M. Iijima and Y. Nagasaki, J. Mater. Chem., 2007, 17, 3720–3725 RSC.
- Y. Tang, S. Y. Liu, S. P. Armes and N. C. Billingham, Biomacromolecules, 2003, 4, 1636–1645 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2010 |